
Abstract
The last decade has witnessed the reawakening of cancer metabolism as a therapeutic target. In particular, inhibition of pyruvate dehydrogenase kinase (PDK) holds remarkable promise. Dichloroacetic acid (DCA), currently undergoing clinical trials, is a unique PDK inhibitor in which it binds to the allosteric pyruvate site of the enzyme. However, the safety of DCA as a drug is compromised by its neurotoxicity, whereas its usefulness as an investigative tool is limited by the high concentrations required to exert observable effects in cell culture. Herein, we report the identification – by making use of saturation-transfer difference NMR spectroscopy, enzymatic assays and computational methods – of furoate and thenoate derivatives as allosteric pyruvate-site-binding PDK2 inhibitors. This work substantiates the pyruvate regulatory pocket as a druggable target.
Introduction
Metabolic reprogramming is essential for the viability of cells trapped in a pathological state of constitutive growth and proliferationCitation1. New findings are shedding light on the mechanisms orchestrating such reprogramming and favour a comprehensive paradigm that recognizes an altered metabolism as a core hallmark of cancerCitation2,Citation3. The signalling and metabolic pathways implicated in cancer bioenergetics are thus (re)emerging as promising therapeutic targetsCitation4.
Cancer cells tend, for instance, to divert glucose towards cytosolic lactic acid production, a phenomenon historically referred to as the Warburg effectCitation5. In turn, inhibitors of protein dehydrogenase kinase (PDK), whose isoforms 1 and 3 are upregulated in several cancersCitation6, have been shown to reverse this phenomenon, hence hampering tumours from meeting their abnormal metabolic requirementsCitation7.
PDK is a tightly regulated mitochondrial kinase that inhibits the activity of the pyruvate dehydrogenase complex (PDC) via reversible phosphorylationCitation8. Several allosteric sites have been identified in its regulatory domain, including the pyruvate-binding pocket, which induces the inactivation of the enzyme as pyruvate concentration raisesCitation9. Synthetic PDK inhibitors have been discovered for all four binding sites that are large enough to host small organic moleculesCitation10–16. However, dichloroacetic acid (DCA) remained to date the only bona fide pyruvate-site PDK synthetic inhibitorCitation10–12.
DCA has already shown significant promise in vivoCitation8, either alone or in combination therapyCitation17. However, the limited clinical studies conducted so far remain insufficient to appraise the therapeutic value of the compoundCitation18. Moreover, several reports caution about its potential neurotoxic side effectsCitation19. The usefulness of DCA as an investigative chemical probe for the study of cancer metabolism is also limited as a consequence of the high concentrations (ranging from 17 to 50 mM) required in order to observe a significant effect in cell cultureCitation20.
The aim of this study was to determine whether or not the pyruvate regulatory site of PDK is a druggable pocket with a view to develop novel inhibitors with improved therapeutic profiles as compared to DCA. Advantages of targeting the pyruvate pocket include the fact that it is a well-defined and largely hydrophobic pocketCitation10 and that inhibitors binding to this site are noncompetitiveCitation7.
Materials and methods
Starting materials and reagents were purchased from Aldrich, Acros, Alfa Aesar or Apollo Scientific. All solvents were reagent-grade and, if necessary, dried and distilled prior to use. All reactions were run under a nitrogen atmosphere unless otherwise stated. Column chromatography was performed using silica gel (Silicycle® SiliaSepTM 40–63 μm). TLC was performed with silica gel 60/Kieselguhr F254. 1H-NMR and 13C-NMR spectra were recorded at 400 MHz on a Varian AMX400 spectrometer (400 MHz for 1H, 101 Hz for 13C) at 25 °C. Chemical shifts (δ) are reported relative to the residual solvent peak (CD3OD, 1H = 3.31; 13C = 49.15. (CD3)2SO, 1H = 2.50; 13C = 39.52). Splitting patterns are indicated as (s) singlet and (d) doublet. High-resolution mass spectra were recorded with an LTQ Orbitrap XL (Thermo Fisher Scientific) mass spectrometer, using ESI as ionization source. FT-IR were measured on PerkinElmer FT-IR spectrometer. Melting points (mp) were measured with a Buchi melting point B-545. Some of the compounds were purchased from Sigma (2), Maybridge (4), Apollo (5, 12, 16), Acros (6, 13, 14), and Alfa Aesar (15) and used without further purification after confirming their identity and purity by 1H-NMR. Compounds 7Citation21, 3Citation22, 17Citation23 were prepared according to literature procedures and their spectral data are consistent with the literature. Proof of purity was obtained for all compounds subjected to biochemical assays (>95% purity by HPLC).
Experimental
Model building
In order to fill the missing regions in the template, a model of human PDK2 was built with Modeller 9v2 using the crystal structure 2BU810 as a template. ADP, DCA, Mg+2 and the coordinated water molecules were kept from the template, whereas a K+ ion bound to the potassium binding site and one coordinated water molecule were added from 3CRK. The added loops were refined with Modeller. The resulting model, solvated within a water box 14 Å larger than the molecular system dimensions and neutralized with 0.15 M KCl, was relaxed using NAMD by means of an alternate series of energy minimizations and molecular dynamics simulations using periodic boundary conditions (PBC) and the canonical ensemble. First, only the bulk solvent and ions were allowed to relax, next, different parts of the system were consecutively freed in the following order: hydrogen atoms, loops, side chains and, finally, the entire system was relaxed. Initial CHARMM general force field parameters for DCA were derived using the SwissParam server and the initial topology was refined on the basis of PCM/MP2/6-31 + g(d,p) calculations carried out with GAMESSCitation24. The quality of the final and intermediate protein models was evaluated with MolProbity.
Calculation of binding enthalpies
The geometry of all the described ligands was optimized at the PCM/MP2/6-31 + g(d,p) level of theory with GAMESSCitation24. The compounds were docked using AutoDock VinaCitation25 into the pyruvate site of the protein structure model built from 2BU810 as described previously. The enthalpies of binding were reevaluated for each complex at the COSMO/PM6-DH+//COSMO/PM6-DH2X level of theory (the full complexes were minimized with PM6-DH+Citation26 and single-point calculations on the minimized structures were then carried out with PM6-DH2XCitation27) using MOPAC2012Citation28. Two alternative poses for each ligand were evaluated. The data reported correspond to that with the most favourable PM6-DH2X binding energy.
Ligand efficiency
Solvent accessible surface areas (ASAs) were computed with ASVCitation29 taking the van der Waals radii from Mantina et al.Citation30
Overexpression and purification of PDC proteins
Recombinant human E1 mutant (the α subunit with only phosphorylation site 1 functional and the serine residues at sites 2 and 3 replaced with alanineCitation31) and wild-type human E3Citation32 were overexpressed in E. coli BL21 cells, purified using Ni2+-NTA-agarose chromatography and eluted with a linear 50–200 mM imidazole gradient. The resulting proteins were ∼95% pure as judged by SDS-PAGE densitometry. Recombinant human E2-E3BP was co-expressed and purified from BL21 cells harbouring pP-DHE2/E3BP as described by Yang et al.Citation33 with minor modificationsCitation34. Human PDK2 was amplified from cDNA made from HEK293 cells and subcloned into pET-28b. Recombinant human PDK2 was purified from BL21 cells transformed with pET-28b expression vector as described previouslyCitation34,Citation35.
PDC activity assay
PDK inhibitors were dissolved in DMSO to the concentration of 100 mM. Subsequent dilutions were done with double distilled water. DMSO had no effect on PDC activity. All the inhibitors were investigated for their ability to alter the PDC activity. None of the inhibitors affected PDC activity when used alone or in the presence of PDK2 (without added ATP). The PDC assay took the advantage of PDK-dependent phosphorylation of the α subunit of E1 resulting in the inhibition of PDC activity. Phosphorylation of mutant E1 (the α subunit with only phosphorylation site 1 functional and the serine residues at sites 2 and 3 replaced with alanine) was measured by inactivation of PDC by PDK2Citation31. Since there was only one functional phosphorylation site (site 1) in the mutant E1, the rate of its phosphorylation correlated with the rate of PDC inactivation. In the two-step assay, the PDK assay was conducted first with the known PDC activity in the presence of PDK2 causing phosphorylation and hence inactivation of PDC. In the second step, the remaining PDC activity is measured using PDC assayCitation31. PDC proteins with final concentrations of: 0.1 mg/mL E1, 0.1 mg/ml E2-E3BP, 0.05 mg/mL dihydrolopoamide dehydrogenase component (E3) and 0.15 mg/mL PDK2 were incubated in a premix (20 mM KHPO4, pH 7.5, 1 mM MgCl2, 0.05 mM EDTA, 1 mM dithiothreitol) for 10 min at 4 °C to allow formation of the PDC complex. Aliquots (10 μL) were taken and 3 μL of 0.25 mM ATP was added to start PDK2 reaction, and after 2 min 10 μL was transferred to PDC assay mixture. PDC activity was measured in 1 mL reaction mixture (50 mM potassium phosphate, pH 7.5, 2 mM MgCl2, 1.4 mM NAD+, 500 mM thiaminpyrophosphate, 4 mM cysteine and 156 μM coenzyme A). The reaction was started by the addition of pyruvate to a final concentration of 2 mM and the change in absorbance at 340 nm/min was measured to calculate PDC activityCitation34.
Dynamic light scattering
Dynamic light scattering (DynaPro NanoStar; Wyatt) was used to characterize the purified protein sample. The sample (5 mg/mL) was centrifuged for 5 min at 14 000×g. The analyses showed two molecular weight populations (two peaks). One, 98% of the mass, represents the PDK2 dimer with a polydispersity of ∼40% based on the mass. The second peak, ∼2% of the mass represents a much larger complex with very high polydispersity such as protein aggregate. Storage for several days on ice in the cold room showed comparable DLS results, indicating that the purified protein sample is stable under these conditions.
STD-NMR binding experiments
All 1H-STD-NMR experiments were performed at 10 °C on a Varian 600 MHz spectrometer. The on-resonance irradiation on PDK2 was set at –1 ppm. In each experiment, one ligand and PDK2 were present. The final concentrations of ligand and PDK2 were 400 μm and 10 μm, respectively, ([PDK2]:[ligand] = 1:40). First, the 1H-NMR spectra of ligands in HEPES buffer (20 mM HEPES of pH 8, 100 mM KCl, 10 mM β-mercaptoethanol and 0.1 M EDTA in deuterated water), in presence of protein (off-resonance 1H NMR spectra) were recorded. Subsequently, on-resonance 1H-NMR spectra were recorded with 1000 scans. 1H-STD-NMR spectra were obtained by subtracting the on-resonance spectrum from the corresponding off-resonance spectrum (Figures S5–S10). 1H-STD-NMR analysis revealed that all six ligands bind to the protein.
1H-STD-NMR competition experiments
To confirm that all the ligands bind to the same pocket of PDK2 as DCA, a competition experiment was performed using 8 (which has the highest activity from all ligands investigated). First, the 1H-STD-NMR of 400 μM 8 was recorded as mentioned above. Subsequently, 400 μM DCA was added to the same NMR tube and a second 1H-STD NMR spectrum was recorded. The second 1H-STD-NMR spectrum showed the appearance of DCA peaks and disappearance of peaks from 8, which indicated that 8 was displaced by DCA from the binding pocket of PDK2 (Figure S11). This experiment confirms that both 8 and DCA bind to the same binding pocket of the enzyme.
Results and discussion
By making use of structure-based design and virtual screening techniques, we have identified a number of chemical scaffolds as potential inhibitors of PDK targeting the pyruvate-binding site. Herein, we report our first enzymatic activity results with furoate and thenoate derivatives. displays the structures of the compounds, which were either synthesized or purchased and subjected to biochemical evaluation.
Figure 1. Structures of pyruvic acid (1) and the tested compounds.
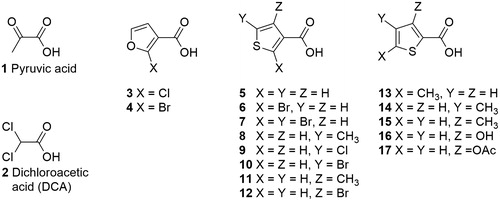
The rationale for privileging such scaffolds at the proof-of-concept phase is two-fold: On the one hand, their structures resemble that of pyruvate but are more versatile in terms of derivatisation; and, on the other hand, furoates are known to be intermediates in the energy metabolism of certain microorganismsCitation36.
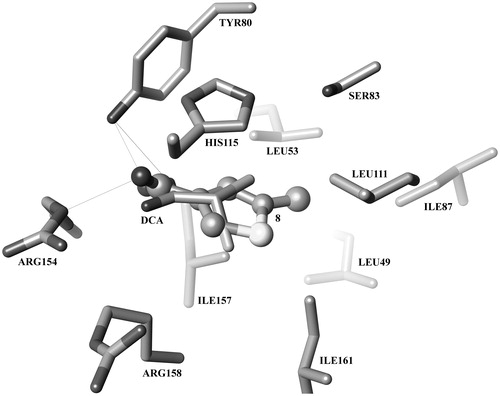
Furthermore, in terms of molecular recognition, molecular docking calculations predict furoates and thenoates to bind to the pyruvate site of PDK2 in a pose resembling that observed for DCA in the crystal structure with PDBID 2BU8Citation10. shows the binding mode of DCA (2) within the pyruvate-binding site of PDK2. The predicted binding pose for the most active compound of the series, 8, is also displayed (balls and sticks). A carboxylate or other negatively charged functional group seems to be fundamental for the initial recognition by gate residues Arg154 and Arg158 (human PDK2 numbering is used, unless otherwise stated), in addition to establishing a hydrogen bond/salt bridge network with Tyr80, Arg154 and, potentially, Arg158. In turn, the aromatic moiety is predicted to establish a π–π-stacking interaction with the side chain of His115. Furthermore, the methyl substituent allows this compound to maintain additional van der Waals interactions with Leu49 and Ile87.
Figure 2. Predicted binding mode of compound 8 (balls and sticks) superimposed with the PDK2/DCA co-crystal structure 2BU8 (sticks).
The inhibitory activity of the reported compounds was determined indirectly by means of the PDC assayCitation31. PDC activity is regulated by phosphorylation/dephosphorylation of the α subunit of the pyruvate dehydrogenase component (E1) on three specific serine residues: SER264 (site 1), SER271 (site2) and SER203 (site 2). PDK2 inhibits PDC by phosphorylating sites 1 and 2, but not 3Citation34. In order to observe a more linear effect between E1α phosphorylation and PDC inactivation, and provided that PDK2 presents the highest affinity towards site 1, sites 2 and 3 were mutated to alanineCitation31.
PDC activity was evaluated, respectively, in the absence of both PDK2 and the inhibitors; in the presence of each of the inhibitors but without PDK2 (this permitted us to exclude a direct effect of our compounds on PDC activity); in the presence of fully-active PDK2 with no inhibitor; and, finally, in the presence of both PDK2 and each of the inhibitors at concentrations ranging from 1 μM to 1 mM. The results are summarized in and represent the percentage of restoration of PDC activity in the presence of the inhibitor, thus [100 × (APDC + PDK2 + Inhibitor – APDC + PDK2)/(APDC – APDC + PDK2)], where APDC is the activity of PDC alone, APDC + PDK2 is the activity of PDC in the presence of PDK2, and APDC + PDK2 + Inhibitor is the activity of PDC in the presence of both PDK2 and the respective inhibitor at a given concentration.
Table 1. Percentage restoration of PDC activity at different concentrations of PDK inhibitors with respect to fully active PDK2. Enthalpies of binding computed at the PM6-DHX2 level of theory. Ligand efficiency.
As it can be seen in , DCA (2) is the most active compound, being able to restore, through PDK2 inhibition, 50% of PDC activity at 0.01 mM and 100% at 0.1 mM. Our most active compound in these series is thenoate 8, which restored 75% of PDC activity at 0.1 mM. In turn, compounds 4, 11, 12 and 15 were able to restore 50% of the basal activity at 0.1 mM and 100% at 1 mM. Finally, compound 5 restored complete PDC activity at 1 mM but showed no inhibitory activity at lower concentrations. None of the thenoates and furoates showed any sizeable inhibitory activity at 0.01 mM.
1H Saturation-transfer difference NMR (1H-STD-NMR) experiments confirmed that 2, 4, 5, 8, 11 and 12 bind directly to PDK2. In addition, we employed 1H-STD-NMR spectroscopy to carry out competition assays between DCA (2) and 8, which revealed that DCA displaces 8 from the binding site of the enzyme. This experiment demonstrated that both DCA and 8 bind to the same pocket of the enzyme. Given that DCA is a bona fide pyruvate-site inhibitorCitation10–12, we conclude that, as predicted by the computational studies, 8 exerts its inhibitory action by binding to the allosteric pyruvate regulatory site of the enzyme.
In terms of structure–activity relationships (SARs), it is noteworthy that four (4, 11, 12 and 15) out of the five most potent compounds bear either a methyl or a bromo substituent in ortho position with respect to the carboxylate moiety and that 5, the only tested compounds with no substituents, was active only at higher concentrations. Comparing the activities of 3 and 4, we observed that while the 2-bromo derivative 4 is active, the 2-chloro derivative 3 is not. However, the thenoate 6 bearing the same substitution pattern as furoate 4 is essentially inactive. Instead, both 4-bromo (12) and 4-methyl (11) substitutions are beneficial, as it is 5-methyl (8). The presence of either a bromine or a chlorine atom at the 5th-position of the series of thenoates (compounds 10 and 9, respectively) causes complete loss of activity. As for the 2-thenoate series, the most active compound, 15, has a methyl group ortho to the carboxylate moiety, whereas 16, with a hydroxyl group at the same position, is less active.
In order to evaluate the enthalpy of binding (ΔHB) for each protein–ligand complex, the missing regions where added to 2BU8 by homology modelling and then the structure was relaxed through a series of energy minimization and molecular dynamics simulations cycles. Each protein–ligand complex was then minimized with different semi-empirical parameterization models, both with the COSMO solvent model and in the gas phase. The enthalpy of binding was computed as [ΔHB=ΔHC− (ΔHP+ΔHL)], where ΔHB is the enthalpy of binding, ΔHC the heat of formation of the protein–ligand complex, ΔHP the heat of formation of the protein and ΔHL the heat of formation of the ligand. Ligand efficiencyCitation37 was then calculated with the formula [–ΔHB× (ASAC/ASAL)], where ΔHB is the computed enthalpy of binding, ASAC is the solvent-ASA of the carbon atom and ASAL is that of the ligand. This is approximately equivalent to the enthalpy of binding per atom adjusted to take atom size into account. Enthalpies of binding and ligand efficiency data are summarized in .
shows the correlation between enzymatic activity and ligand efficiency computed at the COSMO/PM6-DH2X level of theory. It must be noted that the chosen methodology has a number of limitations, which include neglecting the entropic term of the free energy of binding and the inaccurate estimation of the van der Waals interactions with semi-empirical methods.
Figure 3. Correlation between percent restoration of PDC activity at 0.1 mM (x-axis) and computed ligand efficiency (y-axis).
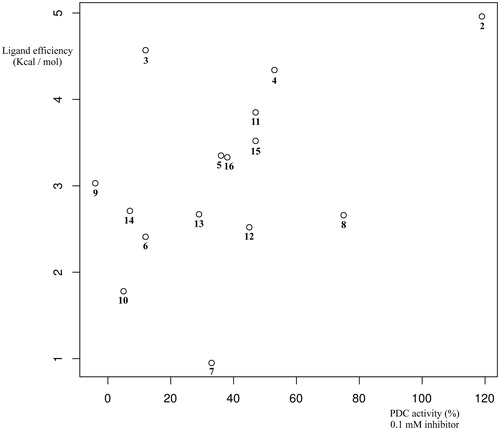
It is, however, noteworthy that both COSMO/PM6-DH + and COSMO/PM6-DH2X correctly predict that DCA is the most efficient ligand. Moreover, the higher level of theory employed, the better the statistical correlation between enzymatic activity and computed ligand efficiency we observe in relative terms. Thus, using the conductor-like screening model COSMO with the dielectric constant of water produced better results in all cases, except PM7, than the corresponding calculations at the homologous level of theory in the gas phase. As for the parameterisation model, plain PM6 and PM7 gave the worst results. Including hydrogen-bond corrections at the PM6-DH + level of theory further improved the statistical correlation, whereas the best results were obtained when both hydrogen bond and halogen bond corrections as implemented in PM6-DH2X were used. Taken together, these results indicate that the predicted binding mode is correct for a significant number of the tested compounds.
Conclusion
Through enzymatic PDC activity assays we have identified several compounds bearing either furoate or thenoate scaffolds as PDK2 inhibitors. 1H-STD-NMR experiments with the five most potent compounds and DCA demonstrated that they bind directly to PDK2. Competition experiments also demonstrated that the most potent compound competes with DCA for PDK2 binding. Taken together, these data indicate that, as predicted theoretically, the reported compounds inhibit PDK2 allosterically via binding to its pyruvate-binding site, thus substantiating this small regulatory site as a druggable target and paving the way towards the design of novel inhibitors with improved pharmacological profiles.
Declaration of interest
The authors declare no competing financial interest. This work has received funding from the National Institutes of Health (M.S.P.) the Netherlands Organization for Scientific Research (A.K.H.H.) and the Dutch Ministry of Education, Culture and Science (A.K.H.H.). The National Institutes of Health (NIH) is acknowledged for Grant DK20478 to M.S.P. The Netherlands Organization for Scientific Research is acknowledged for Grant NWO-CW, ECHO-STIP to A.K.H.H. The Dutch Ministry of Education, Culture and Science for Gravitation Program Grant 024.001.035 to A.K.H.H.
Supplementary material available online
IENZ_1201812_Supplementary_Material.pdf
Download PDF (3.1 MB)References
- Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012;491:364–73
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011;11:85–95
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 2012;21:297–308
- Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 2015;11:9–15
- Kim J, Dang CV. Cancer's molecular sweet tooth and the Warburg effect. Cancer Res 2006;66:8927–30
- Kim J, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3:177–85
- Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K + channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11:37–51
- Patel MS, Nemeria NS, Furey W, Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem 2014;289:16615–23
- Roche TE, Hiromasa Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol Life Sci 2007;64:830–49
- Knoechel TR, Tucker AD, Robinson CM, et al. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry 2006;45:402–15
- Kato M, Li J, Chuang JL, Chuang DT. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure 2007;15:992–1004
- Li J, Kato M, Chuang DT. Pivotal role of the C-terminal DW-motif in mediating inhibition of pyruvate dehydrogenase kinase 2 by dichloroacetate. J Biol Chem 2009;284:34458–67
- Kukimoto-Niino M, Tokmakov A, Terada T, et al. Inhibitor-bound structures of human pyruvate dehydrogenase kinase 4. Acta Crystallogr D Biol Crystallogr 2011;67:763–73
- Tso S-C, Qi X, Gui W-J, et al. Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J Biol Chem 2014;289:4432–43
- Moore JD, Staniszewska A, Shaw T, et al. VER-246608, a novel pan-isoform ATP competitive inhibitor of pyruvate dehydrogenase kinase, disrupts Warburg metabolism and induces context-dependent cytostasis in cancer cells. Oncotarget 2014;5:12862–76
- Mann WR, Dragland CJ, Vinluan CC, et al. Diverse mechanisms of inhibition of pyruvate dehydrogenase kinase by structurally distinct inhibitors. Biochim Biophys Acta 2000;1480:283–92
- Xie Q, Zhang H-F, Guo Y-Z, et al. Combination of Taxol® and dichloroacetate results in synergistically inhibitory effects on Taxol-resistant oral cancer cells under hypoxia. Mol Med Rep 2015;11:2935–40
- Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010;2:31–4
- Brandsma D, Dorlo TPC, Haanen JH, et al. Severe encephalopathy and polyneuropathy induced by dichloroacetate. J Neurol 2010;257:2099–100
- Stockwin LH, Yu SX, Borgel S, et al. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer 2010;127:2510–19
- Pomerantz M, Yang H, Cheng Y. Poly(alkyl thiophene-3-carboxylates). Synthesis and characterization of polythiophenes with a carbonyl group directly attached to the ring. Macromolecules 1995;28:5706–8
- Schnute ME, Brideau RJ, Collier SA, et al. Synthesis of 4-oxo-4,7-dihydrofuro[2,3-b]pyridine-5-carboxamides with broad-spectrum human herpesvirus polymerase inhibition. Bioorg Med Chem Lett 2008;18:3856
- Obrecht D, Ermert P, Oumouch S, et al. Conformationally constrained, fully synthetic macrocyclic compounds. International Patent Application WO/2011/014973, 2011; Polyphor AG
- Schmidt MW, Baldridge KK, Boatz JA, et al. General atomic and molecular electronic structure system. J Comput Chem 1993;14:1347–63
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 2010;31:455–61
- Korth M. Third-generation hydrogen-bonding corrections for semiempirical QM methods and force fields. J Chem Theory Comput 2010;6:3808–16
- Dobeš P, Řezáč J, Fanfrlík J, et al. Semiempirical quantum mechanical method PM6-DH2X describes the geometry and energetics of CK2-inhibitor complexes involving halogen bonds well, while the empirical potential fails. J Phys Chem B 2011;115:8581–9
- Stewart JJP. MOPAC2012 [Internet]. Colorado Springs, CO, USA: Stewart Computational Chemistry; 2012. Available from: http://OpenMOPAC.net
- Petitjean M. On the analytical calculation of van der Waals surfaces and volumes: some numerical aspects. J Comput Chem 1994;15:507–23
- Mantina M, Chamberlin AC, Valero R, et al. Consistent van der Waals radii for the whole main group. J Phys Chem A 2009;113:5806–12
- Korotchkina LG, Patel MS. Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase. Site-specific regulation. J Biol Chem 1995;270:14297–304
- Liu TC, Korotchkina LG, Hyatt SL, et al. Spectroscopic studies of the characterization of recombinant human dihydrolipoamide dehydrogenase and its site-directed mutants. J Biol Chem 1995;270:15545–50
- Yang D, Song J, Wagenknecht T, Roche TE. Assembly and full functionality of recombinantly expressed dihydrolipoyl acetyltransferase component of the human pyruvate dehydrogenase complex. J Biol Chem 1997;272:6361–9
- Korotchkina LG, Patel MS. Probing the mechanism of inactivation of human pyruvate dehydrogenase by phosphorylation of three sites. J Biol Chem 2001;276:5731–8
- Bowker-Kinley MM, Davis WI, Wu P, et al. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998;329:191–6
- Wierckx N, Koopman F, Ruijssenaars HJ, de Winde JH. Microbial degradation of furanic compounds: biochemistry, genetics, and impact. Appl Microbiol Biotechnol 2011;92:1095–105
- Hopkins AL, Keserü GM, Leeson PD, et al. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov 2014;13:105–21