
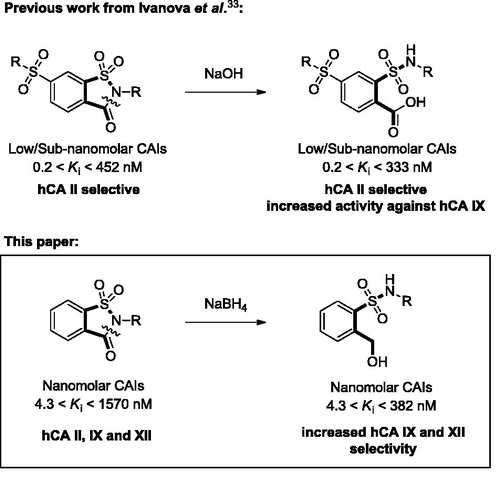
Abstract
A large number of novel secondary sulfonamides based on the open saccharin scaffold were synthesized and evaluated as selective inhibitors of four different isoforms of human carbonic anhydrase (hCA I, II, IX and XII, EC 4.2.1.1). They were obtained by reductive ring opening of the newly synthesized N-alkylated saccharin derivatives and were shown to be inactive against the two cytosolic off-target hCA I and II (Kis > 10 µM). Interestingly, these compounds inhibited hCA IX in the low nanomolar range with Kis ranging between 20 and 298 nM and were extremely potent inhibitors of hCA XII isoenzyme (Kis ranging between 4.3 and 432 nM). Since hCA IX and XII are the cancer-related isoforms recently validated as drug targets, these results represent an important goal in the development of new anticancer candidates. Finally, a computational approach has been performed to better correlate the biological data to the binding mode of these inhibitors.
Introduction
Carbonic anhydrases are a vast family of metalloenzymes (CAs, EC 4.2.1.1.) strongly involved in regulating cell homeostasis and intracellular pH by catalyzing the reversible hydration of carbonic dioxide to bicarbonate ionsCitation1. Fifteen human isoforms are discovered to date and can be differentiated on the basis of cellular localization, tissue distribution, catalytic activity and affinity to various inhibitors, thus representing validated therapeutic targets for the treatment of a wide range of diseasesCitation2,Citation3. In particular, hCA IX and XII were identified as part of the complex machinery that primary and metastatic cancer cell lines use to compensate their dysregulated pH in poorly vascularized microenvironments, like the surroundings of solid tumoursCitation4–6. Targeting these cancer-related isoforms has attracted a lot of attention, hence leading to the proliferation of literature on the subject and the development of structurally diverse inhibitorsCitation7–19. As a result of this superb effort, a potent hCA IX selective compound (SLC-0111) is currently in Phase I clinical trials for the treatment of solid metastatic tumors, consequently boosting the research for new suitable candidatesCitation20,Citation21.
Since the active site of CAs is characterized by the presence of a catalytic zinc ion hold in place by three histidine residues, one of the most successful approaches to design novel CAIs implies introducing a zinc-binding group (ZBG) in the scaffold of potential inhibitorsCitation22. In fact, the most widely prescribed CAIs are characterized by the presence of a primary sulfonamide group, which efficiently engages the positively charged zinc ion upon deprotonationCitation23–26. However, despite primary sulfonamides being preferred “zinc binding group”, they are usually associated with promiscuous profiles and lack of selectivity amongst the different isoforms. On the other side, secondary and tertiary sulfonamides have been lately reported in the literature as efficient and selective inhibitors of the cancer-related hCA IX and XII isoformsCitation27–31, although their binding mode remains a subject of investigationCitation32. In a recent publication, Ivanova et al. reported on the spontaneous ring opening of cyclic tertiary sulfonamides under basic (pH= 9) crystallization conditionsCitation33. Since hCAs do not possess any peptidase activity, they concluded that the base-catalyzed hydrolysis of the saccharin isothiazolone ring happened before the inhibitor entered the active site, and proved their hypothesis by testing both open and closed analogs against a panel of hCAs (hCA I, hCA II, hCA IX and hCA XII). In particular, these open saccharin derivatives determined an increased inhibition of hCA IX, while retaining high activity against hCA II. Such promising results prompted us to investigate the effects of ring opening on our previously reported N-substituted saccharins as selective inhibitors of CA IX and XIICitation15,Citation34. In this paper, we used a reductive ring opening approach to induce the 5-membered isothiazolone ring of saccharin to collapse into its corresponding secondary sulfonamide and benzyl alcohol ()Citation35. The rationale behind this choice could be found in the opportunity of generating two new potential anchoring points for the zinc ion, while introducing several degrees of freedom to the bonds connecting the two hydrophobic phenyl substituents to the polar core of the molecule. In general, the newly synthesized compounds (1–21) proved to be as potent as or slightly less potent than parent inhibitors, while the selectivity for the cancer-related isoforms (hCA IX and XII) over the off-target hCA I and II improved dramatically. In fact, none of the reported compounds inhibited hCA I and II isoforms at concentrations lower than 10 µM, while Ki values spanned from 20 to 298 nM against hCA IX and from 4.3 to 382 nM against hCA XII.
Figure 1. Reductive ring opening approach to the synthesis of novel CAIs.
Chemistry
Saccharin (1.0 eq.) was activated using freshly ground anhydrous potassium carbonate and the corresponding salt was then directly reacted with a proper electrophile (2 eq. of substituted benzyl halide or α-haloacetophenone) by stirring the reaction mixture in N,N-dimethylformamide at 80 °C overnight (Scheme 1). Following these optimized conditions (polar aprotic solvent), we strictly obtained only the more stable regioisomers (N-substituted saccharin derivatives) limiting the Chapman–Mumm thermal rearrangement to the less stable O-substituted counterpartsCitation36. N-substituted saccharin derivatives were then subjected to reductive ring opening with an excess of NaBH4 in dry methanol at room temperature for 2–8 h to give the corresponding secondary sulfonamide compounds 1–21 in discrete yields following a previously reported procedure with slight modificationsCitation35.
Scheme 1. Synthesis and structure of compounds 1–21. For R substituents see .
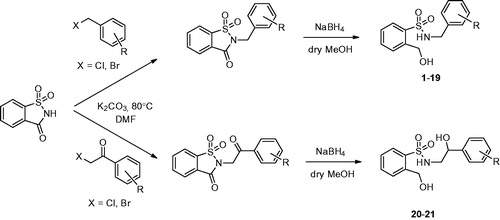
Our choice of NaBH4 as a reducing agent depended on preliminary experiments which suggested that (i) the sulfone group was not affected by these mild reducing conditions leading to the carbonyl group reduction to the alcohol level only with saccharin ring cleavage and that (ii) similar results were obtained for the preparation of o-hydroxymethyl-N-alkyl-benzamides starting from N-alkyl-phthalimidesCitation37.
In their IR spectra, we usually registered for the open saccharin derivatives new but expected signals for the OH and NH stretching at 3240 and 3470 cm−1, respectively, and the disappearance of the C=O stretching at 1735 cm−1.
Biological evaluation
All the synthesized compounds were tested to evaluate their inhibitory activity towards the ubiquitous off-target isoforms, hCA I and II, and the cancer-related ones, hCA IX and XII, by a stopped-flow, CO2 hydrase assay method and their CA inhibition data (Ki) are summarized in .
Table 1. Inhibitory activity of the open saccharin-based derivatives 1–21 and acetazolamide as a reference drug, against selected hCA isoforms by a stopped-flow CO2 hydrase assay.
Experimental protocols
General
Solvents and reagents were used as supplied without further purification. Where mixtures of solvents are specified, the stated ratios are volume:volume. Acetazolamide was purchased by Sigma-Aldrich (Italy) and used in the biological assays without further purification. All synthesized compounds have been fully characterized by analytical and spectral data. Column chromatography was carried out using Sigma-Aldrich® silica gel (high-purity grade, pore size 60 Å, 200–425 mesh particle size). Analytical thin-layer chromatography was carried out on Sigma-Aldrich® silica gel on TLA aluminum foils with fluorescent indicator. Visualization was carried out under UV irradiation (254 nm). 1H-NMR spectra were recorded on a Bruker AV400 (1H: 400 MHz, 13C: 101 MHz). 19F-NMR spectra were recorded on a Bruker AVANCE 600 spectrometer (19F: 564.7 MHz). Chemical shifts are quoted in ppm, based on appearance rather than interpretation, and are referenced to the residual non deuterated solvent peak. In the case of 19F, chemical shifts are referenced to an external standard (CF3COOH, δ − 76.55 ppm). Infrared spectra were recorded on a Bruker Tensor 27 FTIR spectrometer equipped with an attenuated total reflectance attachment with internal calibration. Absorption maxima (νmax) are reported in wavenumbers (cm−1). All melting points were measured on a Stuart® melting point apparatus SMP1 and are uncorrected. Temperatures are reported in °C. Where given, systematic compound names are those generated by ChemBioDraw Ultra® 12.0 following IUPAC conventions. NMR spectra for some representative compounds could be found in the Supplementary material.
Procedures for the synthesis of the new open saccharin derivatives
2-(Hydroxymethyl)-N-(2-methylbenzyl)benzenesulfonamide (1)
(2-Methylbenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 2 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:4). The title compound was a white solid (52% yield); mp 64–66 °C; IR νmax 3459 (ν O–H), 3237 (ν N–H), 3065 (ν Csp2–H), 1319 (νas S=O), 1167 (νs S=O), 712 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.25 (s, 3H, CH3), 2.77 (bs, 1H, OH, D2O exch.), 4.08 (s, 2H, CH2), 5.01 (s, 2H, CH2), 5.43 (bs, 1H, NH, D2O exch.), 7.09–7.19 (m, 4H, CHAr), 7.47–7.61 (m, 3H, CHAr), 8.01–8.04 (m, 1H, CHAr); 13C-NMR (101 MHz, CDCl3) δ 18.7 (CH3), 45.5 (CH2), 64.2 (CH2), 126.2 (Ar), 128.2 (Ar), 128.5 (Ar), 128.9 (Ar), 129.6 (Ar), 129.9 (Ar), 130.5 (Ar), 131.6 (Ar), 133.2 (Ar), 134.1 (Ar), 136.6 (Ar), 138.2 (Ar).
2-(Hydroxymethyl)-N-(2-(trifluoromethyl)benzyl)benzenesulfonamide (2)
(2-Trifluoromethylbenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 4 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product that was purified by column chromatography (EtOAc/n-hexane, 1:3). The title compound was a white solid (47% yield); mp 66–68 °C; IR νmax 3446 (ν O–H), 3214 (ν N–H), 1310 (νas S=O), 1157 (νs S=O), 714 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.81 (t, 1H, J = 5.4 Hz, OH, D2O exch.), 4.17 (d, 2H, J = 6.0 Hz, CH2), 4.91 (s, 2H, CH2), 5.83 (m, 1H, NH, D2O exch.), 7.27 (t, 1H, J = 7.6 Hz, CHAr), 7.35–7.41 (m, 3H, CHAr), 7.46–7.50 (m, 3H, CHAr), 7.89–7.91 (m, 1H, CHAr); 13C-NMR (101 MHz, CDCl3) δ 43.7 (CH2), 63.9 (CH2), 124.2 (C-F, 1JC-F =274.8 Hz, CF3), 125.9 (Ar), 126.0 (Ar), 127.9 (Ar), 128.6 (Ar), 129.7 (Ar), 130.6 (Ar), 131.6 (Ar), 132.3 (Ar), 133.3 (Ar), 135.0 (Ar), 138.0 (Ar), 138.1 (Ar); 19F-NMR (564.7 MHz, CDCl3) δ -56.89 (s, CF3).
2-(Hydroxymethyl)-N-(2-nitrobenzyl)benzenesulfonamide (3)
(2-Nitrobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 6 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give the title compound as a light brown solid (37% yield); mp 103–105 °C; IR νmax 3498 (ν O–H), 3238 (ν N–H), 2973 (ν Csp3–H), 1522 (ν N–O), 1327 (νas S=O), 1165 (νs S=O), 698 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, DMSO-d6) δ 4.34 (d, 2H, J = 5.6 Hz, CH2), 4.88 (s, 2H, CH2), 5.45 (bs, 1H, OH, D2O exch.), 7.39 (t, 1H, J = 7.2 Hz, CHAr), 7.50 (t, 1H, J = 7.6 Hz, CHAr), 7.60–7.68 (m, 3H, 3 x CHAr), 7.74–7.79 (m, 2H, 2 × CHAr), 7.96 (d, 1H, J = 8.4 Hz, CHAr), 8.29 (bs, 1H, NH, D2O exch.); 13C-NMR (101 MHz, DMSO-d6) δ 43.4 (CH2), 60.0 (CH2), 125.1 (Ar), 127.3 (Ar), 128.2 (Ar), 128.5 (Ar), 129.1 (Ar), 130.7 (Ar), 133.0 (Ar), 133.4 (Ar), 134.1 (Ar), 137.0 (Ar), 141.6 (Ar), 148.1 (Ar).
N-(2-bromobenzyl)-2-(hydroxymethyl)benzenesulfonamide (4)
(2-Bromobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 6 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 2:1). The title compound was a white solid (0.05 g, 11% yield); mp 162–164 °C; IR νmax 3273 (ν O–H and ν N–H), 2920 (ν Csp3–H), 1402 (νas S=O), 1065 (νs S=O), 676 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 3.45 (bs, 1H, OH, D2O exch.), 4.14 (d, 2H, J = 6.4 Hz, CH2), 4.93 (s, 2H, CH2), 6.18 (m, 1H, NH, D2O exch.), 7.03 (t, 1H, J = 7.6 Hz, CHAr), 7.15 (t, 1H, J = 7.4 Hz, CHAr), 7.28 (d, 1H, J = 6.8 Hz, CHAr), 7.33–7.49 (m, 4H, CHAr), 7.87 (d, 1H, J = 8.0 Hz, CHAr); 13C-NMR (101 MHz, CDCl3) δ 47.4 (CH2), 63.5 (CH2), 123.4 (Ar), 127.6 (Ar), 128.3 (Ar), 129.4 (Ar), 129.6 (Ar), 130.3 (Ar), 131.3 (Ar), 132.7 (Ar), 133.1 (Ar), 135.6 (Ar), 137.8 (Ar), 138.4 (Ar).
2-(Hydroxymethyl)-N-(3-methylbenzyl)benzenesulfonamide (5)
(3-Methylbenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 7 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:4). The title compound was a white solid (51% yield); mp 79–80 °C; IR νmax 3431 (ν O–H), 3161 (ν N–H), 2936 (ν Csp3–H), 1316 (νas S=O), 1152 (νs S=O), 694 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.27 (s, 3H, CH3), 2.80 (bs, 1H, OH, D2O exch.), 4.07 (bs, 2H, CH2), 4.99 (s, 2H, CH2), 5.72 (bs, 1H, NH, D2O exch.), 6.96 (d, 2H, J = 7.6 Hz, CHAr), 7.04 (d, 1H, J = 8.0 Hz, CHAr), 7.14 (t, 1H, J = 7.4 Hz, CHAr), 7.44–7.49 (m, 2H, CHAr), 7.55–7.59 (m, 1H, CHAr), 7.98 (d, 1H, 7.8 Hz, CHAr); 13C-NMR (101 MHz, CDCl3) δ 21.3 (CH3), 47.5 (CH2), 63.8 (CH2), 124.9 (Ar), 128.4 (Ar), 128.5 (Ar), 128.6 (Ar), 128.7 (Ar), 129.8 (Ar), 131.5 (Ar), 133.1 (Ar), 136.1 (Ar), 138.1 (Ar), 138.2 (Ar), 138.3 (Ar).
2-(Hydroxymethyl)-N-(3-(trifluoromethyl)benzyl)benzenesulfonamide (6)
(3-Trifluoromethylbenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 8 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:3). The title compound was a white solid (71% yield); mp 76–78 °C; IR νmax 3430 (ν O–H), 3278 (ν N–H), 1317 (νas S=O), 1160 (νs S=O), 700 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.98 (t, 1H, J = 4.8 Hz, OH, D2O exch.), 4.06 (d, 2H, J = 6.4 Hz, CH2), 4.91 (d, 2H, J = 4.0 Hz, CH2), 6.07 (m, 1H, NH, D2O exch.), 7.24–7.37 (m, 6H, CHAr), 7.42–7.46 (m, 1H, CHAr), 7.82 (d, 1H, J = 7.6 Hz, CHAr); 13C-NMR (101 MHz, CDCl3) δ 46.9 (CH2), 63.9 (CH2), 123.9 (C-F, 1JC-F =273.4 Hz, CF3), 124.5 (Ar), 124.6 (Ar), 128.6 (Ar), 129.1 (Ar), 129.7 (Ar), 130.7 (Ar), 131.3 (Ar), 131.7 (Ar), 133.3 (Ar), 137.4 (Ar), 137.8 (Ar), 138.0 (Ar); 19F-NMR (564.7 MHz, CDCl3) δ -60.09 (s, CF3).
2-(Hydroxymethyl)-N-(3-nitrobenzyl)benzenesulfonamide (7)
(3-Nitrobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. Excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 6 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product that was purified by column chromatography (EtOAc/n-hexane, 2:1). The title compound was a white solid (37% yield); mp 109–111 °C; IR νmax 3491 (ν O–H), 3164 (ν N–H), 2958 (ν Csp3–H), 1536 (ν N–O), 1326 (νas S=O), 1162 (νs S=O), 695 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, DMSO-d6) δ 4.16 (s, 2H, CH2), 4.87 (s, 2H, CH2), 5.47 (bs, 1H, OH, D2O exch.), 7.33–7.37 (m, 1H, CHAr), 7.51–7.60 (m, 2H, CHAr), 7.64 (d, 1H, J = 7.6 Hz, CHAr), 7.73–7.75 (m, 2H, CHAr), 8.04 (s, 1H, CHAr), 8.06 (s, 1H, CHAr), 8.35 (bs, 1H, NH, D2O exch.); 13C-NMR (101 MHz, DMSO-d6) δ 45.4 (CH2), 59.9 (CH2), 122.5 (Ar), 122.6 (Ar), 127.1 (Ar), 127.9 (Ar), 128.5 (Ar), 130.2 (Ar), 132.9 (Ar), 134.6 (Ar), 137.2 (Ar), 140.8 (Ar), 141.5 (Ar), 148.1 (Ar).
N-(3-fluorobenzyl)-2-(hydroxymethyl)benzenesulfonamide (8)
(3-Fluorobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 5 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:1). The title compound was a light yellow oil (67% yield); IR νmax 3492 (ν O–H), 3283 (ν N–H), 2895 (ν Csp3–H), 1318 (νas S=O), 1252 (ν Csp2–F), 1175 (νs S=O), 689 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.84 (bs, 1H, OH, D2O exch.), 4.05 (s, 2H, CH2), 4.97 (s, 2H, CH2), 6.04 (bs, 1H, NH, D2O exch.), 6.85–6.94 (m, 3H, CHAr), 7.17–7.20 (m, 1H, CHAr), 7.39–7.55 (m, 3H, CHAr), 7.92 (d, 1H, J = 7.6 Hz, CHAr); 13C-NMR (101 MHz, CDCl3) δ 46.6 (CH2), 63.6 (CH2), 114.6 (Ar-F, 2JC-F = 21.1 Hz), 114.8 (Ar-F, 2JC-F = 22.0 Hz), 123.4 (Ar-F, long-rangeJC-F = 2.7 Hz), 128.5 (Ar), 129.7 (Ar), 130.1 (Ar-F, 3JC-F = 8.2 Hz), 131.6 (Ar), 133.2 (Ar), 137.9 (Ar), 138.1 (Ar), 139.0 (Ar-F, 3JC-F = 7.3 Hz), 162.7 (Ar-F, 1JC-F = 247.45 Hz); 19F-NMR (564.7 MHz, CDCl3) δ -110.03 (ddd, JF-H = 9.5 Hz, 8.6 Hz, 5.2 Hz, CF).
N-(3-bromobenzyl)-2-(hydroxymethyl)-benzenesulfonamide (9)
(3-Bromobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 5 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:1). The title compound was a white solid (70% yield); mp 75–76 °C; IR νmax 3454 (ν O–H), 3214 (ν N–H), 2955 (ν Csp3–H), 1319 (νas S=O), 1156 (νs S=O), 694 (δ Csp2-H) cm−1; 1H-NMR (400 MHz, DMSO-d6) δ 4.00 (d, 2H, J = 6 Hz, CH2), 4.87 (d, 2H, J = 5.2 Hz, CH2), 5.42 (bs, 1H, OH, D2O exch.), 7.20 (bs, 2H, CHAr), 7.38 (bs, 3H, CHAr), 7.59–7.63 (m, 1H, CHAr), 7.75–7.78 (m, 2H, CHAr), 8.23 (bs, 1H, NH, D2O exch.); 13C-NMR (101 MHz, DMSO-d6) δ 45.6 (CH2), 59.9 (CH2), 122.0 (Ar), 127.0 (Ar), 127.2 (Ar), 127.9 (Ar), 128.5 (Ar), 130.4 (Ar), 130.7 (Ar), 130.8 (Ar), 132.9 (Ar), 137.2 (Ar), 141.2 (Ar), 141.5 (Ar).
2-(Hydroxymethyl)-N-(4-methylbenzyl)benzenesulfonamide (10)
(4-Methylbenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 6 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:4). The title compound was a white solid (56% yield); mp 100–102 °C; IR νmax 3437 (ν O–H), 3130 (ν N–H), 2930 (ν Csp3–H), 1310 (νas S=O), 1151 (νs S=O), 701 (δ Csp2-H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.31 (s, 3H, CH3), 2.80 (bs, 1H, OH, D2O exch.), 4.07 (d, 2H, J = 5.2 Hz, CH2), 4.99 (s, 2H, CH2), 5.60 (bs, 1H, NH, D2O exch.), 7.06 (bs, 4H, CHAr), 7.44–7.50 (m, 2H, CHAr), 7.55–7.60 (m, 1H, CHAr), 8.00 (d, 1H, 7.6 Hz, CHAr); 13C-NMR (101 MHz, CDCl3) δ 21.1 (CH3), 47.3 (CH2), 63.8 (CH2), 127.9 (Ar), 128.5 (Ar), 129.3 (Ar), 129.9 (Ar), 131.6 (Ar), 133.1 (Ar), 133.2 (Ar), 137.6 (Ar), 138.1 (Ar), 138.2 (Ar).
2-(Hydroxymethyl)-N-(4-(trifluoromethyl)benzyl)benzenesulfonamide (11)
(4-Trifluoromethylbenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 8 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:3). The title compound was a white solid (79% yield); mp 78–80 °C; IR νmax 3417 (ν O–H), 3161 (ν N-H), 3065 (ν Csp2–H), 1321 (νas S=O), 1157 (νs S=O), 688 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 3.03 (bs, 1H, OH, D2O exch.), 4.15 (d, 2H, J = 6.4 Hz, CH2), 5.02 (d, 2H, J = 5.2 Hz CH2), 6.14 (t, 1H, J = 6.4 Hz, NH, D2O exch.), 7.32 (d, 2H, J = 8.0 Hz, CHAr), 7.40–7.49 (m, 4H, CHAr), 7.54–7.58 (m, 1H, CHAr), 7.91 (s, 1H, CHAr); 13C-NMR (101 MHz, CDCl3) δ 46.9 (CH2), 63.9 (CH2), 124.0 (C-F, 1JC-F= 273.1 Hz, CF3), 125.4 (Ar), 125.5 (Ar), 128.1 (Ar), 128.6 (Ar), 129.7 (Ar), 129.8 (Ar), 130.1 (Ar), 131.7 (Ar), 133.3 (Ar), 137.8 (Ar), 138.0 (Ar), 140.4 (Ar); 19F-NMR (564.7 MHz, CDCl3) δ -60.08 (s, CF3).
N-(4-cyanobenzyl)-2-(hydroxymethyl)benzenesulfonamide (12)
(4-Cyanobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 8 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate and concentrated in vacuo to give a crude product, which was purified by column chromatography (EtOAc/n-hexane, 2:1). The title compound was a white solid (67% yield); mp 93–95 °C; IR νmax 3481 (ν O–H), 3187 (ν N–H), 2949 (ν Csp3–H), 2235 CN, 1325 (νas S=O), 1156 (νs S=O), 703 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, DMSO-d6) δ 4.09 (s, 2H, CH2), 4.88 (s, 2H, CH2), 5.46 (bs, 1H, OH, D2O exch.), 7.35–7.41 (m, 3H, CHAr), 7.59–7.62 (m, 1H, CHAr), 7.70–7.77 (m, 4H, CHAr), 8.33 (bs, 1H, NH, D2O exch.); 13C-NMR (101 MHz, DMSO-d6) δ 45.9 (CH2), 59.9 (CH2), 110.3 (Ar), 119.3 (CN), 127.2 (Ar), 127.9 (Ar), 128.5 (Ar), 128.7 (Ar), 132.6 (Ar), 132.9 (Ar), 137.1 (Ar), 141.6 (Ar), 144.3 (Ar).
2-(Hydroxymethyl)-N-(4-nitrobenzyl)benzenesulfonamide (13)
(4-Nitrobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 8 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product, which was purified by column chromatography (EtOAc/n-hexane, 1:1). The title compound was a yellow solid (57% yield); mp 98–99 °C; IR νmax 3493 (ν O–H), 3243 (ν N–H), 1515 (ν N–O), 1318 (νas S=O), 1160 (νs S=O), 690 (δ Csp2-H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.74 (bs, 1H, OH, D2O exch.), 4.21 (bs, 2H, CH2), 5.08 (s, 2H, CH2), 6.13 (bs, 1H, NH, D2O exch.), 7.40–7.59 (m, 5H, CHAr), 7.96–7.97 (m, 1H, CHAr), 8.11–8.13 (m, 2H, CHAr); 13C-NMR (101 MHz, CDCl3) δ 46.7 (CH2), 64.3 (CH2), 123.7 (Ar), 128.5 (Ar), 128.8 (Ar), 129.8 (Ar), 131.6 (Ar), 133.4 (Ar), 137.6 (Ar), 138.7 (Ar), 144.0 (Ar), 147.2 (Ar).
N-(4-fluorobenzyl)-2-(hydroxymethyl)benzenesulfonamide (14)
(4-Fluorobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 8 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 x 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:3). The title compound was a light brown solid (79% yield); mp 99–100 °C; IR νmax 3439 (ν O–H), 3140 (ν N–H), 1312 (νas S=O), 1151 (νs S=O), 696 (δ Csp2-H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 3.18 (bs, 1H, OH, D2O exch.), 4.04 (d, 2H, J = 6.4 Hz, CH2), 4.98 (s, 2H, CH2), 6.00 (bs, 1H, NH, D2O exch.), 6.88–6.93 (m, 2H, CHAr), 7.12–7.15 (m, 2H, CHAr), 7.41–7.57 (m, 3H, CHAr), 7.93 (d, 1H, J = 8.0 Hz, CHAr); 13C-NMR (101 MHz, CDCl3) δ 46.7 (CH2), 63.7 (CH2), 115.4 (Ar-F, 2JC-F =21.2 Hz), 128.5 (Ar), 129.6 (Ar), 129.7 (Ar), 131.6 (Ar), 132.0 (Ar), 132.1 (Ar), 133.2 (Ar), 138.0 (Ar), 138.1 (Ar), 135.4 (Ar-F, 1JC-F =247.3 Hz); 19F-NMR (564.7 MHz, CDCl3) δ -111.71 (tt, JF-H = 8.7 Hz (ortho), 5.3 Hz (meta), CF).
N-(4-chlorobenzyl)-2-(hydroxymethyl)benzenesulfonamide (15)
(4-Chlorobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 7 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:1). The title compound was a white solid (70% yield); mp 103–105 °C; IR νmax 3458 (ν O–H), 3236 (ν N–H), 2876 (ν Csp3–H), 1314 (νas S=O), 1152 (νs S=O), 690 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.90 (bs, 1H, OH, D2O exch.), 4.06 (d, 2H, J = 6.0 Hz, CH2), 5.01 (s, 2H, CH2), 5.88 (bs, 1H, NH, D2O exch.), 7.11 (d, 2H, J = 8.4 Hz, CHAr), 7.21 (d, 2H, J = 8.4 Hz, CHAr), 7.43–7.49 (m, 2H, CHAr), 7.56–7.60 (m, 1H, CHAr), 7.95 (d, 1H, J = 7.6 Hz, CHAr); 13C-NMR (101 MHz, CDCl3) δ 46.8 (CH2), 63.9 (CH2), 128.6 (Ar), 128.7 (Ar), 129.3 (Ar), 129.8 (Ar), 131.6 (Ar), 133.2 (Ar), 133.7 (Ar), 134.8 (Ar), 137.9 (Ar), 138.1 (Ar).
N-(4-bromobenzyl)-2-(hydroxymethyl)benzenesulfonamide (16)
(4-Bromobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 7 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:2). The title compound was a white solid (53% yield); mp 124–125 °C; IR νmax 3461 (ν O–H), 3234 (ν N–H), 3064 (ν Csp2–H), 1314 (νas S=O), 1152 (νs S=O), 690 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.71 (bs, 1H, OH, D2O exch.), 4.06 (bs, 2H, CH2), 5.03 (s, 2H, CH2), 5.78 (bs, 1H, NH, D2O exch.), 7.06–7.08 (m, 2H, CHAr), 7.37–7.59 (m, 5H, CHAr), 7.96–7.97 (m, 1H, CHAr); 13C-NMR (101 MHz, CDCl3) δ 46.9 (CH2), 64.1 (CH2), 121.9 (Ar), 128.6 (Ar), 129.6 (Ar), 129.8 (Ar), 131.7 (Ar), 133.2 (Ar), 137.9 (Ar), 138.3 (Ar).
N-(2,6-difluorobenzyl)-2-(hydroxymethyl)benzenesulfonamide (17)
(2,6-Difluorobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 7 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:1). The title compound was a white solid (67% yield); mp 128–130 °C; IR νmax 3495 (ν O–H), 3159 (ν N–H), 2958 (ν Csp3–H), 1320 (νas S=O), 1159 (νs S=O), 693 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.72 (bs, 1H, OH, D2O exch.), 4.23 (s, 2H, CH2), 4.89 (bs, 2H, CH2), 6.04 (bs, 1H, NH, D2O exch.), 6.79–6.73 (m, 2H, CHAr), 7.10–7.14 (m, 1H, CHAr), 7.26 (s, 1H, CHAr), 7.33–7.48 (m, 2H, CHAr), 7.92–7.94 (m, 1H, CHAr); 13C-NMR (101 MHz, CDCl3) δ 35.1 (CH2), 63.6 (CH2), 111.2 (Ar-F, 2JC-F = 24.8 Hz), 128.3 (Ar), 129.7 (Ar), 129.9 (Ar), 130.0 (Ar), 130.1 (Ar), 131.2 (Ar), 133.1 (Ar), 137.8 (Ar), 138.2 (Ar), 161.2 (Ar-F, 1JC-F = 246.9 Hz); 19F-NMR (564.7 MHz, CDCl3) δ -112.08 (t, JF-H = 6.9 Hz, CF).
N-(3,4-dichlorobenzyl)-2-(hydroxymethyl)benzenesulfonamide (18)
(3,4-Chlorobenzyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 7 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 2:1). The title compound was a light brown solid (81% yield); mp 83–84 °C; IR νmax 3463 (ν O–H), 3192 (ν N–H), 2947 (ν Csp3–H), 1319 (νas S=O), 1158 (νs S=O), 690 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.88 (bs, 1H, OH, D2O exch.), 3.96 (d, 2H, J = 6.4 Hz, CH2), 4.93 (bs, 2H, CH2), 6.00 (t, 1H, J = 6.0 Hz NH, D2O exch.), 6.94–6.96 (bs, 1H, CHAr), 7.17–7.22 (m, 2H, CHAr), 7.33–7.39 (m, 2H, CHAr), 7.46–7.48 (m, 1H, CHAr), 7.83 (d, 1H, J = 7.6 Hz, CHAr); 13C-NMR (101 MHz, CDCl3) δ 46.3 (CH2), 64.0 (CH2), 127.2 (Ar), 128.6 (Ar), 129.7 (Ar), 129.8 (Ar), 130.5 (Ar), 131.7 (Ar), 131.8 (Ar), 132.6 (Ar), 133.3 (Ar), 136.7 (Ar), 137.7 (Ar), 138.1 (Ar).
2-(Hydroxymethyl)-N-(naphthalen-1-ylmethyl)benzenesulfonamide (19)
2-(Naphthalen-1-ylmethyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 6 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:1). The title compound was a white solid (57% yield); mp 80–81 °C; IR νmax 3517 (ν O–H), 3347 (ν N–H), 3061 (ν Csp2–H), 1318 (νas S=O), 1157 (νs S=O), 690 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.68 (bs, 1H, OH, D2O exch.), 4.51 (s, 2H, CH2), 4.91 (s, 2H, CH2), 5.69 (bs, 1H, NH, D2O exch.), 7.29–7.55 (m, 7H, CHAr), 7.74–7.86 (m, 3H, CHAr), 8.05 (s, 1H, CHAr); 13C-NMR (101 MHz, CDCl3) δ 45.6 (CH2), 63.7 (CH2), 123.2 (Ar), 125.2 (Ar), 126.0 (Ar), 126.6 (Ar), 127.0 (Ar), 128.4 (Ar), 128.7 (Ar), 129.0 (Ar), 130.0 (Ar), 131.1 (Ar), 131.4 (Ar), 131.5 (Ar), 133.2 (Ar), 133.7 (Ar), 137.6 (Ar), 138.3 (Ar).
N-(2-hydroxy-2-phenylethyl)-2-(hydroxymethyl)benzenesulfonamide (20)
2-(2-Oxo-2-phenylethyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 8 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 2:1). The title compound was a light yellow oil (49% yield); IR νmax 3291 (ν O–H and ν N–H), 2923 (ν Csp3–H), 1316 (νas S=O), 1153 (νs S=O), 696 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.94 (bs, 1H, CH2), 3.15 (bs, 1H, CH2), 3.42 (bs, 1H, OH, D2O exch.), 4.56–4.64 (m, 2H, CH2), 4.91–5.03 (m, 1H, CH), 5.28 (bs, 1H, OH, D2O exch.), 6.57 (bs, 1H, NH, D2O exch.), 7.13–7.20 (m, 5H, CHAr), 7.38–7.47 (m, 3H, CHAr), 7.91 (d, 1H, J = 6.4 Hz, CHAr); 13C NMR (101 MHz, CDCl3) δ 50.4 (CH2), 62.7 (CH2), 72.4 (CH), 125.8 (Ar), 127.9 (Ar), 128.2 (Ar), 128.5 (Ar), 129.5 (Ar), 131.3 (Ar), 133.3 (Ar), 137.3 (Ar), 138.6 (Ar), 140.8 (Ar).
N-(2-hydroxy-2-(3-methoxyphenyl)ethyl)-2-(hydroxymethyl)benzenesulfonamide (21)
2-(2-Hydroxy-2-(3-methoxyphenyl)ethyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (1.0 eq.) was suspended in 20 mL of anhydrous methanol at room temperature. An excess of NaBH4 was added portionwise and the reaction monitored by TLC. After 8 h the reaction was quenched with water and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The organic layers were reunited, dried over sodium sulfate, and concentrated in vacuo to give a crude product which was purified by column chromatography (EtOAc/n-hexane, 1:2). The title compound was a yellow oil (88% yield); IR νmax 3477 (ν O–H), 3295 (ν N–H), 2935 (ν Csp3–H), 1317 (νas S=O), 1160 (νs S=O), 697 (δ Csp2–H) cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.89–2.95 (m, 1H, CH2), 3.13–3.17 (m, 1H, CH2), 3.64 (s, 3H, OCH3), 4.41 (bs, 2H, CH2), 4.58 (bs, 1H, OH), 4.87–4.91 (m, 1H, CH), 4.97 (bs, 1H, OH, D2O exch.), 6.48 (bs, 1H, NH, D2O exch.), 6.67–6.71 (m, 3H, CHAr), 7.07–7.11 (m, 1H, CHAr), 7.32–7.47 (m, 3H, CHAr), 7.87 (d, 1H, J = 7.6 Hz, CHAr); 13C-NMR (101 MHz, CDCl3) δ 50.4 (CH2), 55.1 (OCH3), 60.6 (CH2), 72.2 (CH), 111.4, (Ar), 113.3 (Ar), 118.1 (Ar), 128.2 (Ar), 129.5 (Ar), 129.6 (Ar), 131.2 (Ar), 133.2 (Ar), 137.3 (Ar), 138.6 (Ar), 142.5 (Ar), 159.5 (ArO).
Enzyme inhibition assays
An applied photophysics stopped-flow instrument has been used for assaying the CA-catalyzed CO2 hydration activity. Phenol red (0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5, for α-CAs) as buffer, and 20 mM NaClO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of each inhibitor (1 µM) were prepared in distilled-deionized water and dilutions up to 0.1 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex or for the eventual active site mediated hydrolysis of the inhibitor. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, and represent the average from at least three different determinations. All recombinant CA isoforms were obtained in-house as previously reportedCitation38,Citation39.
Molecular modeling studies
Preparation of saccharin-based structures
The saccharin-based analogs 1–21 were prepared in 3D with the MOE software package (v2013.08.02, Chemical Computing Group Inc., Montreal, Canada) as previously reportedCitation40. All possible structural isomers of compounds were constructed. Strong acids were deprotonated and strong bases were protonated. Finally, the ligands were energy minimized using a steepest-descent protocol (MMFF94x force field).
Preparation of hCA crystal structures for docking studies
The structures of hCA I (PDB: 3LXE, 1.90 Å), hCA II (PDB: 4E3D, 1.60 Å), hCA IX (PDB: 3IAI, 2.20 Å) and hCA XII (PDB: 1JD0, 1.50 Å) were obtained from the protein databank (PDB). The protein atoms, the active site zinc ions and the zinc-bound water molecule of hCA II were retained and all other atoms were omitted. The remaining structure was protonated using the MOE software package and subsequently the obtained structure was energy-minimized (AMBER99 force field). Finally, the obtained protein models were superposed on the hCA I structure using the backbone Cα-atoms. The zinc-bound water molecule of hCA II coordinated well to the zinc ions of the other hCAs.
Docking studies
The GOLD Suite software package (v5.2, CCDC, Cambridge, UK) and the GoldScore scoring function were used to dock the derivatives into the hCA structures with and without the zinc-bound water molecule (25 dockings per ligand). The binding pocket was defined as all residues within 13 Å of a centroid (x: −17.071, y: 35.081, 43.681; corresponding approximately to the position of the thiadiazole ring of acetazolamide in the 1JD0 structure).
Results and discussion
The analysis of the biological data was accomplished comparing the open saccharin-based derivatives with their corresponding parent compounds in order to evaluate if the ring opening enhanced or reduced their biological activity.
All the tested compounds had no affinity for the common off-target hCA I and II isoforms (Kis > 10000 nM) compared to their corresponding parent drugs. Moreover, our molecules with a benzyl alcohol group, instead of a carboxylic acidCitation33, abolished completely this inhibitory activity improving the biological profile of this scaffold.
The inhibition profile of the open saccharin-based derivatives against the two tumor-related hCA IX and XII isoforms also displayed some important changes compared to parent drugs. Among the new open saccharin derivatives reported here, the best activity was obtained toward hCA XII isoform by compounds 1, 5, 6, 10 and 11, all provided of CH3 or CF3 groups. These molecules exhibited a slightly preference for hCA XII respect to hCA IX isoform, although the inhibition of the latter was also in the nanomolar range. Compound 5, containing a phenyl ring substituted with methyl group in meta position, had the highest inhibitory activity against hCA XII (Ki = 4.3 nM), but also compound 6, containing a meta trifluoromethyl substituent on phenyl ring, exhibited similar inhibitory activity (Ki = 4.4 nM). Comparable profile against hCA XII was observed for compounds 10 (Ki = 5.7 nM) and 11 (Ki = 7.2 nM), which are para substituted regioisomers of 5 and 6, respectively. Compound 1, which had a methyl group at ortho position of phenyl ring, showed similar inhibitory activity (Ki hCA XII = 4.7 nM) with respect to 5 and 6. Other compounds with strong selectivity between hCA IX and XII were 9 (Ki hCA IX = 267 nM, Ki hCA XII = 64 nM) with bromine in meta position of phenyl ring, 12 (Ki hCA IX = 126 nM, Ki hCA XII = 57 nM) containing a cyano group in para position, 15 (Ki hCA IX = 154 nM, Ki hCA XII = 48 nM) which had a para chloro-substituted phenyl ring, 17 and 18 with, respectively, 2,6-difluoro and 3,4-dichloro-substituted phenyl rings.
Conversely, some compounds displayed a good selectivity towards hCA IX isoform. Compounds with nitro substituents in ortho (3), meta (7) or para (13) position of the ring had potent inhibitory activity preferentially against this overexpressed isoform in the hypoxic tumoral niche.
For compounds 20 (Ki hCA IX = 224 nM, Ki hCA XII = 64 nM) and 21 (Ki hCA IX = 31 nM, Ki hCA XII = 355 nM), the reaction with NaBH4 led to the further reduction of exocyclic carbonyl moiety. The presence of this additional group maintained the biological profile with a loss of inhibitory activity against hCA I and II and a preferential selectivity against the cancer-related isoforms.
Collectively, these promising data showed that the reductive ring opening of the saccharin nucleus improved the hCA inhibitory activity with a better selectivity with respect to the off-target isoforms. From the above, we also observed that the inhibition profile was affected positively or negatively by the substitution pattern.
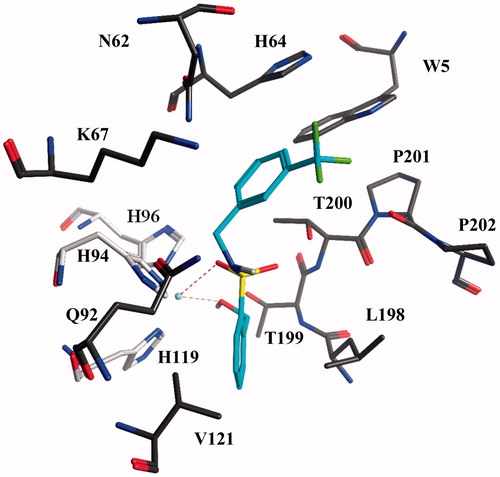
Docking studies into the active site of hCA XII
The open saccharin analogs (compounds 1–21) were endowed with inhibition values in the nanomolar range against hCA XII isoform (Ki values: 4.3–432 nM, , approximately 100-fold difference between lowest and highest Ki values). The interaction of compound 6 with the active site of hCA XII was shown as an example (). Docking studies indicated that the hydroxymethyl group and one of the sulfonamide oxygen atoms could interact simultaneously with the Zn2+-ion, whereas the other sulfonamide oxygen atom was water accessible. This oxygen might also form hydrogen bonds with the backbone of Thr199. The other polar substituents of the molecule were water accessible.
Figure 2. Docked pose of compound 6 in the active site of hCA XII. Hydrogen bonds and interactions to the Zn2+-ion are depicted in red dashed lines. The Zn2+-ion is depicted as a turquoise sphere. The three zinc-binding Histidines (H94, H96 and H119) are depicted in light gray for clarity.
Conclusion
We proposed the design, synthesis, characterization and pharmacological evaluation in vitro of several new secondary sulfonamides based on the open saccharin scaffold as selective inhibitors of four different isoforms of human carbonic anhydrase. They were shown to be inactive against the two cytosolic off-target hCA I and II (Kis > 10 µM); conversely, all these compounds inhibited hCA IX and XII in the low nanomolar range with Kis ranging between 4.3 and 432 nM. The analysis of the Ki values showed as the substituent on phenyl moiety that gives the best outcomes relative to inhibition of hCA XII isoform are methyl and trifluoromethyl groups. The results were also rationalized by means of docking studies into the active site of hCA XII. Since these two cancer-related hCA isoforms were recently validated as drug targets, these results provided the development of new anticancer candidates.
Declaration of interest
The authors declare no conflict of interest.
This work was financed by two FP7 EU projects (Metoxia and Dynano) to CTS.
IENZ_1235040_Supplementary_material.pdf
Download PDF (484.8 KB)Related Research Data
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Winum JY, Scozzafava A, Montero JL, Supuran CT. Therapeutic potential of sulfamides as enzyme inhibitors. Med Res Rev 2006;26:767–92.
- Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 2011;11:671–7.
- De Simone G, Supuran CT. Carbonic anhydrase IX: biochemical and crystallographic characterization of a novel antitumor target. Biochim Biophys Acta 2010;1804:404–9.
- Lock FE, McDonald PC, Lou Y, et al. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene 2013;32:5210–19.
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810.
- Scozzafava A, Menabuoni L, Mincione F, Supuran CT. Carbonic anhydrase inhibitors. A general approach fir the preparation of water-soluble sulfonamides incorporating polyamino-polycarboxylate tails and of their metal complexes possessing long-lasting, topical intraocular pressure-lowering properties. J Med Chem 2002;45:1466–76.
- Innocenti A, Beyza Oztürk Sarikaya S, Gülçin I, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of mammalian isoforms I-XIV with a series of natural products polyphenols and phenolic acids. Bioorg Med Chem 2010;18:2159–64.
- Bayram E, Senturk M, Kufrevioglu OI, Supuran CT. In vitro inhibition of salycilic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg Med Chem 2008;16:9101–5.
- Carradori S, Mollica A, De Monte C, et al. Nitric oxide donors and selective carbonic anhydrase inhibitors: a dual pharmacological approach for the treatment of glaucoma, cancer and osteoporosis. Molecules 2015;20:5667–79.
- Maccallini C, Di Matteo M, Vullo D, et al. Indazole, pyrazole, and oxazole derivatives targeting nitric oxide synthases and carbonic anhydrases. ChemMedChem 2016;11:1695–9.
- Gidaro MC, Alcaro F, Carradori S, et al. Eriocitrin and apigenin as new carbonic anhydrase VA inhibitors, from a virtual screening of Calabrian natural products. Planta Medica 2015;81:533–40.
- De Monte C, Carradori S, Secci D, et al. Cyclic tertiary sulfamates: selective inhibition of the tumor-associated carbonic anhydrases IX and XII by N- and O-substituted acesulfame derivatives. Eur J Med Chem 2014;84:240–6.
- D’Ascenzio M, Carradori S, De Monte C, et al. Design, synthesis and evaluation of N-substituted saccharin derivatives as selective inhibitors of tumor-associated carbonic anhydrase XII. Bioorg Med Chem 2014;22:1821–31.
- Mollica A, Costante R, Akdemir A, et al. Exploring new probenecid-based carbonic anhydrase inhibitors: Synthesis, biological evaluation and docking studies. Bioorg Med Chem 2015;23:5311–18.
- D’Ascenzio M, Carradori S, Secci D, et al. Selective inhibition of human carbonic anhydrases by novel amide derivatives of probenecid: synthesis, biological evaluation and molecular modeling studies. Bioorg Med Chem 2014;22:3982–8.
- De Monte C, Carradori S, Gentili A, et al. Dual cyclooxygenase and carbonic anhydrase inhibition by nonsteroidal anti-inflammatory drugs for the treatment of cancer. Curr Med Chem 2015;22:2812–18.
- Carradori S, Mollica A, Ceruso M, et al. New amide derivatives of Probenecid as selective inhibitors of carbonic anhydrase IX and XII: biological evaluation and molecular modeling studies. Bioorg Med Chem 2015;23:2975–81.
- See more at ClinicalTrials.gov: Safety Study of SLC-0111 in Subjects With Advanced-Solid Tumors-Clinical Trials_gov.mht
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902.
- Supuran CT, Winum JY, eds. Drug design of zinc-enzyme inhibitors: functional, structural, and disease applications. Hoboken, NJ: Wiley; 2009: Part II.
- Langella E, D'Ambrosio K, D’Ascenzio M, et al. A combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes within carbonic anhydrase active site. Chem Eur J 2016;22:97–100.
- D’Ambrosio K, Carradori S, Monti SM, et al. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem Comm 2015;51:302–5.
- Carradori S, De Monte C, D’Ascenzio M, et al. Salen and tetrahydrosalen derivatives act as effective inhibitors of the tumor-associated carbonic anhydrase XII: a new scaffold for designing isoform-selective inhibitors. Bioorg Med Chem Lett 2013;23:6759–63.
- Mollica A, Locatelli M, Macedonio G, et al. Microwave-assisted extraction, HPLC analysis and inhibitory effects on carbonic anhydrase I, II, VA and VII isoforms of fourteen blueberry Italian cultivars. J. Enzyme Inhib. Med. Chem 2016. [Epub ahead of print]. doi: 10.1080/14756366.2016.1214951.
- Wilkinson BL, Bornaghi LF, Houston TA, et al. A novel class of carbonic anhydrase inhibitors: glycoconjugate benzene sulfonamides prepared by “click-tailing”. J Med Chem 2006;49:6539–98.
- Güzel-Akdemir Ö, Akdemir A, Isik S, et al. o-Benzenedisulfonimido–sulfonamides are potent inhibitors of the tumor-associated carbonic anhydrase isoforms CA IX and CA XII. Bioorg Med Chem 2013;21:1386–91.
- Liu F, Martin-Mingot A, Lecornué F, et al. Carbonic anhydrases inhibitory effects of new benzenesulfonamides synthesized by using superacid chemistry. J Enzyme Inhib Med Chem 2012;27:886–91.
- Métayer B, Mingot A, Vullo D, et al. New superacid synthesized (fluorinated) tertiary benzenesulfonamides acting as selective hCA IX inhibitors: toward a new mode of carbonic anhydrase inhibition by sulfonamides. Chem Commun 2013;49:6015–17.
- Coviello V, Marchi B, Sartini S, et al. 1,2-benzisothiazole derivatives bearing 4-, 5-, or 6-alkyl/arylcarboxamide moieties inhibit carbonic anhydrase isoform IX (CAIX) and cell proliferation under hypoxic conditions. J Med Chem 2016;59:6547–52.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- Ivanova J, Leitans J, Tanc M, et al. X-ray crystallography-promoted drug design of carbonic anhydrase inhibitors. Chem Commun (Camb.) 2015;51:7108–11.
- Carradori S, Secci D, De Monte C, et al. A novel library of saccharin and acesulfame derivatives as potent and selective inhibitors of carbonic anhydrase IX and XII isoforms. Bioorg Med Chem 2016;24:1095–105.
- Rice LM, Grogan CH, Reid EE. N-Alkyl saccharins and their reduction products. J Am Chem Soc 1953;75:4304–5.
- Hettler H. Chapman-Mumm rearrangement of pseudosaccharinethers. Tetrahedron Lett 1968;9:1793–6.
- Fichert T, Massing U. A new strategy for the preparation of secondary amines via o-(tetrahydropyranyloxymethyl)-benzamides. Tetrahedron Lett 1998;39:5017–18.
- Bozdag M, Ferraroni M, Nuti E, et al. Combining the tail and the ring approaches for obtaining potent and isoform-selective carbonic anhydrase inhibitors: solution and X-ray crystallographic studies. Bioorg Med Chem 2014;22:334–40.
- Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs – antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzyme Inhib Med Chem 2014;29:379–87.
- Akdemir A, De Monte C, Carradori S, Supuran CT. Computational investigation of the selectivity of salen and tetrahydrosalen compounds towards the tumor-associated hCA XII isozyme. J Enzyme Inhib Med Chem 2015;30:114–18.