
Abstract
The isatin scaffold is the constitutive fragment of several natural and synthetic bioactive molecules. Albeit several benzene sulphonamide-based carbonic anhydrase inhibitors (CAIs) have been reported, only recently isatin benzene sulphonamides have been studied and proposed as CAIs. In this study we have designed, synthesised, and evaluated the biological activity of a series of differently substituted isatin-based benzene sulphonamides which have been designed for the inhibition of carbonic anhydrase isoforms. The activity of all the synthesised compounds was evaluated towards human carbonic anhydrase I, II, IX, and XII isozymes. Our results indicate that the nature and position of substituents on the isatin ring can modulate both activity and isozyme selectivity.
Introduction
The isatin nucleus could be considered as a privileged scaffold for the design of biologically active agents. The discovery and optimisation of isatin-based therapeutic agents have consistently attracted the interest of medicinal chemists and the chemistry, the biological properties and the therapeutic potential of isatin-based agents has been recently reviewedCitation1. Several biological activities could be achieved by decoration of the isatin scaffold such as anti-cancerCitation2–7, anti-oxidantCitation6, HIV reverse transcriptase inhibitionCitation8,Citation9, neuroprotectiveCitation10, anti-fungalCitation11,Citation12, anti-bacterialCitation13,Citation14 and anti-diabeticCitation15. Moreover, the isatin ring has been pointed out as an essential part of anticancer hybrid moleculesCitation16,Citation17. Recently, the design of isatin-based carbonic anhydrase inhibitors (CAIs) has been reportedCitation10,Citation11,Citation18–20. It is common knowledge that carbonic anhydrase isozyme family is involved in several physiological and/or pathological metabolic pathwaysCitation21–26. They catalyse the simple reaction of the reversible hydration of carbon dioxide to bicarbonate and protonsCitation27, which is essential for the regulation of the different chemical species connected with CO2 in the body and its transport across biological membranes such as the inter-, intra- and extra-cellular spacesCitation25,Citation28. Not surprisingly several CAIs have been reported and their therapeutic potential has been directed towards different pathologiesCitation29–31 ().
Figure 1. Carbonic anhydrase inhibitors in clinical use: (1) acetazolamide, (2) (methazolamide, (3) ethoxzolamide, (4) dichlorphenamide, (5) dorzolamide, (6) brinzolamide, and (7) topiramate.
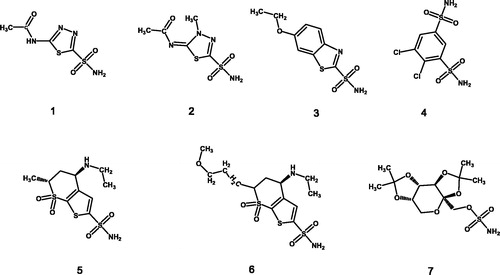
Within CAIs, benzene-sulphonamides are widely representedCitation32–35and their binding on carbonic anhydrase investigatedCitation36. Benzene-sulphonamides are versatile scaffold that can be efficiently substituted to achieve isozyme specificityCitation17,Citation19,Citation20,Citation35,Citation37–39. In this respect, the specific targeting of the tumour associated CA isoforms IX and XII represents an innovative and specific approach for the treatment of tumoursCitation23,Citation40,Citation41. Furthermore, the identification of hybrid molecules, containing both the isatin scaffold and the benzene-sulphonamide moiety, with a multi-pharmacological effect is an attractive target for medicinal chemists, for the treatment of multifactorial pathologies such as cancerCitation41–43. On the basis of the above and with the aim to achieve structure-activity relationships on isatin derived CAIs, we have designed and synthesised a series of new 4-{[5–(2-oxo-2,3-dihydro-1H-indol-3-ylidene)-3-methyl-4-oxo-1,3-thiazolidin-2-ylidene]amino}benzene-1-sulphonamides as potential inhibitors of the tumour associated CA isoforms IX and XII.
Methods
Materials and apparatus
Starting materials and reagents were obtained from commercial suppliers and were used without purification. All melting points were determined on a Stuart SMP11 melting points apparatus (Stone, UK) and are uncorrected. Electron ionisation mass spectra were obtained by a Fisons QMD 1000 mass spectrometer (Danvers, MA) (70 eV, 200 mA, ion source temperature 200 °C). Samples were directly introduced into the ion source. Found mass values are in agreement with theoretical ones. Melting points, yield of reactions and the analytical data of derivatives are reported in .
Table 1. Chemical, analytical, and physical data of derivatives EMAC 10020.
1H-NMR () were registered on a Bruker 400 MHz spectrometer (Billerica, MA) or on a Varian 500 MHz (Palo Alto, CA) (). All samples were measured in DMSO. Chemical shifts are reported referenced to the solvent in which they were measured. Coupling constants J are expressed in hertz (Hz). Elemental analyses were obtained on a Perkin–Elmer 240 B microanalyser (Waltham, MA). Analytical data of the synthesised compounds are in agreement within ±0.4% of the theoretical values. TLC chromatography was performed using silica gel plates (Merck F 254, Billerica, MA), spots were visualised by UV light.
Table 2. 1H NMR and 13C NMR data of derivatives EMAC 10020.
Biological activity
Carbonic anhydrase inhibition assay
The purification of cytosolic CA isoenzymes (CA I and CA II) were previously described with a simple one-step method by a Sepharose-4B-L tyrosine-sulphanilamide affinity chromatographyCitation44. The protein quantity in the column effluents was determined spectrophotometrically at 280 nm. Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) was applied with a Bio-Rad Mini Gel system Mini-PROTEIN® system (Hercules, CA), Bio-Rad Laboratories, Inc., China after purification of both CA isoenzymes. Briefly, it was performed in acrylamide for the running (10%) and the stacking gel (3%) contained SDS (0.1%), respectively. Activities of CA isoenzymes were determined according to a method by Verporte et al.Citation45 The increase in absorbance of reaction medium was spectrophotometrically recorded at 348 nm. Also, the quantity of protein was determined at 595 nm according to the Bradford methodCitation46. Bovine serum albumin was used as standard protein. The IC50 values were obtained from activity (%) versus compounds plotsCitation47. For calculation of KI values, three different concentrations were used. The Lineweaver–Burk curves were drawn and calculations were realisedCitation47. The biological data are reported in .
Table 3. Inhibition data towards hCA I, II, IX, and XII of compounds EMAC 10020.
General procedure for the synthesis of compound EMAC10020
Synthesis of 1-methyl-3–(4-sulphamoylphenyl)thiourea1
4-Aminobenzensulphonamide (1 eq.) was refluxed in 2-propanol until a clear solution is obtained. Then a solution of methylisothiocyanate (1 eq.) in 2-propanol was added dropwise. By adding the isothiocyanate the solution became yellowish. The mixture was stirred until reaction completion (5–6 h) monitored by TLC (ethyl acetate/n-hexane 2/1). The reaction is allowed to cool down at r.t. and the formation of a white foaming precipitate is observed, which was filtered and crystallised from ethanol. White crystals; MW: 245.32 g/mol; yield: 65%; Mp: °C 204–6
1H NMR (400 MHz, DMSO-d6) δ (ppm): 3.15 (3H, s, CH3, N-CH3); 7.08 (d, 2H, CH, J = 8.4, 4-SO2NH2 phenyl); 7.30 (2H, s, NH2, SO2NH2); 7.80 (d, 2H, CH, J = 8.4, 4-SO2NH2 phenyl); 10.01 (s, 2H,NH, thiourea).
Synthesis of 4-(4-oxo-1,3-thiazolidin-2-ylidene)aminobenzene-1-sulphonamide2
An ethanol solution of 1 (1.70 eq.), ethyl bromoacetate (1.90 eq) and anhydrous sodium acetate (6.9 eq.) was refluxed under vigorous stirring till the completion of the reaction (16–20 h), TLC (ethyl acetate/n-hexane 2/1). Then the solution was cooled to 0 °C, and the formed precipitate filtered under vacuum and crystallised from water.
White powder; MW: 285.34 g/mol; yield: 86%; Mp: 179–180 °C
1H NMR (400 MHz, DMSO-d6) δ (ppm): 3.17 (3H, s, CH3, N-CH3); 4.06 (s, 2H, CH2, thiazol.); 7.10 (d, 2H, CH, J = 8.4, 4-SO2NH2 phenyl); 7.31 (2H, s, NH2, SO2NH2); 7.80 (d, 2H, CH, J = 8.4, 4-SO2NH2 phenyl).
Synthesis of 4-(3-methyl-4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylideneamino)benzene-sulphonamides EMAC 10020
A mixture of 4-(3-methyl-4-oxothiazolidin-2-ylideneamino)benzene-sulphonamide (1 eq), the opportune isatin derivative (1 eq), acetic anhydride (1,5 eq), and sodium acetate (2 eq) was refluxed overnight in acetic acid. Once the reaction has come to completion TLC (ethyl acetate/n-hexane 2/1) the hot suspension was filtered. The obtained red/orange solid was washed with water. Compounds EMAC 10020 were purified by column chromatography on silica gel (ethyl acetate/n-hexane 2/1) to obtain the desired compounds whose data are reported in and .
Results and discussion
As a part of our ongoing research in the field of CAIsCitation37 and to achieve a better understanding of the structural requirements for the selective inhibition of the different CA isoforms, we have synthesised a series of 4-(3-methyl-4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylideneamino)-benzenesulphonamides indicated as compounds EMAC 10020 a, c, d, l, m, n, and o. All the synthesised compounds bear a differently substituted isatin scaffold linked, by the interposition of a thiazolidinone spacer, to a benzene-sulphonamide moiety, as zinc binder group. The synthesis of compounds EMAC 10020 was performed as illustrated in .
Figure 2. Synthetic pathway to compounds EMAC 10020. Reagents and conditions: (i) 2-propanol, methyl isothiocyanate; (ii) ethanol, ethyl bromoacetate, dry sodium acetate; (iii) R-isatin, acetic anhydride, dry sodium acetate, acetic acid.
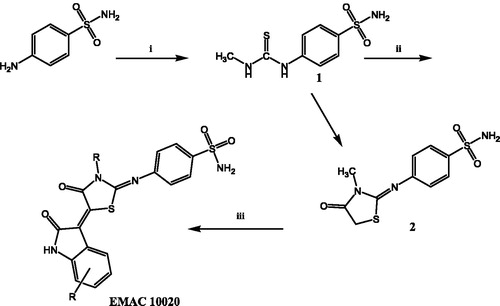
The procedure consists of two steps. The first step is the synthesis of the 4-sulphamoylphenyl-thiourea derivative (1 of ) by simple reaction of the 4-aminobenzensulphonamide with methylisothiocyanate. The second step of the synthetic route consists of the formation of the thiazolidinone spacerCitation2 which can be obtained by reacting 1 with ethyl-bromoacetate. Desired compounds were obtained by reacting compound 2 with the appropriate isatin. We attempted to perform step two and three in a one step one pot reaction, but our efforts only gave poor yields with respect to the three step procedure which was therefore preferred. Compounds EMAC 10020 were submitted to enzymatic assay to evaluate their activity and selectivity towards human CA (hCA) isozymes I, II, IX, XII. The results are illustrated in . As shown in some of the tested compounds could be considered as hCA IX preferential inhibitors. However, the nature and position of the substituents on the isatin scaffold played a crucial role in determining the activity and the isozyme selectivity. In the case of compound EMAC 10020 a, bearing an un-substituted isatin, almost no selectivity can be observed, but for a very poor activity towards XII isozyme. With respect to isozyme IX, the introduction of an electron withdrawing (EW) group (compounds EMAC 10020 c, EMAC 10020 d, and EMAC 10020 n) in the position 5 of the isatin lead to a decrease of activity, albeit some selectivity towards hCA IX could be observed for compound EMAC 10020 c. However, the isosteric replacement of the chlorine atom and of the trifluoro-methyl moiety by a methyl group lead to an increase of activity (KI 3.0 nM) and selectivity. According to these preliminary results, in the case hCA IX, the presence of an EW group in the position 5 of the isatin is not tolerated while the introduction of an electron donating substituent in the same position is beneficial for the activity and selectivity. Also in the case of compound EMAC 10020 m, bearing a fluorine atom in the position 7 of the isatin ring, a good activity (KI 4.1 nM) and selectivity towards hCA IX was observed. Conversely the introduction of a bulkier EW group in the position 7 of the isatin scaffold, as for compound EMAC 10020 o, leads to a decrease of activity towards all isozymes. All together these data corroborate the hypothesis that the hybridisation of isatin scaffold with the benzene-sulphonamide moiety could be advantageous for the design of new therapeutic agents targeting the tumour associated hCA IX isoform.
Conclusions
We have synthesised a small library of isatin/benzene-sulphonamides hybrids for the inhibition of hCA isoforms. The activity of the newly synthesised derivatives has been evaluated towards hCA I, II, IX, and XII. Compounds EMAC 10020 c, EMAC 10020 l, and EMAC 10020 m could be considered as preferential hCA IX inhibitors. Our data indicated that the nature and the position of the substituents play a crucial role in tuning activity and selectivity towards hCA isozyme. Overall these data support the hypothesis that isatin hybrid molecules could represent a valuable starting point for the design of active and selective hCAIs.
Acknowledgements
The authors wish to acknowledge the “Ufficio Valorizzazione dei Risultati della Ricerca” of Sardegna Ricerche Technological Park, Pula (CA) – Italy.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Related Research Data
References
- Rane RA, Karunanidhi S, Jain K, et al. A recent perspective on discovery and development of diverse therapeutic agents inspired from isatin alkaloids. Curr Top Med Chem (Sharjah, United Arab Emirates) 2016;16:1262–89.
- Hou J, Jin K, Li J, et al. LJNK, an indoline-2,3-dione-based aminopeptidase N inhibitor with promising antitumor potency. Anti-Cancer Drugs 2016;27:496–507.
- Pape VFS, Toth S, Furedi A, et al. Design, synthesis and biological evaluation of thiosemicarbazones, hydrazinobenzothiazoles and arylhydrazones as anticancer agents with a potential to overcome multidrug resistance. Eur J Med Chem 2016;117:335–54.
- Chen G, Ning Y, Zhao W, et al. Synthesis, neuro-protection and anti-cancer activities of simple isatin mannich and schiff bases. Lett Drug Des Discov 2016;13:395–400.
- Rana S, Blowers EC, Tebbe C, et al. Isatin derived spirocyclic analogues with α-methylene-γ-butyrolactone as anticancer agents: a structure-activity relationship study. J Med Chem 2016;59:5121–7.
- Premanathan M, Radhakrishnan S, Kulangiappar K, et al. Antioxidant & anticancer activities of isatin (1H-indole-2,3-dione), isolated from the flowers of Couroupita guianensis Aubl. Indian J Med Res 2012;136:822–6.
- Pettersson M, ed. Sunitinib (Sutent): an angiogenesis inhibitor. Hoboken (NJ): John Wiley & Sons, Inc; 2010.
- Corona A, Meleddu R, Esposito F, et al. Ribonuclease H/DNA polymerase HIV-1 reverse transcriptase dual inhibitor: mechanistic studies on the allosteric mode of action of isatin-based compound RMNC6. PLoS One 2016;11:e0147225/1-e/18.
- Meleddu R, Distinto S, Corona A, et al. (3Z)-3-(2-[4-(aryl)-1,3-thiazol-2-yl]hydrazin-1-ylidene)-2,3-dihydro-1H-indol-2-one derivatives as dual inhibitors of HIV-1 reverse transcriptase. Eur J Med Chem 2015;93:452–60.
- Tavari M, Malan SF, Joubert J. Design, synthesis, biological evaluation and docking studies of sulphonyl isatin derivatives as monoamine oxidase and caspase-3 inhibitors. MedChemComm 2016. [Epub ahead of print]. doi: 10.1039/C6MD00228E.
- Akdemir A, Guzel-Akdemir O, Karali N, Supuran CT. Isatin analogs as novel inhibitors of Candida spp. β-carbonic anhydrase enzymes. Bioorg Med Chem 2016;24:1648–52.
- Chundawat TS, Kumari P, Sharma N, Bhagat S. Strategic synthesis and in vitro antimicrobial evaluation of novel difluoromethylated 1-(1, 3-diphenyl-1H-pyrazol-4-yl)-3, 3-difluoro-1, 3-dihydro-indol-2-ones. Med Chem Res 2016. [Epub ahead of print]. doi: 10.1007/s00044-016-1658-z.
- Sahoo S, Mahendrakumar CB, Setty CM. Synthesis, antiinflammatory and antibacterial activities of some substituted isatin and isatin fused with 3-substituted 4-amino-5-mercapto-1, 2, 4-triazoles. Int J Chem Sci 2015;13:613–24.
- Roopan SM, Khan FRN, Selvan NT. Synthesis of 1-[(2-chloroquinolin-3-yl)methyl]indoline-2,3-dione derivatives as potential antimicrobials. J Pharm Res 2010;3:950–2.
- Wang G, Peng Z, He D, Yan C, Liu W, inventors; Jishou University, Peop. Rep. China. assignee. Coumarin-isatin type compound useful in treatment of diabetes mellitus and its preparation patent CN105237521A; 2016.
- Havrylyuk D, Zimenkovsky B, Vasylenko O, et al. Synthesis of new 4-thiazolidinone-, pyrazoline-, and isatin-based conjugates with promising antitumor activity. J Med Chem 2012;55:8630–41.
- Ibrahim HS, Abou-Seri SM, Tanc M, et al. Isatin-pyrazole benzenesulfonamide hybrids potently inhibit tumour-associated carbonic anhydrase isoforms IX and XII. Eur J Med Chem 2015;103:583–93.
- Ozgun DO, Yamali C, Gul HI, Taslimi P, Gulcin I, Yanik T, et al. Inhibitory effects of isatin Mannich bases on carbonic anhydrases, acetylcholinesterase, and butyrylcholinesterase. J Enzyme Inhib Med Chem 2016. [Epub ahead of print]. doi: 10.3109/14756366.2016.1149479.
- Eldehna WM, Fares M, Ceruso M, et al. Amido/ureidosubstituted benzenesulfonamides-isatin conjugates as low nanomolar/subnanomolar inhibitors of the tumour-associated carbonic anhydrase isoform XII. Eur J Med Chem 2016;110:259–66.
- Guzel-Akdemir O, Akdemir A, Karali N, Supuran CT. Discovery of novel isatin-based sulphonamides with potent and selective inhibition of the tumour-associated carbonic anhydrase isoforms IX and XII. Org Biomol Chem 2015;13:6493–9.
- Zhou Y, Mokhtari RB, Pan J, et al. Carbonic anhydrase II mediates malignant behavior of pulmonary neuroendocrine tumors. Am J Respir Cell Mol Biol 2015;52:183–92.
- Imtaiyaz Hassan M, Shajee B, Waheed A, et al. Structure, function and applications of carbonic anhydrase isozymes. Bioorg Med Chem 2013;21:1570–82.
- Supuran CT. Inhibition of carbonic anhydrase IX as a novel anticancer mechanism. World J Clin Oncol 2012;3:98–103.
- Swietach P, Hulikova A, Vaughan-Jones RD, Harris AL. New insights into the physiological role of carbonic anhydrase IX in tumour pH regulation. Oncogene 2010;29:6509–21.
- Boron WF. Evaluating the role of carbonic anhydrases in the transport of HCO3-related species. Biochim Biophys Acta 2010;1804:410–21.
- Swietach P, Vaughan-Jones RD, Harris AL. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev 2007;26:299–310.
- Domsic JF, Avvaru BS, Kim CU, et al. Entrapment of carbon dioxide in the active site of carbonic anhydrase II. J Biol Chem 2008;283:30766–71.
- Geers C, Gros G. Carbon dioxide transport and carbonic anhydrase in blood and muscle. Physiol Rev 2000;80:681–715.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Supuran CT. Carbonic anhydrase inhibitors in the treatment and prophylaxis of obesity. Expert Opin Ther Pat 2003;13:1545–50.
- Picard F. Topiramate reduces energy and fat gains in lean (Fa/?) and obese (fa/fa) Zucker rats. Obes Res 2000;8:656–63.
- Carta F, Scozzafava A, Supuran CT. Sulphonamides: a patent review (2008-2012). Expert Opin Ther Pat 2012;22:747–58.
- Suthar SK, Bansal S, Lohan S, et al. Design and synthesis of novel 4-(4-oxo-2-arylthiazolidin-3-yl)benzenesulfonamides as selective inhibitors of carbonic anhydrase IX over I and II with potential anticancer activity. Eur J Med Chem 2013;66:372–9.
- Supuran Claudiu T. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- Pala N, Micheletto L, Sechi M, et al. Carbonic anhydrase inhibition with benzenesulfonamides and tetrafluorobenzenesulfonamides obtained via click chemistry. ACS Med Chem Lett 2014;5:927–30.
- Alterio V, Di Fiore A, D'Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Meleddu R, Maccioni E, Distinto S, et al. New 4-[(3-cyclohexyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene) amino]benzene-1-sulphonamides, synthesis and inhibitory activity toward carbonic anhydrase I, II, IX, XII. Bioorganic Med Chem Lett 2015;25:3281–4.
- Grandane A, Tanc M, Di Cesare Mannelli L, et al. 6-Substituted sulfocoumarins are selective carbonic anhydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J Med Chem 2015;58:3975–83.
- Bozdag M, Ferraroni M, Nuti E, et al. Combining the tail and the ring approaches for obtaining potent and isoform-selective carbonic anhydrase inhibitors: solution and X-ray crystallographic studies. Bioorg Med Chem 2014;22:334–40.
- Supuran CT, Winum J-Y. Carbonic anhydrase IX inhibitors in cancer therapy: an update. Future Med Chem 2015;7:1407–14.
- De Monte C, Carradori S, Gentili A, et al. Dual cyclooxygenase and carbonic anhydrase inhibition by nonsteroidal anti-inflammatory drugs for the treatment of cancer. Curr Med Chem 2015;22:2812–18.
- Anighoro A, Bajorath J, Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem 2014;57:7874–87.
- Rosini M. Polypharmacology: the rise of multitarget drugs over combination therapies. Future Med Chem 2014;6:485–7.
- Akbaba Y, Akincioglu A, Gocer H, et al. Carbonic anhydrase inhibitory properties of novel sulfonamide derivatives of aminoindanes and aminotetralins. J Enzyme Inhib Med Chem 2014;29:35–42.
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–9.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54.
- Senturk M, Gulcin I, Beydemir S, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9.