
Abstract
In this study, 4-[3-(4-hydroxyphenyl)-5-aryl-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide (1–9) types compounds were synthesized and their chemical structures were confirmed by 1H NMR, 13C NMR and HRMS spectra. Cytotoxic and carbonic anhydrase (CA) inhibitory effects of the compounds were investigated. Cytotoxicity experiments pointed out that compound 4, (4-[5-(4-chlorophenyl)-3-(4-hydroxyphenyl)-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide), exerting the highest tumor selectivity (TS) and potency selectivity expression (PSE) values, can be considered as a lead compound of this study in terms of development of novel anticancer agents. All synthesized sulfonamides showed a good inhibition profile on hCA IX and XII in the range of 53.5–923 nM and 6.2–95 nM, respectively. These compounds were 2.5–13.4 times more selective for the inhibition of hCA XII versus hCA IX, except compound 2 which had similar inhibitory action towards both isoenzymes.
Introduction
Cancer is a disease characterized by uncontrolled cell division which spread throughout the body and cause damage to essential organs. Although there are several strategies for the treatment of cancer, chemotheraphy is the most preferable method for inoperable cancers and medicinal chemists interest chemotheraphy parts. Despite several drugs are available in market, they have several problems such as side effects, stability, selectivity or gained resistance problems. So there is an urgent need to find new drug candidate compounds with high selectivity to the cancer cellsCitation1,Citation2.
Carbonic anhydrases (CAs, EC 4.2.1.1) belong to the family of zinc metalloenzymes found in a diversity of organisms and primarily responsible for catalyzing simple fundamental reaction, i.e. CO2 hydration to bicarbonate (HCO3−) and proton (H+)Citation3,Citation4. Sixteen CA isoenzymes have been identified till now. These enzymes differ in their subcellular localization, catalytic activity and susceptibility to different classes of inhibitors. Some of them are cytosolic (CA I, CA II, CA III, CA VII and CA XIII), others are membrane bound (CA IV, CA IX, CA XII and CA XIV), two are mitochondrial (CA VA and CA VB), and one is secreted in saliva (CA VI)Citation5,Citation6. Different CAs play vital roles in various physiological processes, including respiration, calcification, acid-base balance, bone resorption, etc.Citation7–9. They are also involved in a number of biosynthetic pathways such as gluconeogenesis, ureagenesis, and lipogenesis as well as in pathological disorders including edema, glaucoma, obesity and epilepsyCitation7. Targeting/inhibiting a particular CA is often associated with treatment of a particular disease/syndrome, e.g. CA II for antiglaucoma drug, CA VA/VB for antiobesity drug, CA VII/XIV for anticonvulsant, CA IX/XII for antitumor drug etc.Citation10.
In the past few years, several new tumor cell targets have been identified which led to the emergence of CA isozymes as promising targetCitation11. Since hCA IX and XII have been established to contribute to pH regulation of tumor cells, cell proliferation, cell adhesion and malignant cell invasion, they have been considered as valuable markers for cancer and are being targeted for designing anticancer drugs. In addition, CA IX and XII isoenzymes play a critical role in cell survival of hypoxic tumorsCitation12–14.
The selective inhibition of CA IX and XII provide significant antitumor/antimetastatic effectsCitation6,Citation15,Citation16. Unfortunately, classical CA inhibitors do not selectively target CA IX and XII. They also inhibit other types of CA isoenzymes which have physiological relevance such as CA I and IICitation17,Citation18.
The sulfonamides are an important drug class more than 70 years for their antibacterial, antiCA, diuretic, hypoglycemic, and anticancer activitiesCitation19–27. In addition, sulfonamide derivatives E7010, ER-34410 and E7070 have recently been reported as potent antitumor agents and are in advanced clinical trialsCitation28. Aromatic or heterocyclic compounds containing primary sulfonamide group have been extensively studied as important scaffolds for the development of new carbonic anhydrase inhibitors (CAIs). Sulfonamide derivatives such as acetazolamide (AZA), methazolamide (MZA), ethoxzolamide (EZA), pazopanib etc. () are widely used as CAIs in clinical trialsCitation29–31.
Figure 1. Chemical structures of some carbonic anhydrase inhibitors which are in clinical use.
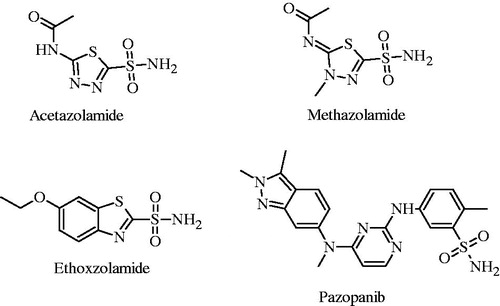
Pyrazol(in)e derivatives were reported their wide range of bioactivities such as anticancer, antiinflammatory, antiinfective, carbonic anhydrase inibitory and analgesic activitiesCitation32–38. Celecoxib, a clinically used nonsteroidal antiinflammatory drug that selectively inhibits COX-2, has sulfonamide and pyrazole scaffolds in its chemical structureCitation39. On the other hand, it was reported that compounds carring phenol moiety had CA inhibitory effectCitation40–45.
Our group recently focused on the synthesis of the compounds having both pyrazole-sulfonamide pharmacophores in a molecule to search for several bioactivitiesCitation32,Citation33,Citation37. To further extend these lines of studies, the present study aims to synthesize of 4-[3-(4-hydroxyphenyl)-5-aryl-4,5-dihydro-pyrazol-1-yl]benzenesulfonamides which has pyrazole, sulfonamide and phenolic pharmacophores all together to investigate their cytotoxic/anticancer activities and also their effects on hCA IX and XII which are tumor associated CA isoenymes, expecting to find out new candidate compound/s for further studies.
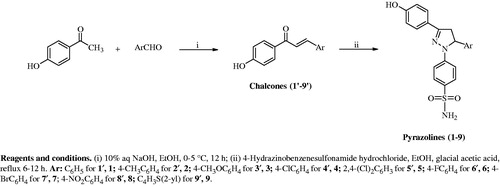
Scheme 1. Synthesis of compounds 1–9.
Materials and methods
Experimental
Melting points were determined using an Electrothermal 9100 (Bibby Scientific Limited, Staffordshire UK) instrument and are uncorrected. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were obtained using a Varian Mercury Plus spectrometer (Palo Alto, CA). Chemical shifts (δ) are reported in ppm. Mass spectra were undertaken on an HPLC-TOF Waters Micromass LCT Premier XE (Milford, MA) mass spectrometer using an electrospray ion source (ESI).
General procedure for the synthesis of chalcones (Scheme 1, 1′–9′)
Aqueous solution of NaOH (10%,10 ml) was added into the ethanol (6 ml) solution of suitable arylaldehyde (20.0 mmol) and 4-hydroxyacetophenone (20.0 mmol). The mixture was stirred overnight at room temperature and then it was poured on ice-water (100 ml) in a beaker. The mixture was neutralized with solution of HCl (10%, 8.5 ml)Citation46. The colored precipitate formed was filtered and crystallized from suitable solvent at room temperature. The crystallization solvent was ethanol-water (1′, 3′, 4′, 6′, 9′) or methanol-water (2′, 5′, 7′, 8′). The yields of the chalcones were in the range of 15–38% [1′ (38%), 2′ (29%), 3′ (18%), 4′ (15%), 5′ (37%), 6′ (17%), 7′ (34%), 8′ (29%), 9′ (27%)].
General procedure for the synthesis of pyrazolines (Scheme 1, 1–9)
A suitable chalcone (1.00 mmol) and 4-hydrazinobenzenesulfonamide hydrochloride (1.10 mmol) were solved in ethanol [25 ml (7), 30 ml (2, 3, 6, 8), 50 ml (1), 60 ml (4), 70 ml (5, 9)] and then catalytic amount of glacial acetic acid was added and the mixture was refluxedCitation32,Citation33,Citation37 [6 h (9), 9 h (2, 7), 10 h (1), 11 h (4, 6), 12 h (3, 5, 8)]. Reactions were followed by thin layer chromotography (TLC). After the reaction was stopped, some of the solvent was removed under vacuum and the mixture was stirred for 12 h. The obtained solid was filtered, dried at room temperature and crystallized from suitable solvent. It was methanol-chloroform (1), methanol (2, 3), methanol-ether (4, 5, 6, 7, 8, 9). Since hydrogens of SO2NH2 exchanged with deuterium of CD3OD, sulfonamide hydrogens were not observed on 1H NMR spectra.
4-[3-(4-Hydroxyphenyl)-5-phenyl-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide (1)
M.p. 208–210 °C. Yield: 47%. 1H NMR (400 MHz, CD3OD, ppm) δ = 7.65 (d, 2H, J = 8.8 Hz), 7.64 (d, 2H, J = 9.0 Hz), 7.36–7.24 (m, 5H), 7.08 (d, 2H, J = 9.0 Hz), 6.84 (d, 2H, J = 8.8 Hz), 5.43 (dd, 1H, J = 12.0, 5.7 Hz), 3.92 (dd, 1H, J = 17.4, 12.0 Hz), 3.13 (dd, 1H, J = 17.4, 5.7 Hz); 13C NMR (100 MHz, CD3OD, ppm) δ = 160.2, 151.5, 148.7, 143.5, 132.8, 130.3, 128.9, 128.8, 128.5, 126.9, 125.0, 116.5, 113.3, 64.5, 44.7; HRMS (ESI-MS): calcd. for C21H20N3O3S [M + H]+ 394.1225; found 394.1217.
4-[3-(4-Hydroxyphenyl)-5-p-tolyl-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide (2)
M.p. 163–164 °C. Yield: 75%. 1H NMR (400 MHz, CD3OD, ppm) δ = 7.62 (d, 2H, J = 8.8 Hz), 7.61 (d, 2H, J = 9.1 Hz), 7.20–7.10 (m, 4H), 7.05 (d, 2H, J = 8.8 Hz), 6.82 (d, 2H, J = 8.8 Hz), 5.37 (dd, 1H, J = 12.1, 5.5 Hz), 3.87 (dd, 1H, J = 17.4, 12.1 Hz), 3.08 (dd, 1H, J = 17.4, 5.5 Hz), 2.28 (s, 3H, –CH3); 13C NMR (100 MHz, CD3OD, ppm) δ = 159.0, 150.3, 147.5, 139.2, 137.4, 131.5, 129.6, 127.7, 127.3, 125.6, 123.8, 115.3, 112.1, 63.1, 43.5, 19.9; HRMS (ESI-MS): calcd. for C22H22N3O3S [M + H]+ 408.1382; found 408.1367.
4-[3-(4-Hydroxyphenyl)-5-(4-methoxyphenyl)-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide (3)
M.p. 176–178 °C. Yield: %23. 1H NMR (400 MHz, CD3OD, ppm) δ = 7.62 (d, 2H, J = 8.8 Hz), 7.61 (d, 2H, J = 8.8 Hz), 7.16 (d, 2H, J = 8.8 Hz), 7.06 (d, 2H, J = 8.8 Hz), 6.86 (d, 2H, J = 8.8 Hz), 6.82 (d, 2H, J = 8.8 Hz), 5.36 (dd, 1H, J = 12.1, 5.5 Hz), 3.86 (dd, 1H, J = 17.2, 12.1 Hz), 3.74 (s, 3H, -OCH3), 3.09 (dd, 1H, J = 17.2, 5.5 Hz); 13C NMR (100 MHz, CD3OD, ppm) δ = 159.5, 159.0, 150.3, 147.6, 134.1, 131.4, 127.7, 127.2, 126.9, 123.9, 115.3, 114.4, 112.1, 62.9, 54.5, 43.5; HRMS (ESI-MS): calcd. for C22H22N3O4S [M + H]+ 424.1331; found 424.1312.
4-[5-(4-Chlorophenyl)-3-(4-hydroxyphenyl)-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide (4)
M.p. 152–154 °C. Yield: 62%. 1H NMR (400 MHz, CD3OD, ppm) δ = 7.64 (d, 2H, J = 8.8 Hz), 7.63 (d, 2H, J = 8.8 Hz), 7.32 (d, 2H, J = 8.4 Hz), 7.26 (d, 2H, J = 8.4 Hz), 7.05 (d, 2H, J = 9.2 Hz), 6.82 (d, 2H, J = 8.8 Hz), 5.45 (dd, 1H, J = 12.1, 5.5 Hz), 3.91 (dd, 1H, J = 17.2, 12.1 Hz), 3.12 (dd, 1H, J = 17.2, 5.5 Hz); 13C NMR (100 MHz, CD3OD, ppm) δ = 159.1, 150.3, 147.3, 141.0, 133.3, 131.9, 129.1, 127.8, 127.5, 127.4, 123.6, 115.3, 112.1, 62.6, 43.3; HRMS (ESI-MS): calcd. for C21H19ClN3O3S [M + H]+ 428.0836; found 428.0824.
4-[5-(2,4-Dichlorophenyl)-3-(4-hydroxyphenyl)-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide (5)
M.p. 246–248 °C. Yield: 74%. 1H NMR (400 MHz, CD3OD, ppm) δ = 7.66 (d, 2H, J = 8.8 Hz), 7.62 (d, 2H, J = 8.8 Hz), 7.56 (d, 1H, J = 2.2 Hz), 7.20 (dd, 1H, J = 8.4, 2.2 Hz), 7.02 (d, 1H, J = 8.4 Hz), 6.97 (d, 2H, J = 8.8 Hz), 6.81 (d, 2H, J = 8.8 Hz), 5.69 (dd, 1H, J = 12.1, 5.5 Hz), 3.97 (dd, 1H, J = 17.4, 12.1 Hz), 3.06 (dd, 1H, J = 17.4, 5.5 Hz); 13C NMR (100 MHz, CD3OD, ppm) δ = 159.2, 150.6, 146.9, 137.6, 134.1, 132.8, 132.2, 129.7, 128.2, 127.9, 127.8, 127.6, 123.4, 115.4, 111.9, 60.0, 41.9; HRMS (ESI-MS): calcd. for C21H17Cl2N3O3S [M–H]− 460.0289; found 460.0282.
4-[5-(4-Fluorophenyl)-3-(4-hydroxyphenyl)-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide (6)
M.p. 243–244 °C. Yield: 72%. 1H NMR (400 MHz, CD3OD, ppm) δ = 7.63 (d, 4H, J = 8.8 Hz), 7.30–7.26 (m, 2H), 7.07–7.03 (m, 4H), 6.82 (d, 2H, J = 8.8 Hz), 5.45 (dd, 1H, J = 12.1, 5.5 Hz), 3.90 (dd, 1H, J = 17.6, 12.1 Hz), 3.11 (dd, 1H, J = 17.6, 5.5 Hz); 13C NMR (100 MHz, CD3OD, ppm) δ = 159.1, 150.3, 147.4, 138.2, 131.8, 127.8, 127.7, 127.3, 123.7, 115.8, 115.6, 115.3, 112.1, 62.6, 43.5; HRMS (ESI-MS): calcd. for C21H19FN3O3S [M + H]+ 412.1131; found 412.1115.
4-[5-(4-Bromophenyl)-3-(4-hydroxyphenyl)-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide (7)
M.p. 174–175 °C. Yield: 38%. 1H NMR (400 MHz, CD3OD, ppm) δ = 7.64 (d, 2H, J = 8.8 Hz), 7.63 (d, 2H, J = 8.8 Hz), 7.48 (d, 2H, J = 8.4 Hz), 7.19 (d, 2H, J = 8.4 Hz), 7.05 (d, 2H, J = 9.1 Hz), 6.82 (d, 2H, J = 8.8 Hz), 5.42 (dd, 1H, J = 12.1, 5.5 Hz), 3.91 (dd, 1H, J = 17.6, 12.1 Hz), 3.12 (dd, 1H, J = 17.6, 5.5 Hz); 13C NMR (100 MHz, CD3OD, ppm) δ = 159.1, 150.3, 147.3, 141.5, 132.1, 131.9, 127.8, 127.7, 127.4, 123.6, 121.2, 115.3, 112.1, 62.7, 43.3; HRMS (ESI-MS): calcd. for C21H19BrN3O3S [M + H]+ 472.0330; found 472.0317.
4-[3-(4-Hydroxyphenyl)-5-(4-nitrophenyl)-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide (8)
M.p. 173–176 °C. Yield: 61%. 1H NMR (400 MHz, CD3OD, ppm) δ = 8.20 (d, 2H, J = 8.8 Hz), 7.65 (d, 2H, J = 9.2 Hz), 7.64 (d, 2H, J = 8.8 Hz), 7.51 (d, 2H, J = 8.8 Hz), 7.05 (d, 2H, J = 9.2 Hz), 6.82 (d, 2H, J = 8.8 Hz), 5.60 (dd, 1H, J = 12.1, 5.5 Hz), 3.97 (dd, 1H, J = 17.6, 12.1 Hz), 3.17 (dd, 1H, J = 17.6, 5.5 Hz); 13C NMR (100 MHz, CD3OD, ppm) δ = 159.2, 150.3, 149.5, 147.7, 147.2, 132.3, 127.9, 127.5, 127.1, 124.2, 123.4, 115.4, 112.1, 62.6, 43.1; HRMS (ESI-MS): calcd. for C21H18N4O5S [M–H]− 437.0920; found 437.0931.
4-[3-(4-Hydroxyphenyl)-5-(thiophen-2-yl)-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide (9)
M.p. 220–221 °C. Yield: 21%. 1H NMR (400 MHz, CD3OD, ppm) δ = 7.66 (d, 2H, J = 9.1 Hz), 7.64 (d, 2H, J = 8.8 Hz), 7.26 (d, 1H, J = 5.0 Hz), 7.17 (d, 2H, J = 8.8 Hz), 7.06 (d, 1H, J = 3.2 Hz), 6.93 (dd, 1H, J = 5.0, 3.2 Hz), 6.83 (d, 2H, J = 8.8 Hz), 5.77 (dd, 1H, J = 11.5, 5.1 Hz), 3.88 (dd, 1H, J = 17.2, 11.5 Hz), 3.26 (dd, 1H, J = 17.2, 5.1 Hz); 13C NMR (100 MHz, CD3OD, ppm) δ = 159.1, 150.5, 147.7, 145.4, 132.1, 127.8, 127.2, 126.7, 124.9, 124.8, 123.7, 115.3, 112.5, 59.3, 43.7; HRMS (ESI-MS): calcd. for C19H18N3O3S2 [M + H]+ 400.0790; found 400.0789.
Biological activity
Cytotoxicity assay
The cytotoxicity of the compounds 1–9 were assayed towards human oral squamous cell carcinoma cell lines derived from gingiva tissue (CA9–22) and tongue (HSC-2, HSC-3, HSC-4), and human normal oral cells (gingival fibroblasts, HGF; periodontal ligament fibroblasts, HPLF; pulp cells, HPC) with some minor modificationsCitation33,Citation43,Citation47–50. In brief, cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). Cells (2.5 × 103 cells/well) were inoculated and incubated for 48 h to achieve complete adherence. Near confluent cells were incubated for a further 48 h in the fresh culture medium containing each test compound (3.12, 6.25, 12.5, 25, 50, 100, 200, 400 μM) or 5-FU (positive control) (7.8, 15.6, 31.2, 62.5, 125, 250, 500, 1000 μM). The viable cell numbers were determined by the MTT method. Cytotoxicity induced by DMSO (0.0078, 0.156, 0.03125, 0.0625, 0.125, 0.25, 0.5 or 1%) was subtracted from each well. The CC50 values were determined from dose-response curves. The tumor selectivity (TS) was calculated by the following equation: TS = mean CC50 against normal cells/mean CC50 against cancer cells [shown as (D/B) or (C/A) in ]. A potency selectivity expression (PSE) was devised which is the product of the reciprocal of average CC50 values towards cancer cell lines and the average SI values towards these cell lines and expressed as a percentageCitation49.
Table 1. Cytotoxic activity of compounds 1–9 against human oral malignant and nonmalignant cells.
Carbonic anhydrase enzyme assay
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration reactionCitation51. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition percantage were obtained by using PRISM 3, as reported earlierCitation52, and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation53.
Results and discussion
The compounds were succesfully synthesized and their chemical structures were elucidated by 1H NMR, 13C NMR, and HRMS spectra as shown in experimental section. The cytotoxic effects of the compounds were assayed towards human oral malignant (Ca9–22, HSC-2, HSC-3 and HSC-4) and nonmalignant (HGF, HPLF and HPC) cells by MTT methodCitation33,Citation43,Citation47–50. The results were shown in .
The first question to be answered is whether the compounds have antineoplastic property or not. CC50 (the concentration of the compound that kills 50% of the cells as mol/L) values of the compounds were in the range of 39.3–99.3 μM while reference compound 5-Fluorouracile (5-FU)’s changed in the range of 13–29 μM. It can be said that the compounds studied here have antineoplastic properties since they are effective at micromolar level. However, they are less cytotoxic than 5-FU towards cancer cell lines ().
The second aspect of the compounds to be considered is whether they are tumor-specific cytotoxins since tumors are surrounded by different types of normal cells in oral cavity. Selectivity index (SI) value, which is the quotient of the average CC50 value of the nonmalignant cells and the CC50 value of a compound towards a specific malignant cell line, was generated in . The compounds which have SI values of >1 can be considered as tumor-specific antineoplastic agentsCitation54. So, it can be said that the most of the compounds have shown tumor specificity against all cancer cell lines, except the compounds 5, 7 and 8 towards HSC-2 cell (). The highest SI value of 2.2 was calculated for the compound 4 towards Ca9–22 cancer cell line.
The TS of the compounds were calculated by two types of calculationsCitation49. The first calculation was made by dividing the average CC50 value towards normal cells into the average CC50 value towards a total of four cancer cell lines (TS = Column D/Column B, ). The second calculation is the comparison of malignant (Ca9–22) and nonmalignant (HGF) cells which has the same tissue origine (gingiva). TS values were calculated by dividing the CC50 value towards HGF cells by the CC50 value towards Ca9–22 cells (TS = Column C/Column A, ). Both types of TS calculations demonstrated that compound 4 showed the highest tumor-specificity (TS = 1.5 and 2.2, respectively).
Lead compound should possess both marked cytotoxic potency and also selective toxicity for tumors. In order to identify such molecule, a PSE was devised which is the product of the reciprocal of average CC50 values towards cancer cell ines and the average SI values towards these cell lines and expressed as a percentageCitation49 (). When PSE values were considered, all compounds had lower PSE values than the reference compound 5-FU. PSE values of the compounds studied were in the range of 1.6–2.6. The chlorine substituted compound 4 had the highest PSE value of 2.6 among the series ().
According to TS and PSE values; it seems that the compound 4, 4-[5-(4-chloro-phenyl)-3-(4-hydroxy-phenyl)-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide, can be considered as a leader compound of this study in terms of cytotoxicity and can be used for further developments.
The compounds 1–9 were also tested in terms of CA inhibition profile on hCA IX and XII which are important isoenzymes taking part important roles in cancer biology, especially at the regulation of extracellular pH of cancer cells. The inhibitory constant (Ki) values of the compounds synthesized were in the range of 53.5–923 nM towards hCA IX while they were in the range of 6.2–68.9 nM towards hCA XII (). The compounds tested were 2.5–13.4 times more selective towards hCA XII isoenzyme than hCA IX isoenzyme, except compound 2.
Table 2. Effects of compounds 1–9 on hCA IX and XII isoenzymes activity.
When the effects of the substituents on CA inhibition were considered the compound 8 which has electron attracting nitro substituent on phenyl ring had the lowest Ki values towards both isoenzymes among the compounds studied 1–8. Compound 8 having nitro substituent had 14.5 and 9.2 times more powerful inhibition potential than the compound 1, which is nonsubstituted phenyl derivative. Replacement of benzene ring by thiophene ring is often used in medicinal chemistry to modify bioactivity of a compound since benzene and thiophene are bioisosteric rings. In this study, replacement of benzene ring by thiophene increased CA inhibitory potential by decreasing the Ki value. When the compounds 1 with benzene and 9 with thiophene were compared, 9 was more potent inhibitor than 1. Inhibitory potential of 9 was 17.3 and 11.1 times more potent than 1 towards hCA IX and XII, respectively.
Any type of substitution on phenyl ring increased the inhibition potential of the compounds by decreasing the Ki values towards both isoenzymes, except the compound 2 towards hCA XII isoenzyme. When halogen bearing compounds were compared, the order of inhibition potency of the compounds was as follows: compound 7 with bromine (Ki = 66.2 nM) > compound 6 with fluorine (Ki = 84.1 nM) > compound 4 with chlorine (Ki = 97.7 nM) towards hCA IX isoenzyme. It was as follows towards hCA XII isoenzyme: compound 7 (Ki = 7.6 nM) > compound 6 (Ki = 8.6 nM) > compound 4 (Ki = 38.8 nM). The potency order of the compounds towards hCA IX an XII the same as 7 > 6 > 4. There was no relation between the electronegativity of halogen and Ki values. Dichlorine substitution was found useful to increase the inhibition potency of the compound towards both isoenzymes in compound 5 comparing to compound 4, which has mono chlorine atom. Inhibition potential increased 7.5 times in compound 5 comparing to compound 1 towards hCA XII isoenzyme while 5 was 10.8 times more potent towards hCA IX isoenzyme than 1. When compounds 2 with methyl substituent and 3 with methoxy substituent were compared, introduction of oxygene into molecule 3 increased the inhibition potential 2.8 times towards hCA XII while there is a slight increase towards hCA IX (1.1 times) by the introduction of oxygene in 3 comparing with 2. The increased inhibition potential may be attributed to the possibility of hydrogen bonding with 3 comparing to 2.
Conclusion
Cytotoxicity results of the synthesized compounds revealed that compound 4, 4-[5-(4-chloro-phenyl)-3-(4-hydroxy-phenyl)-4,5-dihydro-pyrazol-1-yl]benzenesulfonamide, may be considered as a leader compound in terms of cytotoxic/anticancer activity. All studied compounds showed an impressive inhibiton profile on hCA IX and XII, with KIs in the range of 53.5–923 nM and 6.2–95 nM, respectively. Except 2, all compounds were 2.5–13.4 times more selective inhibitor towards hCA XII than hCA IX while compound 2 had similar selectivity towards both isoenzymes. All compounds reported here can be considered as leader compounds to develop new selective hCA XII inhibitors for further detailed studies.
Acknowledgements
The authors Halise Inci Gul and Ebru Mete thank to Ataturk University for financial support. This work was also supported by Grant-in-Aid for Challenging Exploratory Research from The Ministry of Education, Culture, Sports, Science and Technology (Sakagami H. 25670897).
Disclosure statement
The authors report no conflict of interest and are responsible for the contents and writing of the paper.
Related Research Data
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO(2) capture. J Enzyme Inhib Med Chem 2013;28:229–30.
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–89.
- Thiry A, Dogne JM, Masereel B, Supuran CT. Targeting tumor-associated carbonic anhydrase IX in cancer therapy. Trends Pharmacol Sci 2006;27:566–73.
- Taslimi P, Gulcin I, Ozgeris B, et al. The human carbonic anhydrase isoenzymes I and II (hCA I and II) inhibition effects of trimethoxyindane derivatives. J Enzyme Inhib Med Chem 2016;31:152–7.
- Gocer H, Topal F, Topal M, et al. Acetylcholinesterase and carbonic anhydrase isoenzymes I and II inhibition profiles of taxifolin. J Enzyme Inhib Med Chem 2016;31:441–7.
- Supuran CT. Inhibition of carbonic anhydrase IX as a novel anticancer mechanism. World J Clin Oncol 2012;3:98–103.
- Winum JY, Rami M, Scozzafava A, et al. Carbonic anhydrase IX: a new druggable target for the design of antitumor agents. Med Res Rev 2008;28:445–63.
- Svastova E, Hulikova A, Rafajova M, et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 2004;577:439–45.
- Sethi KK, Verma SM, Tanc M, et al. Carbonic anhydrase inhibitors: synthesis and inhibition of the human carbonic anhydrase isoforms I, II, IX and XII with benzene sulfonamides incorporating 4- and 3-nitrophthalimide moieties. Bioorg Med Chem 2014;22:1586–95.
- Grandane A, Tanc M, Zalubovskis R, Supuran CT. Synthesis of 6-aryl-substituted sulfocoumarins and investigation of their carbonic anhydrase inhibitory action. Bioorg Med Chem 2015;23:1430–6.
- Lock FE, McDonald PC, Lou Y, et al. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene 2013;32:5210–19.
- De Simone G, Supuran CT. Carbonic anhydrase IX: Biochemical and crystallographic characterization of a novel antitumor target. Biochim Biophys Acta 2010;1804:404–9.
- Khloya P, Ceruso M, Ram S, et al. Sulfonamide bearing pyrazolylpyrazolines as potent inhibitors of carbonic anhydrase isoforms I, II, IX and XII. Bioorg Med Chem Lett 2015;25:3208–12.
- SitaRam Celik G, Khloya P, et al. Benzenesulfonamide bearing 1,2,4-triazole scaffolds as potent inhibitors of tumor associated carbonic anhydrase isoforms hCA IX and hCA XII. Bioorg Med Chem 2014;22:1873–82.
- Drews J. Drug discovery: a historical perspective. Science 2000;287:1960–4.
- Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors: synthesis of sulfonamides incorporating dtpa tails and of their zinc complexes with powerful topical antiglaucoma properties. Bioorg Med Chem Lett 2001;11:575–82.
- Husain A, Madhesia D, Rashid M, et al. Synthesis and in vivo diuretic activity of some new benzothiazole sulfonamides containing quinoxaline ring system. J Enzyme Inhib Med Chem 2016;31:1682–9.
- Grandane A, Tanc M, Zalubovskis R, Supuran CT. Synthesis of 6-tetrazolyl-substituted sulfocoumarins acting as highly potent and selective inhibitors of the tumor-associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem 2014;22:1522–8.
- Kasimogullari R, Bulbul M, Mert S, Guleryuz H. Synthesis of 5-amino-1,3,4-thiadiazole-2-sulfonamide derivatives and their inhibition effects on human carbonic anhydrase isozymes. J Enzyme Inhib Med Chem 2011;26:231–7.
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74.
- Bayram E, Senturk M, Kufrevioglu OI, Supuran CT. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg Med Chem 2008;16:9101–5.
- Scozzafava A, Menabuoni L, Mincione F, Supuran CT. Carbonic anhydrase inhibitors. A general approach for the preparation of water-soluble sulfonamides incorporating polyamino-polycarboxylate tails and of their metal complexes possessing long-lasting, topical intraocular pressure-lowering properties. J Med Chem 2002;45:1466–76.
- Winum JY, Scozzafava A, Montero JL, Supuran CT. Therapeutic potential of sulfamides as enzyme inhibitors. Med Res Rev 2006;26:767–92.
- Shewach DS, Kuchta RD. Introduction to cancer chemotherapeutics. Chem Rev 2009;109:2859–61.
- Rogez-Florent T, Meignan S, Foulon C, et al. New selective carbonic anhydrase IX inhibitors: synthesis and pharmacological evaluation of diarylpyrazole-benzenesulfonamides. Bioorg Med Chem 2013;21:1451–64.
- Balseven H, Mustafa Isgor M, Mert S, et al. Facile synthesis and characterization of novel pyrazole-sulfonamides and their inhibition effects on human carbonic anhydrase isoenzymes. Bioorg Med Chem 2013;21:21–7.
- Gluszok S, Frederick R, Foulon C, et al. Design, solid-phase synthesis, and biological evaluation of novel 1,5-diarylpyrrole-3-carboxamides as carbonic anhydrase IX inhibitors. Bioorg Med Chem 2010;18:7392–401.
- Mete E, Comez B, Inci Gul H, et al. Synthesis and carbonic anhydrase inhibitory activities of new thienyl-substituted pyrazoline benzenesulfonamides. J Enzyme Inhib Med Chem 2016. [Epub ahead of print]. doi:10.1080/14756366.2016.1181627.
- Gul HI, Tugrak M, Sakagami H, et al. Synthesis and bioactivity studies on new 4-(3-(4-Substitutedphenyl)-3a,4-dihydro-3H-indeno[1,2-c]pyrazol-2-yl) benzenesulfonamides. J Enzyme Inhib Med Chem 2016;31:1619–24.
- Srinivasa Reddy T, Kulhari H, Ganga Reddy V, et al. Synthesis and biological evaluation of pyrazolo-triazole hybrids as cytotoxic and apoptosis inducing agents. Org Biomol Chem 2015;13:10136–49.
- Abdellatif KR, Elsaady MT, Abdel-Aziz SA, Abusabaa AH. Synthesis, cyclooxygenase inhibition and anti-inflammatory evaluation of new 1,3,5-triaryl-4,5-dihydro-1H-pyrazole derivatives possessing methanesulphonyl pharmacophore. J Enzyme Inhib Med Chem 2016;349:801–7.
- Viveka S, Dinesha Shama P, et al. Design and synthesis of some new pyrazolyl-pyrazolines as potential anti-inflammatory, analgesic and antibacterial agents. Eur J Med Chem 2015;101:442–51.
- Kucukoglu K, Oral F, Aydin T, et al. Synthesis, cytotoxicity and carbonic anhydrase inhibitory activities of new pyrazolines. J Enzyme Inhib Med Chem 2016. [Epub ahead of print]. doi:10.1080/14756366.2016.1217852.
- Deng H, Yu ZY, Shi GY, et al. Synthesis and in vitro antifungal evaluation of 1,3,5-trisubstituted-2-pyrazoline derivatives. Chem Biol Drug Des 2012;79:279–89.
- Domiati S, El-Mallah A, Ghoneim A, et al. Evaluation of anti-inflammatory, analgesic activities, and side effects of some pyrazole derivatives. Inflammopharmacology 2016;24:163–72.
- Scozzafava A, Passaponti M, Supuran CT, Gulcin I. Carbonic anhydrase inhibitors: guaiacol and catechol derivatives effectively inhibit certain human carbonic anhydrase isoenzymes (hCA I, II, IX and XII). J Enzyme Inhib Med Chem 2015;30:586–91.
- Karioti A, Ceruso M, Carta F, et al. New natural product carbonic anhydrase inhibitors incorporating phenol moieties. Bioorg Med Chem 2015;23:7219–25.
- Innocenti A, Beyza Ozturk Sarikaya S, Gulcin I, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of mammalian isoforms I-XIV with a series of natural product polyphenols and phenolic acids. Bioorg Med Chem 2010;18:2159–64.
- Gul HI, Yamali C, Yasa AT, et al. Carbonic anhydrase inhibition and cytotoxicity studies of Mannich base derivatives of thymol. J Enzyme Inhib Med Chem 2016;31:1375–80.
- Yamali C, Tugrak M, Gul HI, et al. The inhibitory effects of phenolic Mannich bases on carbonic anhydrase I and II isoenzymes. J Enzyme Inhib Med Chem 2016;31:1678–81.
- Bilginer S, Unluer E, Gul HI, et al. Carbonic anhydrase inhibitors. Phenols incorporating 2- or 3-pyridyl-ethenylcarbonyl and tertiary amine moieties strongly inhibit Saccharomyces cerevisiae β-carbonic anhydrase. J Enzyme Inhib Med Chem 2014;29:495–9.
- Dimmock JR, Kandepu NM, Hetherington M, et al. Cytotoxic activities of Mannich bases of chalcones and related compounds. J Med Chem 1998;41:1014–26.
- Yerdelen KO, Gul HI, Sakagami H, Umemura N. Synthesis and biological evaluation of 1,5-bis(4-hydroxy-3-methoxyphenyl)penta-1,4-dien-3-one and its aminomethyl derivatives. J Enzyme Inhib Med Chem 2015;30:383–8.
- Tugrak M, Yamali C, Sakagami H, Gul HI. Synthesis of mono Mannich bases of 2-(4-hydroxybenzylidene)-2, 3-dihydroinden-1-one and evaluation of their cytotoxicities. J Enzyme Inhib Med Chem 2015; 31:818–23.
- Bilginer S, Gul HI, Mete E, et al. 1-(3-Aminomethyl-4-hydroxyphenyl)-3-pyridinyl-2-propen-1-ones: a novel group of tumour-selective cytotoxins. J Enzyme Inhib Med Chem 2013;28:974–80.
- Sakagami H, Shimada C, Kanda Y, et al. Effects of 3-styrylchromones on metabolic profiles and cell death in oral squamous cell carcinoma cells. Toxicol Rep 2015;2:1281–90.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44.
- D'Ambrosio K, Smaine FZ, Carta F, et al. Development of potent carbonic anhydrase inhibitors incorporating both sulfonamide and sulfamide groups. J Med Chem 2012;55:6776–83.
- Robles-Escajeda E, Das U, Ortega NM, et al. A novel curcumin-like dienone induces apoptosis in triple-negative breast cancer cells. Cell Oncol (Dordr) 2016;39:265–77.