
Abstract
New coumaryl-thiazole derivatives with the acetamide moiety as a linker between the alkyl chains and/or the heterocycle nucleus were synthesized and in vitro tested as acetylcholinesterase (AChE) inhibitors. 2-(diethylamino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6c, IC50 value of 43 nM) was the best AChE inhibitor with a selectivity index of 4151.16 over BuChE. Kinetic study of AChE inhibition revealed that 6c was a mixed-type inhibitor. Moreover, the result of H4IIE hepatoma cell toxicity assay for 6c showed negligible cell death. Molecular docking studies were also carried out to clarify the inhibition mode of the more active compounds. Best pose of compound 6c is positioned into the active site with the coumarin ring wedged between the residues of the CAS and catalytic triad of AChE. In addition, the coumarin ring is anchored into the gorge of the enzyme by H-bond with Tyr130.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease and the most common form of dementia that affects aged peopleCitation1,Citation2. Currently, there is no cure for AD and the cholinergic strategy, in which the acetylcholine (ACh) level in brain has been increased by inhibiting acetylcholinesterase (AChE), remains the most effective therapeutic approach for the treatment of ADCitation3,Citation4.
Acetylcholinesterase (AChE; EC 3.1.1.7) is a hydrolase involved in the termination of impulse transmission at cholinergic synapses by rapid hydrolysis of the neurotransmitter ACh in the central and peripheral nervous system. AChE inhibitors (AChEI) inhibit the hydrolysis of ACh improving both the level and of duration of neurotransmitterCitation5,Citation6. Another cholinesterase, butyrylcholinesterase (BuChE; EC 3.1.1.8), primarily localized in plasma, liver and muscle tissues, able of hydrolyzing ACh and other acylcholines, differs from AChE for tissue distribution and sensitivity to substrates and inhibitorsCitation7. AChE inhibitors such as galantamine, rivastigmine and donepezil are the main stay drugs for the clinical management of AD in the early-to-moderate stage ()Citation8–12. Also, in clinical treatment of AD, selective AChE inhibitors have shown better therapeutic effects, compared with no selective inhibitorsCitation6,Citation13,Citation14. Therefore, the design of selective AChE inhibitors could represent a successful therapeutic strategy for the symptomatic treatment of AD and its progressionCitation15.
The analysis of the 3D structure of AChE revealed the presence of a deep and narrow gorge at the active site mainly composed of dual binding sites: the Ser-His-Glu catalytic site located at the bottom of gorge, and the peripheral anionic binding site (PAS) located at the gorge entranceCitation16–19.
Tricyclic and heterocyclic compounds, such as tacrine, quinolizidinyl and coumarin derivatives, are able to bind both PAS and catalytic anionic site (CAS) of the enzyme by hydrophobic interactions and T-stacking with the aromatic residues of the enzyme gorge in AChECitation20–23. Accordingly, the coumarin scaffold has been considered to design new AChE inhibitorsCitation8,Citation24,Citation25, and many efforts were addressed to synthesize dual binding site inhibitors of AChE by hybridizing a catalytic site interacting moiety with the coumarin scaffold through an appropriate spacerCitation26. Amidic or imidic substituents are key functionalities acting as hydrogen bond donors at the catalytic triad (Ser203-Glu334-His447) of the active site residues of the human acetylcholinesterase by the unpaired electron of N and O atomsCitation27–30. Thiazolo-triazine derivatives are considered efficient acetylcholinesterase inhibitors for the ability to form a hydrogen bond with Tyr124 and T-stacking interaction with Trp286Citation31.
Benzofuran and coumarin derivatives bearing thiazole ring and arylurea/thiourea moieties were synthesized as ChE inhibitors; however, they exhibited moderate inhibitory activities against AChE in our previous studiesCitation32,Citation33. Several studies have presented that benzofuran or coumarin molecules interacted with CAS and PAS of AChE via the steric and T-stacking interactions; the H-bonding and T-stacking interactions were determined between thiazole ring or amide moiety and PAS; the phenyl ring of urea moiety interacted with CAS by T-stackingCitation34,Citation35. It has also been reported that the cation–π interactions were observed between N-alkyl chains or heterocyclic moieties and mid-gorge site of AChECitation22,Citation36.
2-Aminothiazoles can be progressed to generate useful compounds is testified by a number of marketed drugs, including antibiotics such as dopamine agonists for the treatment of Parkinson’s disease, and riluzole, which is the 2-aminobenzothiazole derivative used to treat acute myeloid leukemia (AML)Citation37–39. Conversely, 2-aminothiazoles have displayed cytotoxicity and metabolic instability as antimycobacterial and antiplasmodial agentsCitation39. It has been presented that the 2-amido and heterocyclic thiazoles show little or no binding to any of the proteins (apical membrane antigen AMA1, the E3 ubiquitin ligase adapter protein SPSB2, two DsbA oxidoreductases from different bacterial species (oxidoreductase 1 and 2), carbonic anhydrase II and a kinase) and do not appear to be inherently promiscuousCitation39. However, some docking studies showed that the nitrogen atom of the thiazole ring might form a hydrogen bond and interact with active sites of AChECitation31,Citation32.
Based on the above consideration, we hypothesized that the presence of acetamide moiety, bearing N-alkyl chains and/or heterocycle instead of phenylurea, contributes to inhibitory activity of designed molecules via a cation–π interactions with active sites of AChE (. In this study, a novel series of 20 coumarin derivatives (6a-t) was synthesized, and their inhibitory effects on AChE and BuChE were evaluated. Additionally, kinetic study of AChE inhibition and molecular docking studies were carried out to clarify the inhibition mode of the more active compounds. Moreover, the potential toxicity effect on the hepatoma cell line H4IIE was also examined for the most potent compound 6c.
Methods
Chemistry
All solvents, reagents and starting materials were obtained from commercial sources unless otherwise indicated. Melting points were taken on a Barnstead Electrothermal 9200 (Staffordshire, UK). IR spectra were registered on a Shimadzu Prestige-21 (200 VCE) spectrometer (Columbia, MD). 1H and 13C NMR spectra were registered on a Varian Infinity Plus spectrometer (Palo Alto, CA) at 300 and at 75 Hz, respectively. 1H and 13C chemical shifts are referenced to the internal deuterated solvent. Mass spectra were obtained using MICROMASS Quattro LC-MS-MS spectrometer (Milford, MA). The elemental analyses were carried out with a Leco CHNS-932 instrument (St. Joseph, MI). Spectrophotometric analyses were performed by a BioTek Power Wave XS (Winooski, VT). The electric eel acetylcholinesterase (AChE, Type-VI-S, EC 3.1.1.7, 425.84 U/mg, Sigma) and horse serum butyrylcholinesterase (BuChE, EC 3.1.1.8, 11.4 U/mg, Sigma) were purchased from Sigma (Steinheim, Germany). The other chemicals and solvents were purchased from Fluka Chemie (Taufkirchen, Germany), Merck (Darmstadt, Germany), Alfa Aesar (Ward Hill, MA), and Sigma-Aldrich (Taufkirchen, Germany). Reactions were monitored by thin-layer chromatography (TLC) on 0.25 mm E. Merck silica gel plates (60F-254) and visualized under UV lamp (Camag, Switzerland). Thin-layer chromatography was provided by Merck (Darmstadt, Germany).
General procedures of synthesis and spectral data
3-Acetylcoumarin (2)
A mixture of salicylaldehyde (1) (3 mmol), ethyl acetoacetate (3 mmol) and piperidine (0.1 mmol) was stirred at room temperature for 30 min. After completion of the reaction, the mixture was recrystallized from ethanol to get pure crystalline 3-acetylcoumarin (2) in 95% yields. Spectral data of this compound matched with the literatureCitation40. 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.73 (3H, s), 7.33–7.39 (2H, m), 7.64–7.69 (2H, m), 8.52 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 30.9, 116.9, 118.4, 124.6, 125.2, 130.5, 137.0, 147.8, 155.5, 159.5, 195.7. TLC: Hexane:Ethylacetate (4:1).
3-(Bromoacetyl)coumarin (3)
To a solution of 3-acetylcoumarin (2) (0.01 mol) in 20 mL chloroform, a solution of bromine (0.01 mol) in 5 mL chloroform was added. The mixture was stirred at 50 °C for 15 min and then cooled. The obtained precipitate was filtered and washed with ether. The product was recrystallized from acetic acid. 3-(Bromoacetyl)coumarin (3) was obtained in 85% yields. Spectral data of this compound matched with the literatureCitation41. 1H NMR (DMSO-d6, 300 MHz) δ/ppm: 4.83 (2H, s), 7.28–7.49 (2H, m), 7.60–7.81 (2H, m), 8.56 (1H, s).
3-(2-Amino-1,3-thiazol-4-yl)coumarin (4)
To a solution of 3-(bromoacetyl)coumarin (3) (5 mmol) in boiling ethanol (20 mL), thiourea (5 mmol) was added. The mixture was refluxed for 1 h, then cooled and neutralized with aqueous ammonia. The precipitate was filtered off, washed with ethanol and used directly without re-crystallization or other purification. 3-(2-Amino-1,3-thiazol-4-yl)coumarin (4) was obtained in 80% yields. Spectral data of this compound matched with the literatureCitation42. mp. 226–228 °C; IR: 3376, 3310, 1694, 1642, 1504, 1376, 1093, 758; 1H NMR (DMSO-d6, 300 MHz) δ/ppm: 7.16 (2H, s, NH2), 7.33–7.43 (2H,m), 7.49 (1H,s), 7.59 (1H, t, J = 8.2 Hz), 7.82 (1H, d, J = 7.9 Hz), 8.49 (1H, s); 13C NMR (DMSO-d6, 75 MHz) δ/ppm: 109.4, 116.5, 119.9, 121.1, 125.3, 129.3, 132.1, 138.7, 143.9, 152.8, 159.4, 168.1.
2-Chloro-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (5)
To a solution of 3-(2-amino-1,3-thiazol-4-yl)coumarin (4) (6.5 mmol) in THF (30 mL), chloroacetylchloride (8.125 mmol) was added followed by a catalytic amount of Et3N. The mixture was refluxed for 10 h, then cooled and evaporated in vacuo. The residue was washed with water and dried in a vacuum-oven at 40 °C. Greenish powder, 96% yield, IR: 3171, 3134, 2981, 1707, 1658, 1568, 1316, 1094, 737; 1H NMR (DMSO-d6, 300 MHz) δ/ppm: 4.41 (2H, s), 7.36–7.47 (2H, m), 7.63 (1H, t, J = 7.02 Hz), 7.83 (1H, d, J = 7.9 Hz), 8.04 (1H, s), 8.57 (1H, s), 12.68 (1H, s, NH); LC-MS-MS(ESI-) (m/z) 320 [M+].
N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)-2-(RN-1-yl)acetamide (6a-t)
To a solution of 2-chloro-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (5) (1 mmol) in DMF, 1.25 mmol Et3N and 1.85 mmol amine derivatives were added. The mixture was refluxed overnight, poured on crushed ice then extracted with CH2Cl2. The organic layer was washed with water, dried over Na2SO4 and concentrated in vacuo. The products were recrystallized from ethanol over 95% purity. 6a-t were obtained with 20–80% yields.
2-(Methylamino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6a)
Yellow powder, 60% yield, mp. 168–170 °C; IR: 3291, 3138, 1708, 1689, 1557, 1250, 1178, 1032, 783 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.55 (3H, s), 3.50 (2H, s, CO-CH2-N), 7.28–7.38 (2H, m), 7.50–7.61 (2H, m), 8.15 (1H, s), 8.58 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 36.9, 54.1, 114.8, 116.3, 119.5, 121.0, 124.8, 128.4, 131.6, 138.8, 142.6, 152.9, 156.9, 159.7, 170.5; LC-MS-MS(ESI+) (m/z): 315.08 [M+]. Anal. Calcd for C15H13N3O3S: C, 57.13; H, 4.16; N, 13.33; found: C, 57.10; H, 4.19; N, 13.31.
N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)-2-(propylamino)acetamide (6b)
Light yellow powder, 71% yield, mp. 138–140 °C; IR: 3304, 3178, 2958, 1710, 1653, 1542, 1253, 1091, 784 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 0.99 (3H, t, J = 7.3 Hz), 1.59–1.66 (2H, m), 2.69 (2H, t, J = 7.3 Hz), 3.52 (2H, s, CO-CH2-N), 7.27–7.36 (2H, m), 7.50–7.61 (2H, m), 8.14 (1H, s), 8.57 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 11.8, 23.4, 52.1, 52.5, 115.3, 116.6, 119.7, 121.1, 124.8, 128.5, 131.6, 138.9, 142.7, 153.1, 156.8, 159.9, 170.7; LC-MS-MS(ESI-) (m/z): 343.03 [M+]. Anal. Calcd for C17H17N3O3S: C, 59.46; H, 4.99; N, 12.24; found: C, 59.41; H, 4.95; N, 12.28.
2-(Diethylamino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6c)
Light yellow powder, 59% yield, mp. 150–152 °C; IR: 3269 3148, 2975, 1726, 1703, 1528, 1435, 1269, 1092, 781 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.13 (6H, t, J = 7.02 Hz), 2.70 (4H, q, J = 7.02 Hz), 3.29 (2H, s, CO-CH2-N), 7.27–7.37 (2H, m), 7.52 (1H, t, J = 8.4 Hz), 7.60 (1H, d, J = 7.6 Hz), 8.14 (1H, s), 8.59 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 12.4, 49.1, 57.0, 115.4, 116.6, 119.7, 121.1, 124.8, 128.4, 131.6, 138.9, 142.8, 153.1, 156.8, 159.9, 170.7; LC-MS-MS(ESI-) (m/z): 357.05 [M+]. Anal. Calcd for C18H19N3O3S: C, 60.49; H, 5.36; N, 11.76; found: C, 60.47; H, 5.39; N, 11.72.
2-(Dibutylamino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6d)
Light yellow powder, 70% yield, mp. 166–168 °C; IR: 3289, 3142, 2956, 1724, 1700, 1525, 1269, 1093, 782 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 0.95 (6H, t, J = 7.3 Hz), 1.34–1.56 (8H, m), 2.59 (4H, t, J = 7.3 Hz, N-CH2), 3.30 (2H, s, CO-CH2-N), 7.28–7.37 (2H, m), 7.52 (1H, t, J = 8.2 Hz), 7.61 (1H, d, J = 7.6 Hz), 8.14 (1H, s), 8.59 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 14.2, 20.8, 29.5, 55.7, 58.2, 115.3, 116.6, 119.7, 121.2, 124.8, 128.5, 131.6, 139.0, 142.8, 153.2, 156.7, 159.9, 170.7; LC-MS-MS(ESI-) (m/z): 413.21 [M+]. Anal. Calcd for C22H27N3O3S: C, 63.90; H, 6.58; N, 10.16; found: C, 63.93; H, 6.56; N, 10.14.
2-(Diisopropylamino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6e)
Yellow powder, 20% yield, mp. 160–162 °C; IR: 3342, 3141, 2984, 1717, 1651, 1562, 1468, 1094, 747 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.31–1.42 (8H, m), 3.59–3.66 (6H, m), 4.05 (2H, s, CO-CH2-N), 7.22–7.33 (2H, m), 7.46 (1H, t, J = 7.3 Hz), 7.53 (1H, d, J = 7.3 Hz), 8.06 (1H, s), 8.81 (1H, s); 13C NMR (CDCl3+ DMSO-d6, 75 MHz) δ/ppm: 7.9, 53.7, 61.1, 114.2, 116.0, 120.1, 121.7, 124.5, 128.1, 130.7, 138.2, 141.9, 152.6, 160.4, 166.1, 168.1; LC-MS-MS(ESI+) (m/z): 387.07 [MH+]. Anal. Calcd for C20H23N3O3S: C, 62.32; H, 6.01; N, 10.90; found: C, 62.35; H, 6.04; N, 10.87.
2-(Cyclohexylamino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6f)
Dark yellow powder, 77% yield, mp. 156–159 °C; IR: 3301, 3160, 2988, 1712, 1651, 1557, 1254, 1089, 753 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.14–1.31 (4H, m), 1.67–1.97 (6H, m), 2.45–2.52 (1H, m), 3.54 (2H, s, CO-CH2-N), 7.28–7.39 (2H, m), 7.51–7.63 (2H, m), 8.15 (1H, s), 8.61 (1H, s); 13C NMR (CDCl3+ DMSO-d6, 75 MHz) δ/ppm: 25.1, 26.4, 33.9, 49.8, 58.17, 115.4, 116.6, 119.7, 121.1, 124.8, 128.5, 131.6, 139.0, 142.8, 153.1, 156.8, 159.9, 171.3; LC-MS-MS(ESI-) (m/z): 383.01 [M+]. Anal. Calcd for C20H21N3O3S: C, 62.64; H, 5.52; N, 10.96; found: C, 62.60; H, 5.56; N, 10.93.
N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)-2-(pyrrolidin-1-yl)acetamide (6g)
Yellow powder, 20% yield, mp. 160–161 °C; IR321, 3140, 2953, 1709, 1529, 1268, 1092, 760 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.91 (4H, s, br), 2.74 (4H, s, N-CH2, br), 3.43 (2H, s, CO-CH2-N), 7.27–7.38 (2H, m), 7.51–7.61 (2H, m), 8.15 (1H, s), 8.58 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 24.3, 55.0, 58.0, 115.5, 116.6, 119.7, 121.1, 124.8, 128.4, 131.7, 138.9, 142.7, 153.1, 156.9, 159.9, 169.7; LC-MS-MS(ESI-) (m/z): 355.03 [M+]. Anal. Calcd for C18H17N3O3S: C, 60.83; H, 4.82; N, 11.82; found: C, 60.85; H, 4.84; N, 11.80.
N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)-2-(piperidin-1-yl)acetamide (6h)
Grey powder, 80% yield, mp. 167–170 °C; IR: 3273, 3145, 2931, 1719, 1682, 1545, 1251, 1093, 756 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.52 (2H, s, br), 1.68–1.75 (4H, m), 2.58 (4H, s, N-CH2, br), 3.22 (2H, s, CO-CH2-N), 7.27–7.39 (2H, m), 7.54 (1H, t, J = 8.4 Hz), 7.62 (1H, d, J = 7.9 Hz), 8.15 (1H, s), 8.61 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 23.7, 26.3, 55.3, 62.0, 115.4, 116.6, 119.7, 121.1, 124.8, 128.5, 131.7, 138.9, 142.7, 153.1, 156.8, 159.9, 169.6; LC-MS-MS(ESI-) (m/z): 369.16 [M+]. Anal. Calcd for C19H19N3O3S: C, 61.77; H, 5.18; N, 11.37; found: C, 61.74; H, 5.16; N, 11.39.
2-(4-Methylpiperazin-1-yl)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6i)
Yellow powder, 60% yield, mp. 189–191 °C; IR: 3291, 3144, 2934, 1721, 1705, 1528, 1170, 1088, 753 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.36 (3H, s), 2.58 (4H, s, N-CH2, br), 2.70 (4H, s, N-CH2, br), 3.29 (2H, s, CO-CH2-N), 7.32 (1H, td, J = 1.17, 7.3 Hz), 7.36 (1H, d, J = 8.2 Hz), 7.54 (1H, td, J = 1.75, 7.3 Hz), 7.62 (1H, dd, J = 1.75, 7.9 Hz), 8.15 (1H, s), 8.59 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 46.2, 53.8, 55.2, 61.1, 115.5, 116.6, 119.6, 121.0, 124.8, 128.4, 131.7, 138.9, 142.7, 153.1, 156.7, 159.8, 168.9; LC-MS-MS(ESI-) (m/z): 384.12 [M+]. Anal. Calcd for C19H20N4O3S: C, 59.36; H, 5.24; N, 14.57; found: C, 59.38; H, 5.20; N, 14.53.
2-Morpholino-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6j)
Light yellow powder, 80% yield, mp. 198–200 °C; IR: 3341, 3157, 2930, 1715, 1528, 1270, 1092, 766 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.64 (4H, s, N-CH2, br), 3.29 (2H, s, CO-CH2-N), 3.85 (4H, s, O-CH2, br), 7.26–7.39 (2H, m), 7.55 (1H, t, J = 7.3 Hz), 7.62 (1H, d, J = 7.3 Hz), 8.17 (1H, s), 8.60 (1H, s), 10.27 (1H, s, NH); 13C NMR (CDCl3, 75 MHz) δ/ppm: 54.1, 61.7, 67.1, 115.5, 116.6, 119.6, 121.0, 124.8, 128.5, 131.7, 138.9, 142.8, 153.1, 156.6 159.8, 168.5; LC-MS-MS(ESI-) (m/z): 371.13 [M+]. Anal. Calcd for C18H17N3O4: C, 58.21; H, 4.61; N, 11.31; found: C, 58.24; H, 4.63; N, 11.30.
2-((2,3-Dihydro-1H-inden-2-yl)amino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6k)
Yellow powder, 60% yield, mp. 156–158 °C; IR: 3306, 3137, 2900, 1712, 1692, 1554, 1267, 1101, 742 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.85 (2H, dd, J = 4.68, 15.81 Hz), 3.23 (2H, dd, J = 6.73, 15.81), 3.58 (2H, s, CO-CH2-N), 3.69–3.73 (1H, m), 7.20–7.35 (6H, m), 7.51 (1H, t, J = 7.6 Hz), 7.58 (1H, d, J = 7.6 Hz), 8.12 (1H, s), 8.53 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 40.0, 50.5, 60.2, 115.3, 116.6, 119.7, 121.1, 124.8, 125.2, 127.0, 128.4, 131.6, 139.0, 141.1, 142.8, 153.1, 156.6, 159.9, 170.5; LC-MS-MS(ESI-) (m/z): 416.89 [M+]. Anal. Calcd for C23H19N3O3S: C, 66.17; H, 4.59; N, 10.07; found: C, 66.14; H, 4.57; N, 10.09.
N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)-2-((2-(pyrrolidin-1-yl)ethyl)amino)acetamide (6 l)
Yellow powder, 35% yield, mp. 130–135 °C; IR: 3283, 3152, 2954, 1710, 1654, 1553, 1257, 1091, 757 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.91 (4H, s, br), 2.63–2.67 (6H, m), 2.88 (2H, t, J = 6.1 Hz), 3.56 (2H, s, CO-CH2-N), 7.28–7.38 (2H, m), 7.51–7.57 (2H, m), 8.13 (1H, s), 8.58 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 23.8, 48.7, 52.4, 54.0, 55.3, 115.2, 116.6, 119.7, 121.3, 124.8, 128.3, 131.5, 138.7, 142.7, 153.1, 157.5, 159.9, 171.9; LC-MS-MS(ESI-) (m/z): 398.07 [M+]. Anal. Calcd for C20H22N4O3S: C, 60.28; H, 5.56; N, 14.06; found: C, 60.25; H, 5.54; N, 14.09.
2-((2-Morpholinoethyl)amino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6m)
Yellow powder, 50% yield, mp. 150–153 °C; IR: 3359, 3181, 2934, 1702, 1681, 1536 1253, 1095, 739 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.53 (8H, t, J = 5.8 Hz), 2.85 (2H, t, J = 5.5 Hz), 3.55 (2H, s, CO-CH2-N), 3.86 (2H, t, J = 4.6 Hz), 7.27–7.49 (2H, m), 7.52–7.60 (2H, m), 8.12 (1H, s), 8.57 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 46.5, 52.1, 53.9, 57.9, 66.9, 115.2, 116.5, 119.7, 121.1, 124.8, 128.5, 131.6, 138.9, 142.7, 153.1, 157.1, 159.9, 171.3; LC-MS-MS(ESI-) (m/z): 414.22 [M+]. Anal. Calcd for C20H22N4O4S: C, 57.96; H, 5.35; N, 13.52; found: C, 57.94; H, 5.37; N, 13.54.
2-((2-(Cyclohex-1-en-1-yl)ethyl)amino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6n)
Yellow powder, 40% yield, mp. 164–166 °C; IR: 3295, 3177, 3150, 2987, 1710, 1650, 1550, 1258, 1094, 761 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.64 (4H, s, br), 1.96 (2H, s, br), 2.07 (2H, s, br), 2.20 (2H, t, J = 6.15 Hz), 2.79 (2H, t, J = 6.44 Hz), 3.51 (2H, s, CO-CH2-N), 5.54 (1H, s, br), 7.29–7.39 (2H, m), 7.51–7.61 (2H, m), 8.15 (1H, s), 8.60 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 22.7, 23.0, 25.5, 28.3, 38.4, 48.0, 52.0, 115.5, 119.7, 121.1, 123.9, 124.8, 128.4, 131.6, 134.9, 138.9, 142.8, 153.1, 156.8, 159.9, 170.7; LC-MS-MS(ESI+) (m/z): 411.03 [MH+]. Anal. Calcd for C22H23N3O3S: C, 64.53; H, 5.66; N, 10.26; found: C, 64.50; H, 5.64; N, 10.29.
2-((3,4-Dimethoxyphenethyl)amino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6o)
Yellow powder, 60% yield, mp. 90–92 °C; IR: 3288, 3143, 2993, 1714, 1654, 1513, 1258, 1091, 123, 924, 754 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.82 (2H, t, J = 6.15 Hz), 2.99 (2H, t, J = 6.7 Hz), 3.51 (2H, s, CO-CH2-N), 3.83 (3H, s), 3.87 (3H, s), 6.76–6.85 (3H, m), 7.27–7.35 (2H, m), 7.52 (2H, t, J = 8.4 Hz), 7.70 (1H, d, J = 7.3 Hz), 8.11 (1H, s), 8.61 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 36.0, 51.6, 52.0, 56.0, 56.1, 111.6, 111.8, 115.2, 116.5, 119.8, 120.9, 121.0, 124.8, 128.6, 131.6, 131.8, 139.2, 142.8, 147.9, 149.4, 153.1, 156.6, 159.9, 170.4; LC-MS-MS(ESI-) (m/z): 465.20 [M+]. Anal. Calcd for C24H23N3O5S: C, 61.92; H, 4.98; N, 9.03; found: C, 61.94; H, 4.97; N, 9.05.
2-((Benzo[d][1,3]dioxol-5-ylmethyl)amino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6p)
Yellow powder, 73% yield, mp. 168–170 °C; IR: 3291, 3139, 2887, 1715, 1604, 1535, 1243, 1094, 924, 754 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 3.53 (2H, s, CO-CH2-N), 3.80 (2H, s, N-CH2), 5.95 (2H, s, O-CH2-O), 6.79 (2H, s), 6.88 (1H, s), 7.31–7.37 (2H, m), 7.51 (1H, t, J = 7.0 Hz), 7.61 (1H, dd, J = 1.46, 7.6 Hz), 8.13 (1H, s), 8.59 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 51.31, 54.0, 101.3, 108.5, 108.8, 115.3, 116.5, 119.7, 121.0, 121.8, 124.8, 128.5, 131.6, 132.6, 139.0, 142.7, 147.3, 148.2, 153.1, 156.6, 159.9, 170.2; LC-MS-MS(ESI-) (m/z): 435.18 [M+]. Anal. Calcd for C22H17N3O5S: C, 60.68; H, 3.93; N, 9.65; found: C, 60.65; H, 3.96; N, 9.63.
2-(Morpholinoamino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6q)
Yellow powder, 66% yield, mp. 212–214 °C; IR: 3332, 3140, 2820, 1710, 1534, 1268, 1090, 757 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 3.30–3.33 (8H, m), 3.76 (4H, t, J = 4.6 Hz), 7.06 (1H, s), 7.35–7.46 (2H, m), 7.62 (1H, t, J = 8.4 Hz), 7.80 (1H, d, J = 1.1 Hz), 7.98 (1H, s), 8.62 (1H, s), 11.78 (1H, s, NH); 13C NMR (CDCl3, 75 MHz) δ/ppm: 50.9, 65.9, 70.7, 114.9, 116.6, 119.7, 123.4, 125.4, 129.4, 132.5, 139.1, 142.8, 153.1, 158.2 159.4, 162.9; LC-MS-MS(ESI-) (m/z): 387.06 [M+]. Anal. Calcd for C18H18N4O4S: C, 55.95; H, 4.70; N, 14.50; found: C, 55.93; H, 4.72; N, 14.53.
2-((4-Methylpiperazin-1-yl)amino)-N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (6r)
Yellow powder, 62% yield, mp. 192–194 °C; IR: 3288, 3136, 2946, 1715, 1708, 1520, 1160, 1085, 755 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.79 (3H, s), 2.37 (2H, s, br), 2.60 (4H, t, J = 5.2 Hz), 3.39 (4H, t, J = 4.9 Hz), 6.80 (1H, s), 7.26–7.38 (2H, m), 7.50–7.59 (2H, m), 7.11 (1H, s), 8.56 (1H, s), 9.84 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 46.2, 50.3, 54.0, 115.0, 116.5, 119.7, 121.1, 124.8, 128.4, 131.6, 138.7, 142.7, 153.1, 157.3, 159.9, 162.5; LC-MS-MS(ESI-) (m/z): 401.01 [M+]. Anal. Calcd for C19H21N5O3S: C, 57.13; H, 5.30; N, 17.53; found: C, 57.10; H, 5.34; N, 17.51.
N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)-2-(piperidin-1-ylamino)acetamide (6s)
Yellow powder, 66% yield, mp. 158–159 °C; IR: 3301, 3160, 2988, 1712, 1651, 1557, 1254, 1089, 753 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.63–1.83 (4H, m), 2.17 (2H, s), 3.46–3.52 (2H, m), 3.73–3.77 (2H, m), 4.23 (2H, s), 6.58 (2H,s), 7.29–7.35 (2H, m), 7.48–7.57 (2H, m), 7.97 (1H, s), 8.81 (1H, s); 13C NMR (CDCl3+ DMSO-d6, 75 MHz) δ/ppm: 20.3, 21.6, 63.2, 69.3, 113.7, 116.4, 120.2, 121.8, 125.2, 129.3, 131.7, 138.1, 142.0, 152.8, 159.8, 167.3, 167.9; LC-MS-MS(ESI-) (m/z): 387.05 [M+]. Anal. Calcd for C19H20N4O3S: C, 59.36; H, 5.24; N, 14.57; found: C, 59.38; H, 5.22; N, 14.54.
N-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)-2-((2-(piperazin-1-yl)ethyl)amino)acetamide (6t)
Yellow powder, 64% yield, mp. 151–153 °C; IR: 3359, 3181, 2934, 1702, 1681, 1536 1253, 1095, 739 cm − 1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.32–2.87 (12H, m), 3.28 (2H, s), 7.34–7.44 (2H, m), 7.61 (1H, t, J = 7.0 Hz), 7.82 (1H, d, J = 7.6 Hz) 7.98 (1H, s), 8.58 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 35.2, 53.2, 57.4, 60.9, 114.9, 116.6, 119.7, 121.0, 125.4, 129.5, 132.5, 139.2, 142.7, 153.1, 159.4, 161.7, 169.6; LC-MS-MS(ESI-) (m/z): 415.07 [M+]. Anal. Calcd for C20H23N5O3S: C, 58.09; H, 5.61; N, 16.94; found: C, 58.07; H, 5.63; N, 16.91.
Anticholinesterase activity assays
Acetyl- (AChE) and butyryl-cholinesterase (BuChE) inhibitory activities of the synthesized compounds were determined according to Ellman’s methodCitation43. The IC50 was determined by constructing an absorbance and/or inhibition (%) curve and examining the effect of five different concentrations. IC50 values were calculated for a given inhibitor by determining the concentration needed to inhibit half of the maximum biological response of the substrate. The substrates of the reaction were acetylthiocholine iodide and butyrylthiocholine iodide. 5,5'-dithio-bis(2-nitrobenzoic) acid (DTNB) was used to measure anticholinesterase activity. Stock solutions of the compounds and galanthamine in methanol were prepared at a concentration of 4000 µg/mL. Aliquots of 150 µL of 100 mM phosphate buffer (pH 8.0), 10 µL of sample solution and 20 µL AChE (2.476 × 10 − 4 U/µL) (or 3.1813 × 10 − 4 U/µL BuChE) solution were mixed and incubated for 15 min at 25 °C. About 10 µL of DTNB solution was prepared by adding 2.0 mL of pH 7.0 and 4.0 mL of pH 8.0 phosphate buffers to a mixture of 1.0 mL of 16 mg/mL DTNB and 7.5 mg/mL NaHCO3 in pH 7.0 phosphate buffers. The reaction was initiated by the addition of 10 µL (7.1 mM) acetylthiocholine iodide (or 0.79 mM butyrylthiocholine iodide). In this method, the activity was measured by following the yellow colour produced as a result of the thio anion produced by reacting the enzymatic hydrolysis of the substrate with DTNB. Also, methanol was used as a control solvent. The hydrolysis of the substrates was monitored using a BioTek Power Wave XS at 412 nmCitation43.
Kinetic study of AChE inhibition
The kinetic study of AChE was performed according to Ellman’s methodCitation43 with three different concentrations (20, 40 and 60 nM) of compound 6c. Lineweaver–Burk reciprocal plots were constructed by plotting 1/velocity against 1/[substrate] at varying concentrations of the substrate acetylthiocholine (0.05–0.5 mM). The plots were assessed by a weighted least-squares analysis that assumed the variance of velocity (v) to be a constant percentage of v for the entire data set. The inhibition constant Ki was calculated by plots of the slopes of these reciprocal plots versus the concentrations of compound 6c in a weighted analysis.
In vitro cytotoxicity assay
The cytotoxicity effect of test compound on hepatoma cell H4IIE cells was evaluated by MTT assay according to described methodsCitation44. Briefly, H4IIE cells were seeded in a flat-bottomed 96-well plate at a density of 5 × 104 cells/well in DMEM containing 10% FBS. The plate was incubated at 37 °C with 5% CO2 for 24 h, and then 6c was prepared and added to make a final concentration of 2.5, 1.25, 0.625, 0.312, 0.156, 0.078 µM, respectively, in serum-free DMEM. Cells were further incubated for 24 h at 37 °C with 5% CO2; then, the medium was replaced with DMEM containing 10% FBS. About 10 µL of filter-sterilized MTT (3–(4,5 dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution (5 mg/mL in PBS) was added to each well and further incubated at 37 °C with 5% CO2 for 4 h. At the end of incubation, media was aspirated from the wells and 100 µL of DMSO was added to dissolve insoluble formosan crystals formed. The absorbance was measured at 570 nm using a microtiter plate reader. The relative % cell viability was calculated from the following equation: Relative percent cell viability = (Atest/Acontrol) × 100%. (Atest is the absorbance of the sample treated cells and a control is the absorbance of the untreated cells. Each absorbance was taken to be the mean of triplicate measurements.) The cell viability was represented as a percentage relative to untreated cells as a control.
Docking study
Ligands were sketched and energy-minimized using Sybyl v8.1 (Tripos, Inc., St. Louis, MO) on an Intel (Xeon 4 core, HP Z820) using Linux 6 operating system. Protonation states at physiological pH were calculated and considered during molecular editing procedure; in any case, the most abundant protomer was saved. AutoDock Vina software (version 1.1.2) was used to perform docking (standard options) of coumarin derivatives into 1ACJ, 1EVE and 1P0M crystallographic structures. To validate the docking program, the co-crystallized ligand (donepezil) was redocked on the target enzyme. A RMS (Root Mean Square) value of 0.531 was found for donepzil-bound acetylcholinesterase. A ligand-centred grid box, defined with a size of 50 × 60 × 50 Å3 and a regular space of 0.375 Å, able to cover the whole binding site, was considered for docking. Nine poses (docking solutions) were generated for each ligand into each model and then energetically scored. A total of 9 × 20 × 3 ligand–protein complexes were analyzed to identify the best solution from both a geometrical and energetic point of viewCitation45.
Results and discussion
Chemistry
The synthetic procedures to obtain the target compounds 6a-t are depicted in Scheme 1. 3-acetylcoumarin (2) was synthesized from salicylaldehyde (1) according to the literatureCitation46, and then it was brominated by molecular bromine in chloroform. 3–(2-Amino-1,3-thiazol-4-yl)coumarin (4) was obtained by reacting 3-(bromoacetyl)coumarin (3) with thiourea. The reaction of 4 with chloroacetylchloride in THF gave 2-chloro-N-(4–(2-oxo-2H-chromen-3-yl)thiazol-2-yl)acetamide (5). Coumarylthiazole-substituted acetamido derivatives (6a-t) were obtained by the reaction between compound 5 and various amine derivatives in DMF.
Scheme 1. Synthesis of new coumarylthiazole-substituted acetamide derivatives. Reaction conditions: (i) Ethylacetoacetate, piperidin, rt, 30 min; (ii) Br2, CHCl3, 50 °C, 15 min; (iii) Thiourea, EtOH, 80 °C, 2 h; (iv) Chloroacetylchloride, Et3N, THF, 70 °C, 8 h; (v) RNH2, DMF, 60 °C, 12 h.
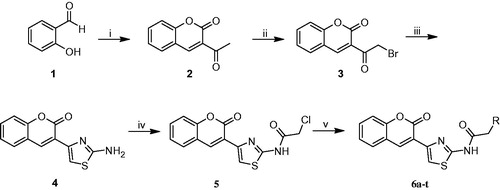
All the new compounds were characterized by 1H NMR, 13C NMR, IR, MS and elemental analysis. 1H NMR, 13C NMR and MS spectra of the synthesized compounds are given in supplementary materials.
Cholinesterase inhibitory activity
The inhibitory activities of the target compounds (6a-t) on AChE and BuChE were determined by the Ellman’s methodCitation43 using galantamine as the reference compound. The IC50 values for AChE and BuChE inhibitions are summarized in . IC50 values against AChE ranged from nanomolar to micromolar units (43 nM-13.53 µM). High AChE selectivity (7.32–4151.16) over BuChE was observed. Compound 6c exhibited the strongest inhibition against AChE with an IC50 value of 43 nM, which was 56-fold more than that of galantamine (IC50 = 2.41 µM). Furthermore, 11 of the synthesized compounds (6d, 6e, 6 g, 6 h, 6i, 6k, 6 l, 6m, 6p, 6r and 6s) exhibited better AChE inhibition (IC50 = 0.09–2.36 µM) than the positive control, galantamine, by 1.4–26.7-fold. Most of the synthesized compounds showed lesser inhibitory activity against BuChE than galantamine, except of four compounds 6i, 6 l, 6r and 6s. Compound 6 l exhibited the strongest inhibition against BuChE with an IC50 value of 2.35 µM, which was 2- and 7.5-fold more than that of donepezil (IC50 = 4.66 µM) and galanthamine (IC50 = 17.38 µM), respectively.
Table 1. In vitro inhibition IC50 values (μM) and selectivity of compounds 6a-t for AChE and BuChE.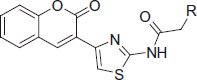
Some acetylcholinesterase inhibitors such as donepezil, rivastigmine and galantamine are currently used to treat the cognitive problems of ADCitation9,Citation22,Citation47. Tacrine is also a well-known class of AChE inhibitors with an IC50 value of 167 nmCitation1. Among the synthesized compounds in this work, 6c showed the strongest inhibition against AChE with an IC50 value of 43 nM, which was almost 4-, and 70-fold more than that of tacrine (IC50 = 167 nM) and rivastigmine (IC50 = 3.01 µM), respectively. Also, it approached that of donepezil (IC50 = 30 nM), which is one of the most used AChE inhibitor. The AChE selectivity of 6c increased 26-, 28828- and 41511-fold when compared to donepezil (26 versus 155.3), tacrine (28828 versus 0.144) and rivastigmine (41511 versus 0.10), respectively ().
A plethora of literature papers has presented the newly synthesized coumarin derivatives as AChE inhibitors. Xie et al.Citation36 and Nam et al.Citation23 designed and synthesized novel tacrine-coumarin hybrids and aminoalkyl-subsitituted coumarin derivatives as cholinesterase inhibitors. In these studies, the strongest inhibitors had IC50 value of 92 nm and 2.87 µM, respectively. These IC50 values are 2- and 67-fold less than that of 6c, which is the best AChE inhibitor in here, respectively. On the other hand, Asadipour et al.Citation18 and Catto et al.Citation47 synthesized various coumarin-3-carboxamide derivatives as potent AChE inhibitors. These new compounds showed potent activity in the range of sub-micromolar concentrations (0.3–633 nmCitation18 and 7.6–6600 nmCitation47).
When compared to the best AChE inhibitors in our previously works, 6c exhibited better AChE inhibition (IC50 = 43 nM) than the benzofuran derivative (PBI-1) (IC50 = 3.85 µM)Citation32 and coumarin derivative (PBI-2) (IC50 = 4.58 µM)Citation33 by almost 90- and 107-fold, respectively. This increase can relate to the various interactions (a cation–π interaction, especially) between each of the fragments at the designed molecules and the catalytic triad of AChE. This finding supports that the interactions with the catalytic triad of AChE play a crucial role for efficient inhibitory activities of the molecules.
The following structure–activity relationship (SAR) observations can be drawn from data of : (i) Replacing the methyl (6a) on the acetamide moiety by a propyl (6b) or a cyclohexyl (6f) did not cause significant changes on the AChE inhibitory activity; (ii) the increase of steric hindrance on the N atom of the acetamide moiety in compound 6c (IC50 = 43 nM) by a dibutyl (6d: IC50 = 1.71 µM) or a propyl (6e: IC50 = 2.29 µM) group led to a decline of the inhibitory activity against AChE; (iii) the expansion of the pyrrolidine ring of compound 6g (IC50 = 1.41 µM against AChE; IC50 = 95.81 µM against BuChE) to a piperidine ring (compound 6h: IC50 = 0.73 µM against AChE; IC50 = 22.90 µM against BuChE) and 4-methylpiperazine ring (6i: IC50 = 0.61 µM against AChE; IC50 = 7.42 µM against BuChE) increased the inhibitory activity against both ChEs. On the contrary, the presence of the morpholine ring (6j: IC50 = 3.78 µM against AChE; IC50 = 182.70 µM against BuChE) strongly decreased the AChE and BuChE inhibitory activity. The reason for this decrease can be explained by a negative inductive effect of the oxygen, on the morpholine moiety which lowers the electron density of the amine nitrogen, finally leading to a decrease of its H-bonding capability;(iv) The presence of an ethyleneamine group as a spacer between the acetamide moiety and the morpholine or pyrrolidine ring positively affected the inhibitory activity against both ChEs. On the contrary, the ethyleneamine group on the piperazine ring caused an opposite effect on the inhibitory activity against both AChE and BuChE as in the case of compound 6i (IC50 = 0.61 µM against AChE; IC50 = 7.42 µM against BuChE) and 6t (IC50 = 8.25 µM against AChE; IC50 = 115.48 µM against BuChE); (v) the presence of an amine group between the acetamide moiety and the piperidine or morpholine ring did not significantly affect AChE and BuChE inhibition as showed by compound 6h (IC50 = 0.73 µM against AChE; IC50 = 22.90 µM against BuChE) when compared to 6s (IC50 = 0.82 µM against AChE; IC50 = 15.20 µM against BuChE). On the contrary, the same amino group caused a decrease in the AChE and BuChE inhibitory activity, as showed by compound 6i (IC50 = 0.61 µM against AChE; IC50 = 7.42 µM against BuChE) on respect to compound 6r (IC50 = 1.12 µM against AChE; IC50 = 8.20 µM against BuChE). An opposite trend was observed for compounds 6j (IC50 = 3.78 and 182.70 µM for AChE and BuChE, respectively) and 6q (IC50 = 2.52 µM against AChE; IC50 =152.25 µM against BuChE) where the introduction of an amine group between the acetamide moiety and the morpholine ring negatively affects the activity against both AChE and BuChE; (vi) The replacement of the ethyleneamine group (6o: IC50 = 13.53 µM against AChE; IC50 = 189.40 µM against BuChE) between the acetamide moiety and the aromatic ring by a methyleneamine (6p: IC50 = 0.74 µM against AchE; IC50 = 60.35 µM against BuChE) and the acetalization of the dimethoxy group to a phenyl ring led to an effective increase on the activity against both ChEs.
The obtained IC50 values against BuChE demonstrated that all compounds act as selective AChE inhibitors, more than galantamine. The amine functional group on alkyl side chain or lipophilic moieties and a tertiary amino group can represent key requirement for an anti-AChE activity.
Besides inhibitory activity of coumarin compounds against AChE, they are a well-known class of the carbonic anhydrase inhibitorsCitation48–50. Supuran et al. investigated that both the simple lead coumarin for its interaction with the CA active site by means of high resolution X-ray crystallography as well as a series of coumarin derivatives possessing various moieties substituting the coumarin ring in the 3-, 6-, 7-, 3,6-, 4,7-, 3,8-, 6,7- and 7,8- positionsCitation51,Citation52. According to these studies, the 3-substituted coumarin derivatives with bulky moieties had a poor CA inhibitory activity and the best positions would be 4-, 5-, 6-, 7-, 8- and 7,8-. These coumarin derivatives were highly active as CAIsCitation51,Citation52. Our previous studiesCitation33,Citation53 showed that 3-substituted coumarin derivatives bearing thiazole ring and urea moieties had higher inhibitory activities against AChE than CA I and II and the moderate AChE selectivity (almost 1–10-fold) with respect to CA I and II was observed. Many studies have also supported that most of synthesized compounds as AChE and CA inhibitors have high AChE selectivity with respect to CAsCitation30,Citation54–56.
Kinetic study of AChE inhibition
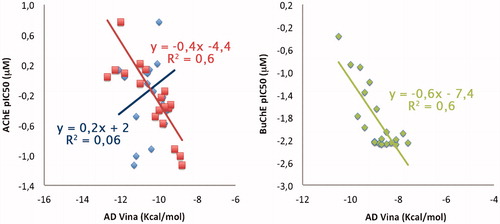
In order to explore the inhibition mechanism of compound 6c with AChE, an enzyme kinetic study was carried out. Graphical analysis of the reciprocal Lineweaver–Burk plot () showed both increased slopes (decreased Vmax) and intercepts (higher Km) at higher inhibitor concentration. This pattern indicated a mixed-type inhibition and therefore revealed that compound 6c might be able to bind to the catalytic active site (CAS) and peripheral anionic site (PAS) as well as catalytic triad of AChE, which were consistent with our design strategy. The Ki value of 31 nM was determined by plots of the slopes of the Lineweavere–Burk reciprocal plots versus concentrations of 6c.
Docking study
Docking studies have been performed in order to analyze the binding profile of the new synthetized coumarin derivatives into AChE and BuChE enzymes. Both cholinesterases (AChE and BuChE) are structurally similar and they share 65% of the amino acid sequenceCitation45. The main difference substantially is on the replacement of aromatic with aliphatic amino acids in BuChE, which is crucial for the selectivity against different inhibitors of the two enzymesCitation22.
The two PDB structures 1ACJ of TcAChE co-crystallized with tacrine and 1EVE of TcAChE co-crystallized with Donepezil have been taken into account to model the binding to AChE. The choice of these two structures is not casual since they are representative of two peculiar ligand-induced local conformational changes (see )Citation57.
Analysis of the crystallographic structures for TcAChE deposited in PDB reveals two important conformational changes located at CAS and PAS, which can drastically affect ligand accommodation during docking. In 1EVE crystal structure (), the conformation adopted by Phe330 (CAS) and Trp279 (PAS) opens the gorge allowing the placement of dual binding site inhibitors as Donepezil (E20). Conversely, in 1ACJ, Phe330 is stacked against tacrine thus closing the gorge.
Proceeding through the species, analysis of pairwise sequence alignment reveals the replacement of Phe330 with Tyr337 in hAChE (see ). However, superposition of folded structures for Torpedo californica and human AChE (; 1EVE in blue and 4EY7 in brown) allows to see that Tyr337 in hAChE (4EY7) assumes the same orientation of Phe330 in TcAChE (1EVE). Considering the structure of our ligands and the low accuracy in energy predictions of scoring functions, we can suppose that the additional hydroxyl in Tyr337 should not affect significantly our docking simulations.
In addition, given the limited “crystallographic” mobility of aminoacids within the binding site emerged by structural analysis, we can conclude that a satisfactory covering the conformational ensemble for AChE would be reached by using the two structures 1ACJ and 1EVE which are indicative of the most important changes in side-chain orientation of residues Phe330 and Trp279. For this, both the two structures of TcAChE (1ACJ and 1EVE) have been selected for docking studies. Moreover, the impact of Phe330 and Trp279 side-chains orientation on ligand binding and ranking performances has been analyzed in detail.
The BuChE system
Comparative analysis of the binding site for TcAChE (EVE in blue) and hBuChE (P0M in gold) allows seeing important changes in aminoacid sequence generally identifiable as aromatic-to-aliphatic switch. As shown in , modifications affect both CAS and PAS. Among them, Phe330-to-Ala328 (CAS) and Trp279-to-Ala277 (PAS) mutations seem to remarkably contribute to differentiate proteins fingerprint, thus helping in explanation of ligand selectivity profile.
Figure 1. Structures of well-known cholinesterase inhibitors and designed compounds.
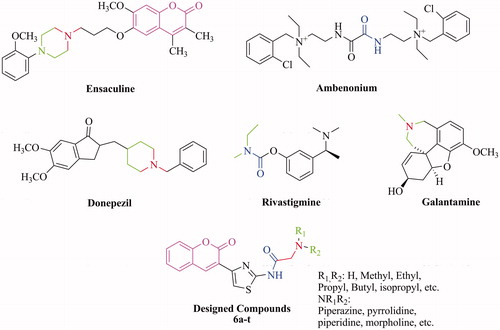
Figure 2. Development strategy and interactions of fragment of the synthesized compounds.
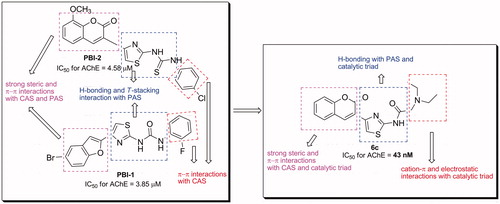
Figure 3. Kinetic study on the mechanism of AChE inhibition by compound 6c. Overlaid Lineweaver–Burk reciprocal plots of AChE initial velocity at increasing substrate concentration (0.05–0.50 mM) in the absence of inhibitor and in the presences of different concentrations of 6c are shown.
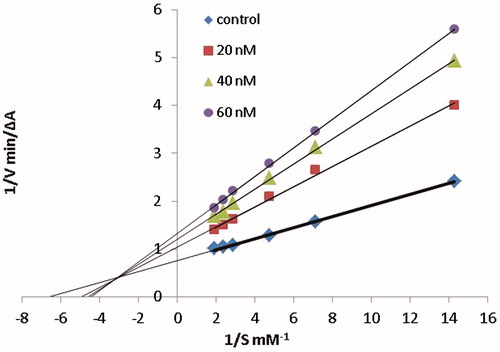
Figure 4. (A) Superposition of crystallographic structures for TcAChE co-crystallized with tacrine (1ACJ, in green) and Donepezil (1EVE, in blue). (B) Superposition of TcAChE (1EVE residue Phe330, Ile287, in blue) and hAChE (4EY7 residue Tyr337, Val294, in brown), both in complex with Donepezil. Red circles highlight the most important ligand-induced conformational changes. Pairwise sequence alignment for T. californica (PDB ID: 1ACJ, 1EVE) and human (PDB ID: 4EY7) AChE is also provided. The catalytic triad is reported in red.
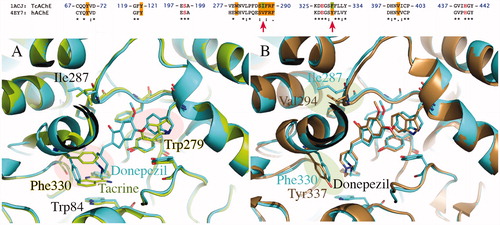
Figure 5. Comparison of TcAChE (1EVE residue Ile287, Phe288, Phe290, Val400, Trp279, Tyr70, Phe330, in blue) and hBuChE (1P0M residue Pro285, Leu286, Val288, Phe398, Ala277, Asn68, Ala328, in gold). The most important changes in aminoacids sequence are highlighted by blue, green and red areas. As shown, modifications generally affect both CAS (1, in blue) and PAS (2, in green and 3, in red).
For docking purposes, the PDB structure 1P0M of hBuChE bound to a substrate analogue (CHT) has been selected.
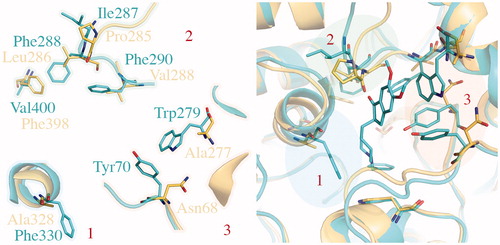
A qualitative representation of correlation between binding energies coming from docking (expressed as kcal/mol) and experimental data (reported here as pIC50; µM) is given in . Keeping in mind the (i) limitations of docking in energetic evaluations and (ii) the limited range of activity for AChE (2.5 logarithmic units) and BuChE (about 2 logarithmic units) inhibitors, this representation aims to be purely indicative and wants to show the impact of the structural model on docking results.
Figure 6. Qualitative representation of correlation between binding energies coming from docking and experimental data. Regarding the TcAChE model, correlation obtained by using 1ACJ/1EVE is reported in blue (R2 = 0.06)/red (R2 = 0.6). Correlation for hBuChE (1P0M) is reported in green.
As expected from the previously performed structural analysis of the two structures of TcAChE, best results in term of qualitative correlation between in silico-determined binding affinities and experimental data are obtained by using the 1EVE structure (: R2 of 0.6; in red) as structural model with respect to 1ACJ (: R2 of 0.06; in blue), with binding affinities that range between −9 and −12 kcal/mol. This is not surprising given the Donepezil-like features of our coumarin derivatives.
A good correlation between experimental and in silico-predicted data is also observed for the BuChE model (: 1P0M, in green; R2 of 0.6) with predicted binding affinities that range between −7 and −10 kcal/mol.
Binding modes for dual binding site inhibitors 6i, 6j, 6o and 6p into TcAChE (1EVE model, A–D) and hBuChE (1P0M model, E–H) are reported in . Compound 6i (; in green), one of the most potent AChE inhibitors of the tested coumarin series, shows a good disposition into the binding site, being able to give H-bonding interactions with Phe288, Arg289 main chains and Tyr121. Moreover, an interesting cation–π interaction can be observed between the protonated piperazine and Trp84. Loss of cation–π due to replacement of piperazine with morfoline moiety could explain the reduction in inhibitory activity for compound 6j (; in pink). T-stacking interaction between the coumarin moiety and Trp279 is also observed in both cases.
Figure 7. Docking poses for compound 6i, 6j, 6o and 6p into TcAChE (1EVE; A–D) and BuChE (1P0M; E–H) binding sites. Aminoacids relevant for ligand interactions are indicated and reported in white sticks.
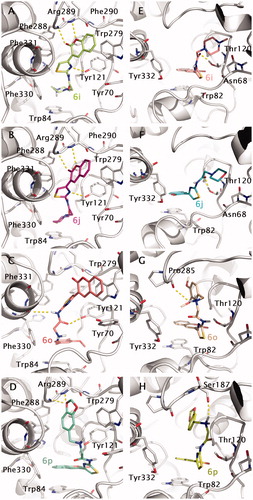
Going to the less active compound 6o (; in light red), a double T-stacking interaction can be observed for the coumarin and the 3,4-di-MeO-phenyl- moiety with Trp279 (PAS) and Trp84 (CAS) residues, respectively. H-bonding interactions also involve Tyr121 and Phe330 (main chain). Another interesting case is represented by compound 6p (; in blue marine). For this compound, an inversion of the binding mode is observed. The coumarin moiety is stacked against Trp84 (CAS) and the 2H-1,3-benzodioxo- moiety with this last also involved in H-bonding with the backbone nitrogens of Phe288 and Arg289.
Docking-predicted protein–ligand complexes for the same compounds into the hBuChE binding site have been also reported in ) for comparative purposes. In principle, the binding profile of coumarin derivatives into BuChE seems to be generally less stable than that observed for AChE. In this regard, the replacement of Phe330 and Trp279 with respectively Ala328 and Ala277 in hBuChE could be in part responsible for inversion of the binding profile observed in case of AChE (the coumarin moiety stacked against Trp82 in CAS) and lowering in binding affinity.
H4IIE hepatoma cell toxicity
On basis of the screening results above, the most potent compound 6c was selected to further examine the potential toxicity effect on the hepatoma cell line H4IIE. After exposing the cells to this compound for 24 h, the cell viability was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay. The result indicated that 6c caused negligible cell death and was non-toxic to H4IIE cell at 0.07–2.5 µM (cell viability: 0.07 µM: 130 ± 5.1%; 0.15 µM: 106 ± 2.4%; 0.3 µM: 101 ± 0.9%; 0.6 µM: 78 ± 7.3%; 1.25 µM: 56 ± 5.0%; 2.5 µM: 38 ± 4.1%).
Conclusions
A series of 20 novel acetamide substituted coumarylthiazoles derivatives (6a-t) was synthesized, and their inhibitory activities on AChE and BuChE were evaluated. All the synthesized compounds are selective inhibitors of AChE. Among them, 6c exhibited the strongest inhibition against AChE with an IC50 value of 43 nM, which was 4-, 56- and 70-fold more than that of tacrine (IC50 = 167 nM), galantamine (IC50 = 2.41 µM) and rivastigmine (IC50 = 3.01 µM), respectively. The selectivity of 6c towards AChE is 26-, 575-, 28826- and 41511-fold compared with donepezil, galantamine, tacrine and rivastigmine, respectively. Kinetic study of AChE inhibition revealed that 6c was a mixed-type inhibitor. Moreover, the result of H4IIE hepatoma cell toxicity assay indicated that 6c caused negligible cell death and was non-toxic at 0.07–2.5 µM
The presence of N-alkyl and/or heterocyclic moiety instead of an aryl group increased the AChE inhibition by almost 107-fold through the cation–π interactions with the catalytic triad of AChE. This finding can provide guidance for researches to design new efficient ChEs inhibitors in the future works.
Overall these derivatives could be recommended as new chemotypes to develop new AChE inhibitors for the treatment of AD disease by suitably modulating the substitution pattern also in the perspective of multifunctional anti AD agents.
Disclosure statement
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
Funding
This work was supported by the Sakarya Research Fund of the Sakarya University, 10.13039/501100004473. Project Number: 2014-28-00-001.
IENZ_1250753_Supplementary_Material.pdf
Download PDF (2.2 MB)Related Research Data
References
- Ruiz PM, Rubio L, Palomero EG, et al. Design, synthesis, and biological evaluation of dual binding site acetylcholinesterase inhibitors: new disease-modifying agents for Alzheimer’s disease. J Med Chem 2005;48:7223–33.
- Bertram L, Tanzi RE. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci 2008;9:768–78.
- Holzgrabe U, Kapkova P, Alptuzun V, et al. Targeting acetylcholinesterase to treat neurodegeneration. Expert Opin Ther Targets 2007;11:161–79.
- Scozzafava A, Kalın P, Supuran CT, et al. The impact of hydroquinone on acetylcholine esterase and certain human carbonic anhydrase isoenzymes (hCA I, II, IX, and XII). J Enzyme Inhib Med Chem 2015;30:941–6.
- Citron M. Strategies for disease modification in Alzheimer's disease. Nat Rev Neurosci 2004;5:677–85.
- Gocer H, Akincioglu A, Goksu S, et al. Carbonic anhydrase and acetylcholinesterase inhibitory effects of carbamates and sulfamoylcarbamates. J Enzyme Inhib Med Chem 2015;30:316–20.
- Pohanka M, Holas O. Evaluation of 2,6-dichlorophenolindophenol acetate as a substrate for acetylcholinesterase activity assay. J Enzyme Inhib Med Chem 2015;30:796–9.
- Anand P, Singh B, Singh N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer's disease. Bioorg Med Chem 2012;20:1175–80.
- Guillou C, Mary A, Renko DZ, et al. Potent acetylcholinesterase inhibitors: design, synthesis and structure-activity relationships of alkylene linked bis-galanthamine and galanthamine-galanthaminium salts. Bioorg Med Chem Lett 2000;10:637–9.
- Camps P, el-Achab R, Gorbig DM, et al. Synthesis, in vitro pharmacology, and molecular modeling of very potent tacrine-huperzine a hybrids as acetylcholinesterase inhibitors of potential interest for the treatment of Alzheimer's disease. J Med Chem 1999;42:3227–42.
- Pohanka M. Acetylcholinesterase inhibitors: a patent review (2008-present). Expert Opin Ther Pat 2012;22:871–86.
- Leonetti F, Catto M, Nicolotti O, et al. Homo- and hetero-bivalent edrophonium-like ammonium salts as highly potent, dual binding site AChE inhibitors. Bioorg Med Chem 2008;16:7450–6.
- Cheng Y, An LK, Wu N, et al. Synthesis, cytotoxic activities and structure-activity relationships of topoisomerase I inhibitors:indolizinoquinoline-5,12-dione derivatives. Bioorg Med Chem 2008;16:4617–25.
- Torre P, Saavedra LA, Caballero J, et al. A Novel Class of Selective Acetylcholinesterase Inhibitors: Synthesis and Evaluation of (E)-2-(Benzo[d]thiazol-2-yl)-3-heteroarylacrylonitriles. Molecules 2012;17:12072–85.
- Razavi SF, Khoobi M, Nadri H, et al. Synthesis and evaluation of 4-substituted coumarins as novel acetylcholinesterase inhibitors. Eur J Med Chem 2013;64:252–9.
- Sussman JL, Harel M, Frolow F, et al. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science 1991;253:872–9.
- Mary A, Renko DZ, Guillou C, Thal C. Potent acetylcholinesterase inhibitors: Design, synthesis, and structure-activity relationships of bis-interacting ligands in the galanthamine series. Bioorg Med Chem 1998;6:1835–50.
- Asadipour A, Alipour M, Jafari M, et al. Novel coumarin-3-carboxamides bearing N-benzylpiperidine moiety as potent acetylcholinesterase inhibitors. Eur J Med Chem 2013;70:623–30.
- Wyman IW, Macartney DH. Host-Guest complexes and pseudorotaxanes of cucurbit[7]uril with acetylcholinesterase inhibitors. J Org Chem 2009;74:8031–8.
- Mao F, Li J, Wei H, et al. Tacrine-propargylamine derivatives with improved acetylcholinesterase inhibitory activity and lower hepatotoxicity as a potential lead compound for the treatment of Alzheimer’s disease. J Enzyme Inhib Med Chem 2015;30:995–1001.
- Bolognesi ML, Cavalli A, Valgimigli L, et al. Multi-Target-Directed drug design strategy: from a dual binding site acetylcholinesterase inhibitor to a trifunctional compound against alzheimer’s disease. J Med Chem 2007;50:6446–9.
- Tasso B, Catto M, Nicolotti O, et al. Quinolizidinyl derivatives of bi- and tricyclic systems as potent inhibitors of acetyl- and butyrylcholinesterase with potential in Alzheimer's disease. Eur J Med Chem 2011;46:2170–84.
- Nam SO, Park DH, Lee YH, et al. Synthesis of aminoalkyl-substituted coumarin derivatives as acetylcholinesterase inhibitors. Bioorg Med Chem 2014;22:1262–7.
- Kumar A, Gupta MH, Kumar M. An efficient non-ionic surfactant catalysed multicomponent synthesis of novel benzylamino coumarin derivative via Mannich type reaction in aqueous media. Tetrahedron Lett 2011;52:4521–5.
- Parvez A, Meshram J, Tiwari V, et al. Pharmacophores modeling in terms of prediction of theoretical physico-chemical properties and verification by experimental correlations of novel coumarin derivatives produced via Betti’s protocol. Eur J Med Chem 2010;45:4370–8.
- Piazzi L, Rampa A, Bisi A, et al. 3-(4-{[Benzyl(methyl)amino]-methyl}phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation: a dual function lead for Alzheimer's disease therapy. J Med Chem 2003;46:2279–82.
- Mohamed T, Osman W, Tin G, Rao PPN. Selective inhibition of human acetylcholinesterase by xanthine derivatives: in vitro inhibition and molecular modelling investigations. Bioorg Med Chem Lett 2013;23:4336–41.
- Kaboudin B, Arefi M, Emadi S, Hasani VS. Synthesis and inhibitory activity of ureidophosphonates, against acetylcholinesterase: pharmacological assay and molecular modelling. Bioorg Chem 2012;41–42:22–7.
- Anand P, Singh B. Synthesis and evaluation of novel carbamate-substituted flavanone derivatives as potent acetylcholinesterase inhibitors and anti-amnestic agents. Med Chem Res 2013;22:1648–59.
- Yılmaz S, Akbaba Y, Özgeriş B, et al. Synthesis and inhibitory properties of some carbamates on carbonic anhydrase and acetylcholine esterase. J Enzyme Inhib Med Chem 2016;31:1484–91.
- Liu S, Shang R, Shi L, et al. Synthesis and biological evaluation of 7H-thiazolo[3,2-b]-1,2,4-triazin-7-one derivatives as dual binding site acetylcholinesterase inhibitors. Eur J Med Chem 2014;81:237–44.
- Zengin-Kurt B, Gazioglu I, Basile L, et al. Potential of aryl-urea-benzofuranylthiazoles hybrids as multitasking agents in Alzheimer's disease. Eur J Med Chem 2015;102:80–92.
- Zengin-Kurt B, Gazioglu I, Sonmez F, Kucukislamoglu M. Synthesis, antioxidant and anticholinesterase activities of novel coumarylthiazole derivatives. Bioorg Chem 2015;59:80–90.
- Luo W, Su YB, Hong C, et al. Design, synthesis and evaluation of novel 4-dimethylamine flavonoid derivatives as potential multi-functional anti-Alzheimer agents. Bioorg Med Chem 2013;21:7275–82.
- Thiratmatrakul S, Yenjai C, Waiwut P, et al. Synthesis, biological evaluation and molecular modeling study of novel tacrine-carbazole hybrids as potential multifunctional agents for the treatment of Alzheimer's disease. Eur J Med Chem 2014;75:21–30.
- Xie SS, Wang XB, Li JY, et al. Design, synthesis and evaluation of novel tacrine-coumarin hybrids as multifunctional cholinesterase inhibitors against Alzheimer’s disease. Eur J Med Chem 2013;64:540–53.
- Mjambili F, Njoroge M, Naran K, et al. Synthesis and biological evaluation of 2-aminothiazole derivatives as antimycobacterial and antiplasmodial agents. Bioorg Med Chem Lett 2014;24:560–4.
- Huth JR, Song D, Mendoza RR, et al. Toxicological evaluation of thiol-reactive compounds identified using a la assay to detect reactive molecules by nuclear magnetic resonance. Chem Res Toxicol 2007;20:1752–9.
- Devine SM, Mulcair MD, Debono CO, et al. Promiscuous 2-aminothiazoles (PrATs): a frequent hitting scaffold. J Med Chem 2015;58:1205–14.
- Sugino T, Tanaka K. Solvent-Free coumarin synthesis. Chem Lett 2001;30:110–11.
- Koelsch CF. Bromination of 3-acetocoumarin. J Am Chem Soc 1950;72:2993–5.
- Hamama WS, Berghot MA, Baz EA, Gouda MA. Synthesis and antioxidant evaluation of some new 3-substituted coumarins. Arch Pharm 2011;344:710–18.
- Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95.
- Xie SS, Wang X, Jiang N, et al. Multi-target tacrine-coumarin hybrids: cholinesterase and monoamine oxidase B inhibition properties against Alzheimer's disease. Eur J Med Chem 2015;95:153–65.
- Meng FC, Mao F, Shan WJ, et al. Design, synthesis, and evaluation of indanone derivatives as acetylcholinesterase inhibitors and metal-chelating agents. Bioorg Med Chem Lett 2012;22:4462–6.
- Karade NN, Gampawar SV, Shinde SV, Jadhav WN. L-Proline catalyzed solvent-free Knoevenagel condensation for the synthesis of 3-substituted coumarins. Chin J Chem 2007;25:1686–9.
- Catto M, Pisani L, Leonetti F, et al. Design, synthesis and biological evaluation of coumarin alkylamines as potent and selective dual binding site inhibitors of acetylcholinesterase. Bioorg Med Chem 2013;21:146–52.
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62.
- Carta F, Maresca A, Scozzafava A, Supuran CT. Novel coumarins and 2-thioxo-coumarins as inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem 2012;20:2266–79.
- Karatas MO, Alici B, Cakir U, et al. Synthesis and carbonic anhydrase inhibitory properties of novel coumarin derivatives. J Enzyme Inhib Med Chem 2013;28:299–304.
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44.
- Maresca A, Scozzafava A, Supuran CT. 7,8-Disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8.
- Zengin-Kurt B, Sonmez F, Gokce B, et al. In vitro inhibition effects on erythrocyte carbonic anhydrase I and II and structure-activity relationships of cumarylthiazole derivatives. Russian J Bioorg Chem 2016;42:506–11.
- Turan B, Sendil K, Sengul E, et al. The synthesis of some β-lactams and investigation of their metal-chelating activity, carbonic anhydrase and acetylcholinesterase inhibition profiles. J Enzyme Inhib Med Chem 2016. [Epub ahead of print]. doi: 10.3109/14756366.2016.1170014.
- Sujayev A, Garibov E, Taslimi P, et al. Synthesis of some tetrahydropyrimidine-5-carboxylates, determination of their metal chelating effects and inhibition profiles against acetylcholinesterase, butyrylcholinesterase and carbonic anhydrase. J Enzyme Inhib Med Chem 2016;31:1531–9.
- Ozgun DO, Yamali C, Gul HI, et al. Inhibitory effects of isatin Mannich bases on carbonic anhydrases, acetylcholinesterase, and butyrylcholinesterase. J Enzyme Inhib Med Chem 2016;31:1498–501.
- Camps P, Formosa X, Galdeano C, et al. Novel donepezil-based inhibitors of acetyl- and butyrylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation. J Med Chem 2008;51:3588–98.