
Abstract
Four multicaulin and miltirone-like phenanthrene derivatives were synthesised and evaluated as antituberculosis agents. The crucial step of the synthesis was Pschorr coupling of 4-(3-isopropyl-4-methoxyphenyl)-2-(2-aminophenyl)ethane (13) to give 2-isopropyl-3-methoxy-9,10-dihydrophenanthrene (9) and 4-isopropyl-3-methoxy-9,10-dihydrophenanthrene (9a). Compound 9 was converted to multicaulin and miltirone-like phenanthrene derivatives by further reactions. The best antituberculosis activity was exhibited by 2-isopropylphenanthrene-3-ol (11).
Introduction
Tuberculosis (TB) has historically been one of the most dangerous diseases. For centuries its cause was not known, but Robert Koch found in the 1880s that the real reason for TB was Mycobacterium tuberculosis. He also noted that 1/7 of all humans died of TB. Even today, 1/3 of all people in the world are infected with TBCitation1. According to the World Health Organization, 10.4 million new TB cases were reported in 2015 and there were an estimated 1.8 million TB deathsCitation2. In addition, multidrug-resistant and extremely drug-resistant TB strains are still considered as a problem for medicine to overcomeCitation3. The mortality rate for TB remains high despite the availability of antibiotics (isoniazide, ethambutol, pyrazinamide, rifampicin, and streptomycin) for its treatmentCitation4. Recently, Kimpe et al. synthesised some new heterocycles and reported their strong antimycobacterial activitiesCitation5–8. In this context, there is an urgent need to develop new drugs to overcome the newer forms of TB. Conventionally, natural products have played an important role in the development of new drugs for the treatment of many diseasesCitation9. Today, nearly one-third of the top-selling drugs in the world are natural products or their synthetic derivativesCitation10. Plants of the genus Salvia, which is a member of the family Lamiaceae, are used worldwide as conventional medicines with various biological activities such as antibacterial, antioxidant, antidiabetic, antitumor, and antituberculousCitation11,Citation12. Ulubelen et al. reported that these species have also been used for the treatment of many ailments, including haemorrhaging, menstrual disorders, miscarriage, heart disease, and hepatitisCitation13.
Phytochemical studies of Salvia species have been conducted by many research groups, and compounds including diterpenoids, sesquiterpenoids, sesterterpenoids, steroids, and polyphenols have been identified as key constituents of these plants. The diterpenoids found in these species, particularly tanshinones (1, 2, 3) and miltirone (4), which are 20-norditerpenes with an abietane-type skeleton that includes a quinone moiety in the C-ring (), have been widely studiedCitation14,Citation15. These compounds behave as antioxidants against lipid peroxidation in vitro and in vivo. A study showed the potential anticancer activity of tanshinone IIA in vitro and in vivo against both estrogen receptor (ER)-positive and ER-negative breast cancersCitation16. Chang et al. also reported the most potential activity of miltirone among 10 diterpene quinones isolated from Salvia miltiorrhiza in the central benzodiazepine receptor binding assay (IC50 = 0.3 pM)Citation17.
Figure 1. Structures of abietane-type diterpenes 1–4, which include a quinone moiety in the C-ring.
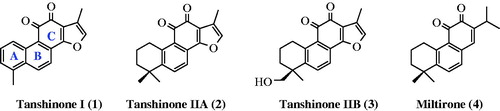
Ulubelen et al.Citation13 isolated an additional four aromatic norabietanes (5–8) with structural and biological properties similar to those of the highly active diterpenoids tanshinones and miltirone (). These compounds exhibit strong antituberculous activity, with minimal inhibitory concentration (MIC) values ranging from 0.46 to 7.3 μg/mlCitation13. Previously, we achieved the first total synthesis of the antituberculous agents multicaulin (5) and O-demethylmulticaulin (6) based on an oxidative photochemical reaction of the corresponding stilbeneCitation18.
Figure 2. Structures of aromatic norabietanes 5–8.
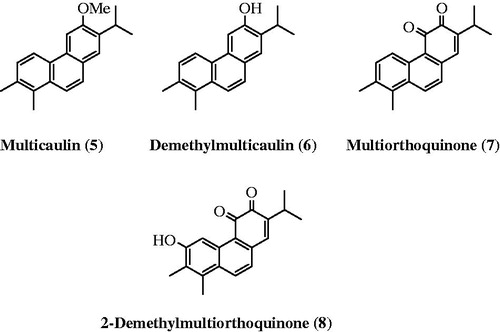
Figure 3. Four C-ring functionalised abietane-like compounds (9–12).
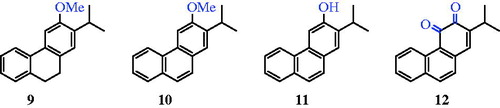
Herein, we describe a new method for four C-ring functionalised abietane-like compounds (9–12) based on a direct chemical synthesis and the evaluation of their antimycobacterial activities (). To the best of our knowledge, among compounds 9–12, there was only one article describing a synthesis of phenanthrene derivative 10 in 1957, in which Sengupta et al.Citation19 prepared compound 10 starting from ethyl 2-methoxybenzoate in 11 steps and with a yield of less than 1%.
A retrosynthetic analysis of abietane-like compounds 9–12 is shown in Scheme 1. The key step in this strategy is chosen as the connection of the A and C rings via the Pschorr reaction of 13.
Scheme 1. Retrosynthetic analysis for compounds 9–12.
Experimental
General experimental procedures
Commercially available reagents and solvents were of analytical grade or were purified by standard procedures prior to use. Reactions were monitored via thin-layer chromatography (TLC). The 1H NMR and 13C NMR spectra were recorded on a 200(50) MHz and 400(100) MHz Varian spectrometer using CDCl3. Chemical shifts (δ) are reported in parts per million (ppm) relative to either a tetramethylsilane (TMS) internal standard or solvent signals. Interchangeable hydrogens and carbons were assigned with the letter. High-resolution mass spectrometry (HRMS) were recorded in Bruker Daltonics microTOF-Q instrument by using atmospheric pressure chemical ionization-electrospray ionization (APCI-ESI) ion source. Column chromatography was performed on silica gel 60 (70–230 mesh ASTM), and TLC was carried out on silica gel (254–366 mesh ASTM). The purity of biologically tested compounds was determined by Q NMR (purity >98%)Citation20–23.
4-Bromo-2-isopropylanisole (19)
Compound 19 was synthesised according to the procedure reported in our previous studyCitation18.
3-Isopropyl-4-methoxybenzonitrile (20)
CuCN (7.60 g; 84.8 mmol) was placed into a 250-ml flask and a solution of 4-bromo-2-isopropylanisole (19) (6.48 g, 28.3 mmol) in dimethylformamide (DMF) (40 ml) was added under N2. The reaction mixture was stirred at 140 °C for 12 h. The reaction mixture was allowed to cool to rt and EtOAc (100 ml) was added. The mixture was washed with a 10% solution of FeCl3 (150 ml). The organic layer was dried with MgSO4. The solvent was removed under reduced pressure to afford 3-isopropyl-4-methoxybenzonitrile (20) as a brown liquid (4.44 g, 90%). Rf = 0.5 (1:9 EtOAc-hexanes).
1H NMR (200 MHz, CDCl3) δ 7.46 (dd, 1H, H-6, J = 8.6 Hz, J = 2.1 Hz); 7.45 (d, 1H, H-2, J = 2.1 Hz); 6.87 (d, 1H, H-5, J = 8.6 Hz); 3.87 (s, 3H, OMe); 3.29 (septet, 1H, CHMe2, J = 6.8 Hz); 1.18 (d, 6H, CHMe2, J = 6.8 Hz).
13C NMR (50 MHz, CDCl3) δ 159.1 (C-4); 138.3 (C-3); 131.4 (C-2 or C-6); 129.9 (C-2 or C-6); 119.6 (CN); 110.5 (C-5); 103.7 (C-1); 55.5 (OMe); 26.6 (CHMe2); 22.1 (CHMe2).
Ethyl 3-isopropyl-4-methoxybenzoate (21)
To a solution of 20 (4.22 g, 24.1 mmol) in EtOH (50 ml), H2SO4 (98%, 7.16 ml, 145 mmol) was carefully added. The reaction mixture was refluxed for 12 h. The mixture was then allowed to cool to rt, and the excess EtOH was removed under reduced pressure. The crude product was dissolved in EtOAc (100 ml) and washed with H2O (2 × 100 ml). The organic layer was dried with MgSO4, and the solvent was removed under reduced pressure to afford ethyl 3-isopropyl-4-methoxybenzoate (21) as a brown liquid (4.50 g, 84%). Compound 21 was used in the next step without further purification.
1H NMR (200 MHz, CDCl3) δ 7.90 (s, 1H, H-2); 7.88 (d, 1H, H-6, J = 8.3 Hz); 6.82 (d, 1H, H-5, J = 8.3 Hz); 4.34 (q, 2H, OCH2CH3, J = 7.0 Hz); 3.85 (s, 3H, OMe); 3.31 (septet, 1H, CHMe2, J = 7.0 Hz); 1.37 (t, 3H, OCH2CH3, J = 7.0 Hz); 1.22 (d, 6H, CHMe2, J = 7.0 Hz).
13C NMR (50 MHz, CDCl3) δ 166.5 (CO); 160.5 (C-4); 136.8 (C-1); 128.8 (C-6); 127.6 (C-2); 122.6 (C-3); 109.5 (C-5); 60.3 (OCH2CH3); 55.3 (OMe); 26.7 (CHMe2); 22.2 (CHMe2); 14.2 (OCH2CH3).
3-Isopropyl-4-methoxybenzyl alcohol (22)Citation24
To a suspension of LiAlH4 (2.37 g, 62.4 mmol) in tetrahydrofuran (THF) (20 ml), a solution of 21 (4.11 g, 18.5 mmol) in THF (20 ml) was added dropwise at 0 °C under N2. The reaction mixture was stirred for 4 h at rt. After monitoring with TLC, the reaction mixture was cooled to 0 °C, and a saturated solution of NH4Cl (10 ml) was added, followed by the addition of EtOAc (30 ml). The precipitate was filtered, and then the organic phase was washed with water (2 × 20 ml) and dried with MgSO4. Finally, the solvent was removed under reduced pressure to yield 3-isopropyl-4-methoxybenzyl alcohol (22) as a yellow liquid (3.03 g, 91%). Rf = 0.23 (1:4 EtOAc-hexanes).
1H NMR (200 MHz, CDCl3) δ 7.24 (d, 1H, H-2, J2,6 = 2.0 Hz); 7.18 (dd, 1H, H-6, J = 8.1 Hz, J = 2.0 Hz); 6.84 (d, 1H, H-5, J = 8.1 Hz); 4.59 (s, 2H, CH2OH); 3.85 (s, 3H, OMe); 3.36 (septet, 1H, CHMe2, J = 7.0 Hz); 2.43 (bs, 1H, OH); 1.26 (d, 6H, CHMe2, J = 7.0 Hz).
13C NMR (50 MHz, CDCl3) δ 156.2 (C-4); 137.0 (C-3); 132.8 (C-1); 125.4 (C-2 or C-6); 125.2 (C-2 or C-6); 110.2 (C-5); 65.0 (CH2OH); 55.3 (OMe); 26.6 (CHMe2); 22.5 (CHMe2).
The 1H NMR and 13C NMR spectra are in agreement with the reported data of Burnell and CaronCitation24.
3-Isopropyl-4-methoxybenzyl bromide (23)Citation24
To a solution of 22 (3.50 g, 19.4 mmol) in CH2Cl2 (50 ml), PBr3 (5.79 g, 2.01 ml, 21.4 mmol) was added dropwise at 0 °C. The reaction mixture was stirred for 12 h at rt. After monitoring with TLC, water (50 ml) was added to the mixture. The organic layer was then separated and dried with MgSO4, and the solvent was removed under reduced pressure to afford 3-isopropyl-4-methoxybenzylbromide (23) as a red liquid (4.35 g, 92%).
1H NMR (200 MHz, CDCl3) δ 7.29 (d, 1H, H-2, J = 2.2 Hz); 7.26 (dd, 1H, H-6, J = 8.2 Hz, J = 2.2 Hz); 6.84 (d, 1H, H-5, J = 8.2 Hz); 4.56 (s, 2H, CH2Br); 3.86 (s, 3H, OMe); 3.36 (septet, 1H, CHMe2, J = 6.9 Hz); 1.28 (d, 6H, CHMe2, J = 6.9 Hz).
13C NMR (50 MHz, CDCl3) δ 156.9 (C-4); 137.4 (C-3); 129.7 (C-1); 127.4 (C-2 or C-6); 127.1 (C-2 or C-6); 110.4 (C-5); 55.4 (OMe); 34.5 (CH2Br); 26.8 (CHMe2); 22.5 (CHMe2).
The 1H NMR and 13C NMR spectra are in agreement with the reported data of Burnell and CaronCitation24.
(3-isopropyl-4-methoxybenzyl)triphenylphosphonium bromide (16)
To a solution of 23 (3.41 g, 14.0 mmol) in CH3CN (100 ml), PPh3 (4.04 g, 15.4 mmol) was added, and the reaction mixture was refluxed for 24 h. After monitoring by TLC, the solvent was removed under reduced pressure to afford (3-isopropyl-4-methoxy)triphenylphosphonium bromide (16) as a white solid (7.06 g). The salt was used for the next step without further purification.
(E/Z)-1-(3-isopropyl-4-methoxyphenyl)-2-(2-nitrophenyl)ethene (14)
To a suspension of NaH (2.39 g, 99.6 mmol) in CH2Cl2 (20 ml) under N2, 16 (7.06 g, 14.0 mmol) in CH2Cl2 (40 ml) was added at 0 °C, and the mixture was stirred for 15 min at the same temperature. After the mixture was stirred for 15 min, 2-nitrobenzaldehyde (15) (2.51 g, 16.6 mmol) in CH2Cl2 (40 ml) was added to the mixture, and the reaction mixture was stirred for 16 h at rt. After monitoring with TLC, a saturated solution of NH4Cl (20 ml) was added dropwise to the mixture to quench the excess NaH. The organic layer was separated and dried with MgSO4. The solvent was removed under reduced pressure, and the crude product was purified by silica gel chromatography (1:4 EtOAc-hexanes) to yield an isomeric mixture of stilbene (E/Z)-14 (4.0 g) as a yellow liquid. This mixture was used for the next step without further purification.
1H NMR of (E)-isomer (400 MHz, CDCl3) δ 7.94 (dd, 1H, H-3''a, J = 8.1 Hz, J = 1.5 Hz); 7.76 (dd, 1H, H-6''a, J = 8.1 Hz, J = 1.5 Hz); 7.57 (bt, 1H, H-5''b, J = 8.1 Hz); 7.46 (d, 1H, H-2, J = 16.1 Hz); 7.38 (s, 1H, H-2'); 7.37 (d, 1H, H-6', J = 9.1 Hz); 7.36 (bt, 1H, H-4''b, J = 8.1 Hz); 7.08 (d, 1H, H-1, J = 16.1 Hz); 6.86 (d, 1H, H-5', J = 9.1 Hz); 3.86 (s, 3H, OMe); 3.33 (septet, 1H, CHMe2, J = 6.6 Hz); 1.24 (d, 6H, CHMe2, J = 6.6 Hz).
HRMS m/z (by using APCI-ESI ion source) 298. 1436 [M + H] + (calcd. for C18H19NO3 298.1438).
1-(3-Isopropyl-4-methoxyphenyl)-2-(2-aminophenyl)ethane (13)
Pd-C (400 mg) was placed into a 250-ml flask and cooled MeOH (40 ml) was carefully added. A solution of 14 (4.00 g) in MeOH (60 ml) was then added to the mixture. The reaction was stirred under H2 (balloon, 1 atm) for 2 h at rt. The catalyst was filtered and the solvent was removed under reduced pressure to afford 1-(3-isopropyl-4-methoxyphenyl)-2-(2-aminophenyl)ethane (13) (3.23 g, 12.0 mmol) as a pale brown liquid. Overall yield from 23 to 13 was 78%.
1H NMR (400 MHz, CDCl3) δ 7.16–7.01 (m, 4H, H-4'', H-6'', H-2', H-6'); 6.86 (d, 1H, H-5', J = 9.0 Hz); 6.84 (dt, 1H, H-5'', J = 7.4 Hz, J = 1.2 Hz); 6.74 (bd, 1H, H-3'', J = 7.8 Hz); 3.89 (s, 3H, OMe); 3.44 (bs, 2H, NH2); 3.40 (septet, 1H, CHMe2, J = 7.0 Hz); 3.00–2.83 (m, 4H, A2B2 system, 2xH-1, 2xH-2); 1.29 (d, 6H, CHMe2, J = 7.0 Hz).
13C NMR (100 MHz, CDCl3,) δ 155.1 (C-4'); 144.2 (C-2''); 136.8 (s); 133.6 (s); 129.4 (d); 127.0 (d); 126.3 (s); 126.2 (d); 126.1 (d); 118.8 (d); 115.6 (d); 110.4 (d); 55.4 (OMe); 34.7 (C-1a); 33.6 (C-2a); 26.6 (CHMe2); 22.6 (CHMe2).
HRMS m/z (by using APCI-ESI ion source) 270.1850 [M + H] + (calcd. for C18H23NO 270.1852).
2-Isopropyl-3-methoxy-9,10-dihydrophenanthrene (9)
To a solution of 13 (0.46 g, 1.71 mmol) in acetone (20 ml), H2SO4 (98%, 0.18 ml, 3.5 mmol) and isopentyl nitrite (0.40 g, 0.45 ml, 3.41 mmol) were added carefully at 0 °C. The reaction mixture was stirred at 0–10 °C for 1 h; then H2O (10 ml) was added and the mixture was stirred further for 1 h. After 2 h, a saturated NaHSO3 solution (5 ml) was added to the mixture, and the mixture was poured into H2O (50 ml). The mixture was extracted with CHCl3 (2 × 50 ml), and the combined organic layers were separated and dried with MgSO4. The solvent was removed under reduced pressure, and the crude product was purified by silica gel chromatography (3:7 EtOAc-hexanes) to yield 2-isopropyl-3-methoxy-9,10-dihydrophenanthrene (9) (77 mg, 18%) and 4-isopropyl-3-methoxy-9,10-dihydrophenanthrene (9a) (25 mg, 6%).
1H NMR of 2-isopropyl-3-methoxy-9,10-dihydrophenanthrene (9) (400 MHz, CDCl3) δ 7.71 (bd, 1H, H-5, J = 7.7 Hz); 7.29 (ddd, quasi dt, 1H, H-6, J = 7.7 Hz, J = 6.9 Hz, J = 1.8 Hz); 7.23 (s, 1H, H-1); 7.22–7.18 (m, 2H, H-7 and H-8); 7.01 (s, 1H, H-4); 3.91 (s, 3H, OMe); 3.33 (septet, 1H, CHMe2, J = 7.0 Hz); 2.88–2.78 (m, A2B2 system, 4H, 2xH-9 and 2xH-10); 1.24 (d, 6H, CHMe2, J = 7.0 Hz).
13C NMR of 2-isopropyl-3-methoxy-9,10-dihydrophenanthrene (9) (100 MHz, CDCl3) δ 156.0 (C-5); 137.3 (s); 136.5 (s); 134.7 (s); 132.5 (s); 129.5 (s); 128.1 (d); 127.0 (d); 126.8 (d); 125.9 (d); 123.3 (d); 106.0 (d); 55.7 (OMe); 29.5 (C-9a); 28.3 (C-10a); 26.7 (CHMe2); 22.8 (CHMe2).
1H NMR of 4-isopropyl-3-methoxy-9,10-dihydrophenanthrene (9a) (400 MHz, CDCl3) δ 7.49 (bd, 1H, H-5, J = 7.7 Hz); 7.29–7.19 (m, 3H, H-6, H-7, H-8); 7.06 (d, 1H, H-1, J = 8.2 Hz); 6.77 (d, 1H, H-2, J = 8.2 Hz); 3.85 (s, 3H, OMe); 3.70 (septet, 1H, CHMe2, J = 7.0 Hz); 2.73–2.63 (m, A2B2 system, 4H, 2xH-9 and 2xH-10); 1.41 (d, 6H, CHMe2, J = 7.0 Hz).
2-isopropyl-3-methoxyphenanthrene (10)
To a solution of 9 (50 mg, 0.20 mmol) in toluene (2 ml), a solution of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (49.5 mg, 0.22 mmol) in toluene (5 ml) was added. The reaction mixture was refluxed for 24 h. After monitoring by TLC, the mixture was allowed to reach rt and then the solvent was removed under reduced pressure. The crude product was purified by TLC eluting with hexane to yield 2-isopropyl-3-methoxyphenanthrene (10) (21 mg, 42%) as a colourless liquid and 3-methoxy-2-(prop-1-en-2-yl)phenanthrene (10a) (14 mg, 28%) as a colourless liquid.
1H NMR of 2-isopropyl-3-methoxyphenanthrene (10) (400 MHz, CDCl3) δ 8.60 (bd, 1H, H-5, J = 8.1 Hz); 7.98 (s, 1H, H-1); 7.87 (d, 1H, H-8, J = 7.7 Hz); 7.69 (s, 1H, H-4); 7.68 (d, 1H, H-9a, J = 8.8 Hz); 7.62 (bt, 1H, H-6 overlapped with H-10); 7.61 (d, 1H, H-10a, J = 8.8 Hz); 7.56 (dt, 1H, H-7, J = 7.7 Hz, J = 1.1 Hz); 4.08 (s, 3H, OMe); 3.48 (septet, 1H, CHMe2, J = 6.6 Hz); 1.35 (d, 6H, CHMe2, J = 6.6 Hz).
13C NMR of 2-isopropyl-3-methoxyphenanthrene (10) (100 MHz, CDCl3) δ 156.6 (C-3); 138.3 (s); 132.1 (s); 129.8 (s); 129.5 (s); 128.6 (d); 126.7 (s); 126.6 (d); 126.1 (d); 125.9 (d); 125.7 (d); 124.4 (d); 122.4 (d); 101.8 (d); 55.5 (OMe); 27.1 (isopropyl CH); 22.8 (2xCH3).
1H NMR of 3-methoxy-2-(prop-1-en-2-yl)phenanthrene (10a) (400 MHz, CDCl3) δ 8.60 (d, 1H, H-5, J = 8.4 Hz); 8.00 (s, 1H, H-1); 7.87 (d, 1H, H-8, J = 7.7 Hz); 7.70 (s, 1H, H-4); 7.67 (d, 1H, H-9, J = 9.2 Hz); 7.63 (t, 1H, H-6 overlapped with H-10); 7.62 (d, 1H, H-10, J = 9.2 Hz); 7.58 (t, 1H, H-7, J = 7.7 Hz); 5.26 (bs, 1H, C = CH2); 5.22 (bs, 1H, C = CH2); 4.07 (s, 3H, OMe); 2.22 (bs, 3H, =C-CH3).
2-Isopropylphenanthren-3-ol (11)
To a solution of 10 (100 mg, 0.40 mmol) in CH2Cl2 (30 ml), BBr3 (110 mg, 0.04 ml, 0.44 mmol) was added dropwise under N2 at 0 °C. The reaction mixture was stirred at rt for 12 h; then MeOH (10 ml) was added to the mixture and the solvent was removed under reduced pressure. The crude product was dissolved in EtOAc (30 ml) and washed with H2O (2 × 30 ml). The organic layer was dried with Na2SO4 and removed under reduced pressure to afford 2-isopropylphenanthren-3-ol (11) (88 mg, 93%) as a yellow solid. Rf = 0.70 (1:4 EtOAc-hexanes), Mp =141–143 °C.
1H NMR (400 MHz, CDCl3) δ 8.48 (d, 1H, H-5, J = 7.6 Hz); 7.94 (s, 1H, H-1); 7.85 (bd, 1H, H-8, J = 8.0 Hz); 7.70 (s, 1H, H-4); 7.67 (A part of AB system, d, 1H, H-9, J = 9.3 Hz); 7.59 (B part of AB system, d, 1H, H-10, J = 9.3 Hz); 7.59–7.52 (m, 2H, H-6 and H-7); 5.19 (s, 1H, OH); 3.40 (septet, 1H, CH(CH3)2, J = 7.0 Hz); 1.40 (d, 3H, CH3, J = 7.0 Hz); 1.39 (d, 3H, CH3, J = 7.0 Hz).
13C NMR (100 MHz, CDCl3) δ 152.5 (C-3); 136.0 (s); 132.0 (s); 129.7 (s); 129.4 (s); 128.5 (d); 127.0 (s); 126.6 (d); 126.2 (d); 126.2 (d); 126.0 (d); 124.4 (d); 122.5 (d); 106.9 (d); 27.5 (CHMe2); 22.6 (2xCHMe2).
HRMS m/z (by using APCI-ESI ion source) 237.1272 [M + H] + (calcd. for C17H16O 237.1274).
2-Isopropylphenanthren-3,4-dione (12)
To a solution of 11 (100 mg, 0.42 mmol) in CH2Cl2 (20 ml), Dess–Martin periodinane (269 mg, 0.63 mmol) was added dropwise at 0 °C. The reaction mixture was stirred at rt for 12 h. After monitoring by TLC, the reaction mixture was washed with 1 M NaOH solution (20 ml). The organic layer was separated and dried with Na2SO4, and the solvent was removed under reduced pressure to afford 2-isopropylphenanthren-3,4-dione (12) (64.1 mg, 61%) as a red liquid. Rf = 0.40 (1:4 EtOAc-hexanes).
1H NMR (400 MHz, CDCl3) δ 9.37 (d, 1H, H-5, J = 8.6 Hz); 8.08 (d, 1H, H-8, J = 8.6 Hz); 7.80 (bd, 1H, H-10, J = 8.2 Hz); 7.69 (ddd, quasi bt, 1H, H-6 b, J = 8.6 Hz, J = 6.9 Hz, J = 1.4 Hz); 7.52 (ddd, quasi bt, 1H, H-7 b, J = 8.6 Hz, J = 6.9 Hz, J = 0.9 Hz); 7.35 (d,1H, H-9, J = 8.2 Hz); 7.18 (d, 1H, H-1, J = 0.9 Hz); 3.07 (septet, 1H, CH(CH3)2, J = 6.9 Hz); 1.21 (d, 6H, CH(CH3)2, J = 6.9 Hz).
13C NMR (100 MHz, CDCl3) δ 182.3 (s); 181.5 (s); 146.9 (s); 139.7 (d); 137.5 (d); 134.3 (s); 132.5 (s); 131.3 (d); 129.1 (d); 127.7 (d); 127.1 (d); 126.9 (d); 125.0 (s); 27.4 (d); 21.7 (q).
HRMS m/z (by using APCI-ESI ion source) 251.1066 [M + H] + (calcd. for C17H14O2 251.1067).
Results and discussion
Synthesis
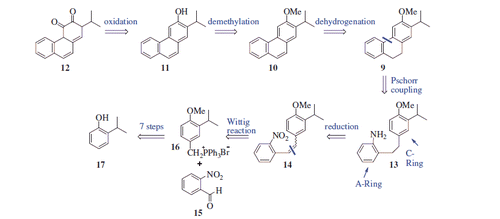
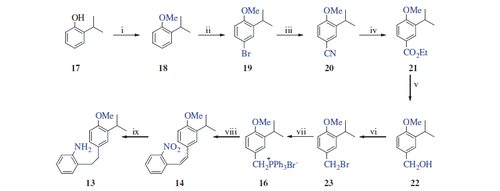
2-Isopropylphenol (17) was used as the starting material for the preparation of 13. The synthesis of 19 with a sequence of 17 → 18 → 19 was described in our previous study in two stepsCitation18. Following our previous procedure, compound 17 was converted to 19 by O-methylation followed by selective bromination with CAN/LiBr. Preparation of benzyl bromide 23 from compound 19 was as described by Burnell and CaronCitation24. Following this procedure with a slight modification, we prepared compound 23. For this purpose, treatment of compound 19 with CuCN provided benzonitrile 20 in 90% yield, which was then esterified with EtOH to give ester 21 in the presence of H2SO4 (94% yield). Reduction of ester 21 using LiAlH4 gave alcohol 22 (81% yield). Treatment of this alcohol with PBr3 afforded benzyl bromide 23 in 92% yield. Phosphonium salt 16 was prepared from benzyl bromide 23 by reaction with PPh3 and used in the next step without further purification. The Wittig reaction of 16 with 2-nitrobenzaldehyde (15) resulted in an (E/Z) mixture of stilbene 14, which was then hydrogenated on Pd-C to give aniline 13 in 86% overall yield in three steps from 23 (Scheme 2).
Scheme 2. Reaction conditions and reagents: (i) Me2SO4, NaOH (aq), 90 °C, 4 h, 93%; (ii) LiBr/(NH4)2Ce(NO3)6, CH3CN, 20 °C, 2 h, 97%; (iii) CuCN, DMF, 140 °C, 12 h, 90%; (iv) EtOH, H2SO4, reflux, 12 h, 84%; (v) LiAlH4, THF, 0 °C, 4 h, 91%; (vi) PBr3, DCM, 0 °C, 12 h, 92%; (vii) PPh3, CH3CN, reflux, 24 h; (viii) NaH, DCM, 0 °C, 15 min; and 2-nitrobenzaldehyde (15), 20 °C, rt, 16 h; and (ix) H2, Pd/C, MeOH, 20 °C, 2 h.
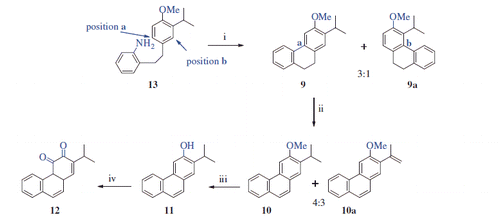
The most important step in our synthetic strategy was the conversion of amine 13 to tricyclic compound 9 via an intramolecular Pschorr coupling reaction. The Pschorr reaction has been known for over a century and proceeds through aryldiazonium salt of biaryl to give tricyclic arenesCitation25. In this context, Caronna et al. synthesised 4-methoxy-9,10-dihydrophenanthrene via Pschorr couplingCitation26. By a similar approach, the intramolecular Pschorr reaction of 13 gave the desirable compound 9 along with side product 9a in a yield of 24% (9:9a = 3:1). Thus, after diazotisation of 13, two different ring closure reactions occurred at position a and position b (Scheme 3). In this work, we suppose that amine 13 should give a phenanthrene ring by a similar approach. Assignment of the structures of the two isomers was performed via 1H NMR analysis. The 1H NMR spectrum of 9 displayed two singlets for the H-1 (δ 7.23 ppm) and H-4 (δ 7.01 ppm) protons. On the other hand, the 1H NMR spectrum of 9a displayed an AB system for H-1 and H-2 at δ 7.06 and δ 6.77 ppm, respectively. To obtain phenanthrene 10, compound 9 was treated with powerful oxidant DDQ. As a result of oxidation of 9, the desired product 10 was obtained in a yield of 42% along with a 29% yield of side product 10a (4:3 ratio, Scheme 3). This mixture was successfully separated using column chromatography, and their structures were evaluated by NMR spectra. Signals for the CH2CH2 at the C-9 and C-10 positions that were present in the 1H NMR spectrum of 9 were not detected in the 1H NMR spectra of 10 and 10a, indicating that both compounds were aromatised. In addition, the 1H NMR spectrum of 10a displayed two broad singlets for olefinic protons at δ 5.26 and δ 5.22 ppm, indicating that the isopropyl group was also oxidised during the reaction. The formation of 10a is thought to proceed via a stable carbenium ion at C-2 of isopropyl (Scheme 3). Next, O-demethylation of 10 via treatment with BBr3 yielded phenanthrene 11 in 93% yield. In the 1H NMR spectrum of 11, no singlet for the methoxy protons (δ 4.07 in the 1H NMR spectrum of 10) was detected. Finally, compound 12 was prepared in 61% yield via the Dess–Martin periodinane oxidation of 11. The 1H NMR spectrum of 12 displayed four doublets for H-5 (δ 9.37), H-8 (δ 8.08), H-9 (δ 7.35) and H-10 (δ 7.80); two quasi-broad triplets for H-6 (δ 7.69) and H-7 (δ 7.52); and one doublet for H-1 (δ 7.18) with 4 J = 0.9 Hz (Scheme 3).
Scheme 3. Synthesis of 11 and 12a. aReaction conditions and reagents: (i) isopentyl nitrite, H2SO4, acetone, 0–10 °C, 2 h, 24%; (ii) DDQ, toluene, reflux, 24 h, 70%; (iii) BBr3, DCM, N2 atm., 0 °C, 12 h, 93%; (iv) DMP, DCM, 0 °C, 12 h, 61%.
Biological assessment – antimycobacterial activity
The chemical compounds were tested against standard bacterial strains Mycobacterium smegmatis ATCC14468, Mycobacterium bovis, Mycobacterium szulgai ATCC35799, Mycobacterium gastri ATCC15754, Mycobacterium simiae ATCC25275 and M. tuberculosis H37Ra for the determination of their antimycobacterial activities (). The MIC studies were performed as described by Sokmen et al.Citation27 In brief, 100 μl of Middlebrook 7H9 Broth was dispersed into the wells, and 100 μl of the chemical compounds was added into the first wells. After being pipetted, 100 μl of the first wells was transferred to the second wells. In this way, serial dilutions were performed. After dilutions, 95 μl of Middlebrook 7H9 Broth was added into the wells, and then 5 μl of the inoculum (with 0.5 McFarland turbidity) was added. Thus, the final volume of each well was 200 μl. The concentration range of chemical compounds was 0–1000 μl/ml. Streptomycin was used as a positive control. Since the chemical compounds were dissolved in ethanol, it was used as a negative control. The plates were incubated at 37 °C until bacterial growth was observed in the wells including 0 μl/ml chemical compounds. The lowest concentrations of the chemical compounds that inhibited the growth of the bacteria were considered as the MIC values. Each experiment was replicated three times for the determination of the MIC.
Table 1. In vitro antimycobacterial activity of synthesised compounds (9–12) expressed as the minimum inhibitory concentration (μm).
Conclusions
In this study, stilbene 14 was prepared from 2-isopropylphenol in eight steps in a total yield of 49%. Catalytic hydrogenation of 14 gave 13, which underwent a Pschorr coupling reaction via diazotisation to afford dihydrophenanthrenes 9 and 9a. Oxidation of 9 with DDQ gave 10 as the main product. Demethylation of 10 gave 11 as a multicaulin analogue, and then oxidation of 11 gave 12 as a miltirone analogue.
Compounds 9–12 were tested for their in vitro antimycobacterial activity against M. smegmatis, M. bovis, M. szulgai, M. gastri, M. simiae, and M. tuberculosis (H37Ra). The preliminary studies revealed that multicaulin analogues (9, 10 and 11) were able to impair the growth of mycobacterial strains. Multicaulin analogues exhibited a considerable antimycobacterial activity; interestingly compound 11 showed a wider activity than the other compounds. The compound 11 showed higher or equal activity (except for M. smegmatis and M. bovis) than 9, 10 and 12.
MIC value of the most active compound (11) determined against the M. tuberculosis (H37Ra) was 8.26 μm when measured under in vitro condition. Compound 11 was also found to be equally effective against M. bovis with 9, and 12; against M. szulgai with 10 and 12.
Compound 9 was totally inactive against all mycobacteria strains utilised. None of the compounds reported here was appreciably active against the M. smegmatis ATCC 14468 and M. bovis ATCC 35734.
Ulubelen et al. reported the activities of the compounds 5–8 against M. tuberculosis strain (H37Rv) with MIC values 6 (0.46 μg/ml), 8 (1.2 μg/ml), 7 (2.0 μg/ml) and 5 (5.6 μg/ml). Thus, 2-isopropylphenanthrene-3-ol structured compound 6 showed the strongest activity. Interestingly, among our synthesised compounds 9–12 the best biological activity was shown by 2-isopropylphenantherene-3-ol (11). These results imply that strong antituberculosis agents can be developed based on the structure of 2-isopropylphenantherene-3-ol (11).
In conclusion, we showed a new example of Pschorr reaction to synthesise multicaulin and miltirone analogues. Biological evaluation of the compounds suggests encouraging results for developing antituberculosis drugs based on phenanthrene derivatives.
IENZ_1337758_Supplementary_Material.pdf
Download PDF (725 KB)Acknowledgements
The synthetic part of this article is a part of PhD dissertation of Serdar Burmaoğlu. We thank The Scientific and Technological Research Council of Turkey (Tubitak, grant number: 108T115) for financial support. We thank Dr. Oztekin Algul (Mersin University) for encouragement and critical reading for the manuscript. Hasan Seçen thanks Leslie Demir for her critical reading of this article.
Disclosure statement
The authors confirm that this article content has no conflict of interest.
Additional information
Funding
Related Research Data
References
- Tiwari R, Moraski GC, Krchňák V, et al. Thiolates chemically induce redox activation of BTZ043 and related potent nitroaromatic anti-tuberculosis agents. J Am Chem Soc 2013;135:3539–49.
- WHO Global Tuberculosis Report. (2016). Available from: http://who.int/tb/publications/global_report/en/ [last accessed 14 May 2017].
- Addla D, Jallapally A, Gurram D, et al. Design, synthesis and evaluation of 1,2,3-triazoleadamantylacetamide hybrids as potent inhibitors of Mycobacterium tuberculosis. Bioorg Med Chem Lett 2014;24:1974–9.
- Naik M, Ghorpade S, Jena LK, et al. Phenylindole and arylsulphonamide: novel scaffolds bactericidal against Mycobacterium tuberculosis. ACS Med Chem Lett 2014;5:1005–9.
- Cappoen D, Jacobs J, Nguyen Van T, et al. Straightforward palladium-mediated synthesis and biological evaluation of benzo[j]phenanthridine-7,12-diones as anti-tuberculosis agents. Eur J Med Chem 2012;48:57–68.
- Claes P, Cappoen D, Mbala BM, et al. Synthesis and antimycobacterial activity of analogues of the bioactive natural products sampangine and cleistopholine. Eur J Med Chem 2013;67:98–110.
- Cappoen D, Claes P, Jacobs J, et al. 1,2,3,4,8,9,10,11-octahydrobenzo[j]phenanthridine-7,12-diones as new leads against Mycobacterium tuberculosis. J Med Chem 2014;57:2895–907.
- Claes P, Cappoen D, Uythethofken C, et al. 2,4-Dialkyl-8,9,10,11-tetrahydrobenzo[g]pyrimido[4,5-c]isoquinoline-1,3,7,12(2H,4H)-tetraones as new leads against Mycobacterium tuberculosis. Eur J Med Chem 2014;77:409–21.
- Dias DA, Urban S, Roessner U. A historical overview of natural products in drug discovery. Metabolites 2012;2:303–36.
- Strohl WR. The role of natural products in a modern drug discovery program. Drug Discov Today 2000;5:39–41.
- Ulubelen A. Cardioactive and antibacterial terpenoids from some Salvia species. Phytochemistry 2003;64:395–9.
- Somer NU, Sarikaya B, Erac B, et al. Chemical composition and antimicrobial activity of essential oils from the aerial parts of Salvia pinnata L. Rec Nat Prod 2015;9:614–8.
- Ulubelen A, Topcu G, Johansson CB. Norditerpenoids and diterpenoids from Salvia multicaulis with antituberculous activity. J Nat Prod 1997;60:1275–80.
- Wu YB, Ni ZY, Shi QW, et al. Constituents from Salvia species and their biological activities. Chem Rev 2012;112:5967–6026.
- Hatipoglu S, Zorlu D, Dirmenci NT, et al. Determination of volatile organic compounds in fourty five Salvia species by thermal desorption-GC-MS technique. Rec Nat Prod 2016;10:659–700.
- Wang X, Wei Y, Yuan S, et al. Potential anticancer activity of tanshinone IIA against human breast cancer. Int J Cancer 2005;116:799–807.
- Chang HM, Chui KY, Tan FW, et al. Structure-activity relationship of miltirone, an active central benzodiazepine receptor ligand isolated from Salvia miltiorrhiza Bunge (Danshen). J Med Chem 1991;34:1675–92.
- Seçinti H, Burmaoğlu S, Altundaş R, Seçen H. Total syntheses of multicaulins via oxidative photocyclization of stilbenes. J Nat Prod 2014;77:2134–7.
- Sengupta SK, Biswas RN, Bhattacharyya BK. Friedel–Crafts reaction. IV. Synthesis of 7-isopropyl-3-oxo-6-methoxy-1,2,3,9,10,10a-hexahydrophenanthrene. J Indian Chem Soc 1959;36:659–68.
- Holzgrabe U. Quantitative NMR spectroscopy in pharmaceutical applications. Prog Nucl Magn Reson Spectrosc 2010;57:229–40.
- Malz F, Jancke H. Validation of quantitative NMR. J Pharm Biomed Anal 2005;38:813–23.
- Saito T, Ihara T, Koike M, et al. New traceability scheme for the development of international system-traceable persistent organic pollutant reference materials by quantitative nuclear magnetic resonance. Accredit Qual Assur 2009;14:79–86.
- Bostan MS, Senol M, Cig T, et al. Controlled release of 5-aminosalicylicacid from chitosan based pH and temperature sensitive hydrogels. Int J Biol Macromol 2013;52:177–83.
- Burnell RH, Caron S. Approach to the synthesis of candelabrone and synthesis of 3,7-diketo-12-hydroxyabieta-8,11,13-triene. Can J Chem 1992;70:1446–54.
- Smith MB. March’s advanced organic chemistry, 6th ed. New Jersey: Wiley; 2007.
- Caronna T, Ferrario F, Servi S. Intermolecular and intramolecular phenol phenylation: regioselective synthesis of unsymmetrically substituted biphenyls and 9,10-dihydrophenanthrenes. Tetrahedron Lett 1979;20:657–60.
- Sokmen A, Gulluce M, Akpulat HA, et al. The in vitro antimicrobial and antioxidant activities of the essential oils and methanol extracts of endemic Thymus spathulifolius. Food Control 2004;15:627–34.