
Abstract
A series of benzo[d]thiazole-5- and 6-sulfonamides has been synthesized and investigated for the inhibition of several human (h) carbonic anhydrase (CA, EC 4.2.1.1) isoforms, using ethoxzolamide (EZA) as lead molecule. 2-Amino-substituted, 2-acylamino- and halogenated (bromo-and iodo-derivatives at the heterocyclic ring) compounds led to several interesting inhibitors against the cytosolic hCA I, II and VII, as well as the transmembrane, tumor-associated hCA IX isoforms. Several subnanomolar/low nanomolar, isoform-selective sulfonamide inhibitors targeting hCA II, VII and IX were detected. The sharp structure–activity relationship for CA inhibition with this small series of derivatives, with important changes of activity observed even after minor changes in the scaffold or at the 2-amino moiety, make this class of scarcely investigated sulfonamides of particular interest for further investigations.
Introduction
The carbonic anhydrases (CAs, EC 4.2.1.1) are a superfamily of metalloenzymes which catalyze the interconversion between CO2 and bicarbonate by using a metal hydroxide nucleophilic mechanismCitation1–9. Seven distinct genetic CA families are known to date, the α-θ-CAs, which differ in their preference for metal ions used within the active site for performing the catalysis, their oligomerization state, but most importantly the three-dimensional fold of the proteinCitation1–12. In all cases, the apoenzymes are devoid of catalytic activity, the presence of the metal ion being essential both for catalysis as well as binding of inhibitors, many of which have biomedical applicationsCitation10–29.
Sulfonamides are the most important class of CA inhibitors (CAIs)Citation10–19, with several compounds such as acetazolamide (AAZ), methazolamide (MZA), ethoxzolamide (EZA), sulthiame (SLT), dichlorophenamide (DCP), dorzolamide (DZA), brinzolamide (BRZ), sulpiride (SLP), zonisamide (ZNS), topiramate (TPM) (a sulfamate, not sulfonamide), celecoxib (CLX) and valdecoxib (VLX) ().
Figure 1. Clinically used CAIs of the sulfonamide and sulfamate typeCitation10–19.
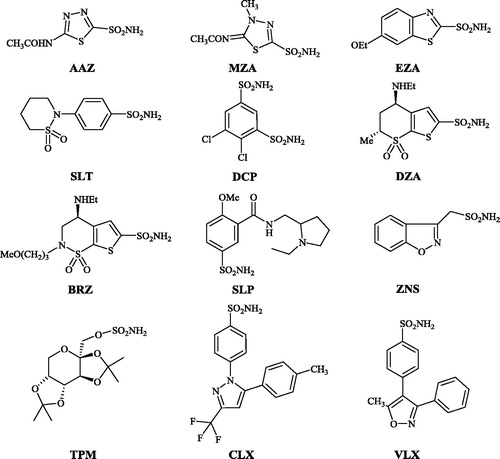
Compounds AAZ–VLX may be considered as first/second generation CAIs. Their main problem is that they indiscriminately inhibit most of the human isoforms known to dateCitation30–39. Indeed, 16 such isozymes were described in non-primates, CA I–XV with two V-type isoforms, CA VA and CA VB, and 15 isoforms are known in primates, as CA XV is not expressed in these mammalsCitation1–9. The nonselective inhibition of most CA isoforms by the first/second generation sulfonamide CAIs is the reason why a large number of new such derivatives are constantly and permanently reportedCitation20–39.
Materials and methods
Chemistry
Anhydrous solvents and all reagents were purchased from Sigma-Aldrich, Alfa Aesar and TCI. All reactions involving air- or moisture-sensitive compounds were performed under a nitrogen atmosphere using dried glassware and syringes techniques to transfer solutions. Nuclear magnetic resonance (1H NMR, 13C NMR) spectra were recorded using a Bruker Avance III 400 MHz spectrometer in DMSO-d6. Chemical shifts are reported in parts per million (ppm), and the coupling constants (J) are expressed in Hertz (Hz). Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet; brs, broad singlet; dd, double of doublets, and dt, double of triplets. The assignment of exchangeable protons (OH and NH) was confirmed by the addition of D2O. Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel F-254 plates. Flash chromatography purifications were performed on Merck Silica gel 60 (230–400 mesh ASTM) as the stationary phase, and ethyl acetate (EtOAc)/n-hexane were used as eluents. Melting points (m.p.) were carried out in open capillary tubes and are uncorrected.
4-Thioureido-benzenesulfonamide (1)
Sulfanilamide (2.0 g, 1.0 eq) was dissolved in a freshly prepared 3.5 M hydrochloric acid aqueous solution under gentle warming. The solution was cooled down to r.t. and potassium thiocyanate (1.0 eq) was added to reaction mixture then the mixture was heated at 90 °C for 3 h, cooled to r.t. to form precipitate which was filtered-off, washed with water, and dried under vacuum to afford the titled compound.
White solid, 93% yield; δH (400 MHz, DMSO-d6) 7.30 (2H, s), 7.70 (2H, d, J 8.8), 7.77 (2H, d, J 8.8), 10.01 (1H, s, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 122.7, 127.2, 139.8, 143.4, 182.2; m/z (ESI positive) 232.01 [M + H]+. Experimental in agreement with reported dataCitation40.
3-Thioureidobenzenesulfonamide (3)
3-Aminobenzensulfonamide (5.0 g, 1 eq) was dissolved in a freshly prepared 3.5 M hydrochloric acid aqueous solution by gentle warming, followed by treatment with potassium thiocyanate (1.0 eq) at r.t. and then heated to 90 °C for 12 h. The reaction mixture was cooled-down to r.t. and extracted with EtOAc (3 × 5.0 ml). The combined organic layers were washed with H2O (3 × 5.0 ml), dried over Na2SO4, filtered and concentrated to obtain a residue that was purified by silica gel column chromatography eluting with EtOAc/n-Hexane 70% v/v, followed by trituration with dichloromethane (DCM) to afford the titled compound.
White solid, 47% yield; δH (400 MHz, DMSO-d6) 7.39 (2H, s, exchange with D2O, SO2NH2), 7.56 (2H, m), 7.74 (1H, dt, J 1.8, 7.8), 7.96 (1H, d, J 1.8), 9.97 (1H, s, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 120.5, 122.0, 126.7, 130.0, 140.8, 145.2, 182.4; m/z (ESI positive) 232.0 [M + H]+. Experimental in agreement with reported dataCitation41.
2-Aminobenzo[d]thiazole-6-sulfonamide (2)
A suspension of 4-thioureido-benzenesulfonamide 1 (1.0 mmol, 1.0 eq) in CHCl3 (4.0 ml) was treated with Br2 (1.5 eq) drop-wise. The mixture was heated to 70 °C for 4.5 h, cooled-down to r.t. and the solvents were removed under reduced pressure to give a solid that was dissolved in H2O (5.0 ml). The aqueous solution was treated with NH4OH and stirred at 90 °C for 1 h. The formed precipitate was filtered-off, washed with H2O and dried under vacuum to afford the titled compound.
White solid, 80% yield; δH (400 MHz, DMSO-d6) 7.23 (2H, s, exchange with D2O, SO2NH2), 7.45 (1H, d, J 8.4), 7.69 (1H, dd, J 8.4, 1.8), 7.89 (2H, s, exchange with D2O, NH2), 8.15 (1H, d, J 1.8); δC (100 MHz, DMSO-d6) 118.0, 120.1, 124.6, 131.8, 137.2, 156.0, 170.3; m/z (ESI positive) 230.00 [M + H]+. Experimental in agreement with reported dataCitation41.
2-Aminobenzo[d]thiazole-5-sulfonamide (4)
A suspension of 3 (1.2 g, 1.0 eq.) in CHCl3 (15.0 ml) was treated with Br2 (1.5 eq) in CHCl3 (1.0 ml) drop-wise. The mixture was heated to 70 °C for 12 h, cooled down to r.t., the solvent eliminated in vacuum to give a residue that was dissolved in H2O (5.0 ml) and treated with NH4OH, followed by 1 h stirring at 90 °C. The cooled reaction mixture was filtered, washed with water and dried under vacuum to afford the titled compound.
White solid, 45% yield; δH (400 MHz, DMSO-d6) 7.42 (1H, t, J 8.0), 7.49–7.56 (4H, m, 2H exchange with D2O, SO2NH2), 7.69 (2H, s, exchange with D2O, NH2); δC (100 MHz, DMSO-d6) 120.39, 121.4, 126.7, 128.5, 137.7, 155.3, 169.7; m/z (ESI positive) 230.00 [M + H]+.
2-Amino-4-bromobenzo[d]thiazole-6-sulfonamide (5)
A suspension of 2 (0.75 g, 1 eq) in chloroform (15.0 ml) was treated with a solution of Br2 (8.0 eq) in chloroform (2.5 ml) drop-wise. The mixture was heated to 70 °C for 4 h. After cooling to r.t. the solvents were removed under reduced pressure. The obtained solid was dissolved in water (5.0 ml) and treated with ammonium hydroxide (pH =10), then the reaction mixture stirred for 1 h at 90 °C. The precipitated solid was filtered under vacuum, washed with H2O (3 × 5.0 ml), then with n-Hexane (3 × 3.0 ml) and dried to afford the titled compound.
Orange solid, 68% yield; δH (400 MHz, DMSO-d6) 7.35 (2H, s, exchange with D2O, SO2NH2), 7.89 (1H, s), 8.17 (1H, s), 8.26 (2H, s, exchange with D2O, NH2); δC (100 MHz, DMSO-d6) 110.7, 119.7, 127.6, 132.6, 138.1, 154.5, 170.9; m/z (ESI positive) 307.9 [M + H]+.
2-Amino-4-bromobenzo[d]thiazole-5-sulfonamide (6)
A suspension of 4 (0.2 g, 1.0 eq) in chloroform (4.0 ml) was treated with a solution of Br2 (6.0 eq) in chloroform (1.0 ml) drop-wise. The mixture was heated to 70 °C for 12 h. After cooling to r.t. the solvents were removed under reduced pressure. The obtained solid was dissolved in water (5.0 ml) and treated with ammonium hydroxide (pH =10), then the reaction mixture stirred for 1 h at 90 °C. After cooling, the reaction mixture was extracted with EtOAc (3 × 5 ml). The combined organic layers were washed with H2O (3 × 5.0 ml), dried over Na2SO4, filtered and concentrated to obtain a residue that was purified by silica gel column chromatography eluting with EtOAc/n-Hexane 70% v/v to afford the titled compound.
Orange solid, 19% yield; δH (400 MHz, DMSO-d6) 7.40 (1H, d, J 8.4), 7.66 (2H, s, exchange with D2O, SO2NH2), 7.69 (1H, d, J 8.4), 8.08 (2H, s, exchange with D2O, NH2); δC (100 MHz, DMSO-d6) 114.3, 120.6, 128.8, 129.8, 137.1, 152.9, 170.1; m/z (ESI negative) 305.7 [M-H]−.
2-Amino-4-iodobenzo[d]thiazole-6-sulfonamide (7)
A solution of 2 (0.3 g, 1.0 eq) in methanol (3.0 ml) was treated with iodine monochloride (4.0 eq) in methanol (1.0 ml) drop-wise. The mixture was heated to reflux temperature for 12 h. After cooling to room temperature, the reaction mixture was extracted with EtOAc (3 × 5.0 ml). The combined organic layers were washed with H2O (3 × 5.0 ml), dried over Na2SO4, filtered and concentrated to obtain a residue that was purified by silica gel column chromatography eluting with EtOAc/n-Hexane 70% v/v to afford the titled compound.
Dark orange solid, 31% yield; δH (400 MHz, DMSO-d6) 7.31 (2H, s, exchange with D2O, SO2NH2), 8.06 (1H, d, J 2.0), 8.16 (1H, d, J 2.0), 8.21 (2H, s, exchange with D2O, NH2); δC (100 MHz, DMSO-d6) 84.4, 119.9, 129.9, 133.1, 138.3, 157.0, 169.7; m/z (ESI positive) 355.9 [M + H]+.
2-Amino-4-iodobenzo[d]thiazole-5-sulfonamide (8)
A solution of 4 (0.2 g, 1.0 eq) in methanol (3.0 ml) was treated with a solution of iodine monochloride (4.0 eq) in methanol (1.0 ml) drop-wise. The mixture was heated to reflux temperature for 12 h. After cooling to room temperature, the reaction mixture was extracted with EtOAc (3 × 5 ml). The combined organic layers were washed with H2O (3 × 5.0 ml), dried over Na2SO4, filtered and concentrated to obtain a residue that was purified by silica gel column chromatography eluting with EtOAc/n-Hexane 70% v/v to afford the titled compound.
Dark orange solid, 16% yield; δH (400 MHz, DMSO-d6) 7.25 (1H, d, J 8.0), 7.63 (2H, s, exchange with D2O, SO2NH2), 7.87 (1H, d, J 8.0), 8.03 (2H, s, exchange with D2O, NH2); δC (100 MHz, DMSO-d6) 84.5, 119.9, 129.9, 133.2, 138.3, 157.0, 169.7; m/z (ESI positive) 355.8 [M + H]+.
N-(6-sulfamoylbenzo[d]thiazol-2-yl)acetamide (9)
A solution of 2 (1.0 g, 1.0 eq) in acetic acid (2.0 ml) was cooled to 0 °C followed by drop-wise addition of acetic anhydride (1.2 eq). The reaction mixture was refluxed for 3 h then excess of solvents were removed under reduced pressure to obtain a residue which was washed with Et2O (3 × 5 ml) to obtain titled compound.
White solid, 96% yield; δH (400 MHz, DMSO-d6) 2.27 (3H, s), 7.39 (2H, s, exchange with D2O, SO2NH2), 7.90 (2H, d, J 1.2), 8.50 (1H, t, J 1.2), 12.59 (1H, s, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 23.7, 121.0, 121.4, 124.8, 132.4, 139.9, 151.7, 161.9, 170.7; m/z (ESI positive) 272.0 [M + H]+.
N-(4-bromo-6-sulfamoylbenzo[d]thiazol-2-yl)acetamide (10)
A solution of 5 (0.1 g, 1.0 eq) in acetic acid (0.5 ml) was cooled to 0 °C followed by drop-wise addition of acetic anhydride (1.2 eq) then the mixture was refluxed for 3 h. The excess of solvents was removed under reduced pressure. The obtained solid was treated with sodium bicarbonate (1 N, 3.0 ml), then the reaction mixture was extracted with EtOAc (3 × 5 ml). The combined organic layers were washed with H2O (3 × 5.0 ml), dried over Na2SO4, filtered and concentrated under reduced pressure to afford the titled compound.
Orange solid, 93% yield; δH (400 MHz, DMSO-d6) 2.24 (3H, s), 7.44 (2H, s, exchange with D2O, SO2NH2), 8.05 (1H, s), 8.50 (1H, s), 12.59 (1H, s, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 23.6, 114.1, 120.5, 127.5, 133.2, 140.8, 149.8, 162.7, 171.0; m/z (ESI positive) 349.8 [M + H]+.
N-(5-sulfamoylbenzo[d]thiazol-2-yl)acetamide (11)
A solution of 4 (0.1 g, 1.0 eq) in acetic acid (0.5 ml) was cooled to 0 °C followed by drop-wise addition of acetic anhydride (1.2 eq) then the mixture was refluxed for 3 h. The excess of solvents was removed under reduced pressure. The obtained solid was treated with sodium bicarbonate (1 N, 3 ml), then the reaction mixture was extracted with EtOAc (3 × 5 ml). The combined organic layers were washed with H2O (3 × 5.0 ml), dried over Na2SO4, filtered and concentrated under reduced pressure to afford the titled compound.
White solid, 89% yield; δH (400 MHz, DMSO-d6) 2.27 (3H, s), 7.66 (1H, t, J 7.8), 7.71 (2H, s, exchange with D2O, SO2NH2), 8.80 (1H, dd, J 7.8, 1.0), 7.98 (1H, dd, J 7.8, 1.0), 12.45 (1H, s, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 23.6, 122.7, 124.6, 127.1, 128.8, 138.6, 150.9, 161.5, 170.6; m/z (ESI positive) 272.0 [M + H]+.
2-((6-Sulfamoylbenzo[d]thiazol-2-yl)carbamoyl)benzoic acid (12)
A solution of 2 (0.3 g, 1.0 eq) in dry DMF (3.0 ml) was treated with phthalic anhydride (1.0 eq), then the mixture was refluxed for 4 h. The reaction mixture was extracted with EtOAc (3 × 5.0 ml). The combined organic layers were washed with H2O (3 × 5.0 ml), dried over Na2SO4, filtered and concentrated under reduced pressure to afford the corresponding pure mixture of 2 isomers in (50:50) as evidenced by 1H NMR integration.
White solid, 100% yield; δH (400 MHz, DMSO-d6) 7.41 (2H, s, exchange with D2O, SO2NH2), 7.51 (2H, s, exchange with D2O, SO2NH2), 7.67–7.77 (4H, m), 7.93–8.03 (7H, m), 8.11–8.13 (2H, m), 8.22 (1H, d, J 8.4), 8.56 (1H, s, exchange with D2O, NH), 8.71 (1H, d, J 1.6, exchange with D2O, NH); δC (100 MHz, DMSO-d6) 121.0, 121.2, 121.5, 123.4, 124.8, 125.0, 125.1, 129.0, 130.6, 130.8, 131.3, 131.9, 132.5, 132.9, 133.2, 136.5, 137.0, 140.0, 141.5, 151.7, 151.8, 156.2, 162.1, 165.2, 167.8, 169.6; m/z (ESI positive) 377.9 [M + H]+.
CA inhibition
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activityCitation42. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled–deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together at room temperature (15 min) prior to assay, in order to allow for the formation of the E–I complex. Data from were obtained after 15 min incubation of enzyme and inhibitor, as for all sulfonamides reported earlierCitation43–50. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation51–55 and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation51–60.
Table 1. Inhibition data of human CA isoforms hCA I, II, VII and IX with compounds 1–12 in comparison with the standard sulfonamide inhibitors AAZ and EZA by a stopped flow CO2 hydrase assayCitation42.
Results and discussion
Chemistry
Most of the CAIs generated in our group over the last two decades were designed by using the tail approachCitation15,Citation32,Citation33. By choosing various functionalities that are appended on the scaffold of aromatic/heterocyclic sulfonamides in such a way as to interact with the middle and rim parts of the CA active site, a large number of isoform-selective CAIs were obtainedCitation15–36. Here on the other hand, we decided to explore a variant of the ring approachCitation1,Citation4, using ethoxzolamide (EZA) () as lead molecule. A series of benzothiazole-6-sulfonamides are reported here, which differ from EZA mainly by the position of the sulfamoyl moiety and by the presence of various substituents at the heterocyclic ring, in various positions (Scheme 1).
Scheme 1. Preparation of sulfonamides 1–12 investigated in this article, starting from sulfanilamide SA or metanilamide MA.
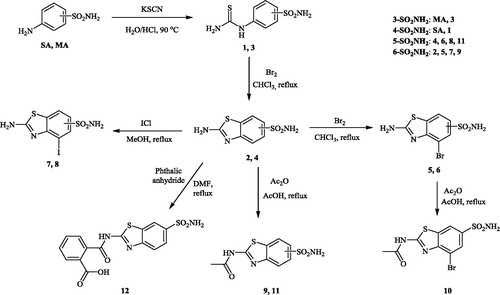
Sulfanilamide (SA)/metanilamide (MA) were reacted with potassium isocyanate in the presence of HCl, leading to the corresponding isothiocyanato-benzenesulfonamides 1 and 3, respectively. Bromination of these key intermediates led to the ring closure and formation of the regiomeric benzothiazole sulfonamides 2 and 4, respectively (Scheme 1). These compounds were acetylated and/or halogenated, leading to the small library of derivatives shown in Scheme 1 (see Experimental for details). Four of these derivatives have the sulfamoyl moiety in the 5 position of the benzothiazole ring, whereas the remaining ones in the 6 position (Scheme 1).
Carbonic anhydrase inhibition
The synthesized compounds 1–12 were investigated for their inhibitory effects against four physiological relevant isoforms, i.e. hCA I, II, VII and IX, by means of a stopped flow CO2 hydrase assayCitation42.
The following structure–activity relationship (SAR) can be drawn from data of :
hCA I was inhibited by all these sulfonamides, with inhibition constants ranging between 84.1 and 2327 nM. Two compounds, 2 and 12, had KIs < 100 nM, and they have both 2-amino-benzothiazole-6-sulfonamide derivatives. However 2 has no substituents on the amino functionality, whereas 12 has the bulky phthaloyl-monoamide functionality, proving thus that the SAR for inhibiting this isoform with sulfonamides investigated here is rather complex. Both these compounds were around three–four times less effective hCA I inhibitors compared to EZA (KI of 25 nM). Introduction of halogens on the benzothiazole scaffold of acetylation of the amino group led to compounds with less effective hCA I inhibitory properties compared to 2. The same was true for the compounds from the benzothiazole-5-sulfonamide series. For the simple derivatives, generally the 6-sulfamoyl derivatives were more effective CAIs compared to the corresponding 5-sulfamoyl ones (e.g. compare 2 and 4) whereas for the halogeno-substituted ones the behavior was not so clear-cut, with the bromoderivatives 5 and 6 behaving like the parent aminoderivatives, whereas an opposite effect was observed for the iododerivatives 7 and 8, case in which the 5-sulfonamide was a better inhibitor compared to the isomeric 6-sulfonamide ().
hCA II was effectively inhibited by sulfonamides investigated here, with KIs in the range of 7.8–369 nM. The best inhibitors were 2, 5–9, 11 and 12, with inhibition constants in the range of 7.8–51.5 nM. They belong to both the 5- as well as 6-sulfonamide series. The 2-amino-benzothiazole-6-sulfonamide derivative 2 was already an effective hCA II inhibitor, and its derivatization (acetylation and mono-phthaloylation) led to even better inhibitors (compare 9, 12, and 2). Halogenation of 2 led to very effective inhibitors, with both the bromo-and iodo-derivatives 5, 7, having KIs of 8.7 and 15.1 nM, respectively. However, bromination of the acetylated derivative 9 led to a strong loss of inhibitory effects in the halogenated derivative 10. For the 5-sulfonamide series, the situation was rather different. The parent compound, 2-amino-benzothiazole-5-sulfonamide derivative 4 was a modest hCA II inhibitor, with an inhibition constant of 369 nM. Its derivatization by introduction of halogeno atoms on the heterocyclic ring, as in 6 and 8, or the acetylation of the amino moiety, as in 11, led to a potent increase in the inhibitory power, with the bromo-derivative 6 being one of the best inhibitors on the series (KI of 7.8 nM, being more effective than AAZ or EZA, see ).
Effective inhibition was observed also for the brain-associated, cytosolic isoform hCA VII, a recently validated target for neuropathic painCitation61,Citation62. The sulfonamides investigated here showed KIs in the range of 0.8–92.3 nM. Most of these derivatives were in fact medium potency inhibitors, with inhibition constants of 42.2–92.3 nM, except 6 (KI of 0.8 nM) and 5 (KI of 31.1 nM). Both of them are the bromine derivatives of the isomeric 2-amino-benzothiazole-sulfonamides, with the 5-sulfonamide derivative 6 being 38.8 times a better hCA VII inhibitor compared to the 6-sulfonamide one 5 (). Compound 6 was equipotent to EZA for inhibiting this isoform.
The tumor-associated, transmembrane isoform hCA IX was also effectively inhibited by these sulfonamides, with KIs in the range of 3.7–295.6 nM (). The most effective inhibitors were 2, 4–6, and 9–12, with KIs in the range of 3.7–38.2 nM, the same range as the clinically used, standard inhibitors AAZ and EZA (). By comparing the two amino derivatives 2 and 4, it may be observed that in this case the 6-sulfonamide 2 was around 10 times a better hCA IX inhibitor compared to the isomeric 5-sulfonamide 4. Halogenation of 2 generally led to a decrease of the inhibitory potency, whereas acylation of the amino group had the same effect (but the loss of potency was smaller). Rather similar effects were observed for the 5-sulfonamide series, except that the bromination led to a slight increase in the hCA IX inhibitory power (compare 4 and 6).
Some of the reported sulfonamides tended to show some selectivity for inhibiting one CA isoform over the remaining ones. Examples in this sense are 6, which showed a good hCA VII selective inhibition profile, 9, 10, 11, and 12, which were effective hCA II and IX inhibitors, but weaker hCA I and VII inhibitors. However, these compounds possess a rather compact scaffold that probably binds deep within the CA active site, where most amino acid residues are conserved among the various isoforms. This is probably the reason why they show a rather low isoform-selective inhibition profile, a problem they share with most inhibitors of the first and second generation, which have been designed by the ring approach. As we stressed here and in other papersCitation1,Citation2,Citation5, this issue has been resolved by the using tail approach, which led to many classes of isoform-selective CAIsCitation63–65.
Conclusions
A small series of benzo[d]thiazole-5- and 6-sulfonamides has been synthesized by following literature procedures, and investigated for the inhibition of several hCA isoforms, using ethoxzolamide as lead molecule. 2-Amino-substituted, 2-acylamino- and halogenated (bromo-and iodo-derivatives at the heterocyclic ring) compounds led to several interesting inhibitors against the cytosolic hCA I, II, and VII, as well as the transmembrane, tumor-associated hCA IX isoforms. Several subnanomolar/low nanomolar, isoform-selective sulfonamide inhibitors targeting hCA II, VII and IX were detected. The sharp structure–activity relationship for CA inhibition with this small series of derivatives, with important changes of activity observed even after minor changes in the scaffold or at the 2-amino moiety, make this class of scarcely investigated sulfonamides of particular interest for further investigations.
Acknowledgements
Morteza Abdoli would like to acknowledge the financial support from Lorestan University for his living expenses in Italy for doing chemical synthesis in Florence University.
Disclosure statement
The authors report no conflict of interest.
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32.
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72.
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74.
- Del Prete S, Vullo D, De Luca V, et al. Biochemical characterization of recombinant beta-carbonic anhydrase (PgiCAb) identified in the genome of the oral pathogenic bacterium Porphyromonas gingivalis. J Enzyme Inhib Med Chem 2015;30:366–70.
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnologic use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30.
- Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs – antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzyme Inhib Med Chem 2014;29:379–87.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res 2011;71:3364–76.
- Casini A, Scozzafava A, Mincione F, et al. Carbonic anhydrase inhibitors: water-soluble 4-sulfamoylphenylthioureas as topical intraocular pressure-lowering agents with long-lasting effects. J Med Chem 2000;43:4884–92.
- Alterio V, Di Fiore A, D'Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates strongly inhibit carbonic anhydrases and show antiglaucoma action in vivo. J Med Chem 2012;55:1721–30.
- Scozzafava A, Menabuoni L, Mincione F, Supuran CT. Carbonic anhydrase inhibitors. A general approach for the preparation of water soluble sulfonamides incorporating polyamino-polycarboxylate tails and of their metal complexes possessing long lasting, topical intraocular pressure lowering properties. J Med Chem 2002;45:1466–76.
- Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47.
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902.
- Winum JY, Scozzafava A, Montero JL, Supuran CT. Therapeutic potential of sulfamides as enzyme inhibitors. Med Res Rev 2006;26:767–92.
- Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem. Commun. (Camb.) 2010;46:8371–3.
- Carta F, Garaj V, Maresca A, et al. Sulfonamides incorporating 1,3,5-triazine moieties selectively and potently inhibit carbonic anhydrase transmembrane isoforms IX, XII and XIV over cytosolic isoforms I and II: solution and X-ray crystallographic studies. Bioorg Med Chem 2011;19:3105–19.
- Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II and IX with sulfonamides incorporating 1,2,4-triazine moieties. Bioorg Med Chem Lett 2004;14:5427–33.
- Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors. Novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumor-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8.
- Carta F, Scozzafava A, Supuran CT. Sulfonamides (RSO2NH2): a patent review 2008-2012. Expert Opin Ther Pat 2012;22:747–58.
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16.
- Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008–2013). Expert Opin Ther Pat 2013;23:737–49.
- Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91.
- Supuran CT. Carbonic anhydrase inhibitors as emerging drugs for the treatment of obesity. Expert Opin Emerg Drugs 2012;17:11–15.
- Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35.
- Supuran CT. The safety and clinical efficacy of acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6.
- Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810.
- Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors. Part 74. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring? J Med Chem 1999;42:2641–50.
- Borras J, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors. Part 73. Synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406.
- Winum JY, Supuran CT. Recent advances in the discovery of zinc-binding motifs for the development of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:321–4.
- Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996;31:1001–10.
- Supuran CT, Carbonic anhydrase inhibitors. In: Puscas I, ed. Carbonic anhydrase and modulation of physiologic and pathologic processes in the organism. Timisoara: Helicon, 1994:29–111.
- Clare BW, Supuran CT. Carbonic anhydrase activators. Part 3. Structure-activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73.
- Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. J Enzyme Inhib Med Chem 2016;31:894–9.
- Kalinin S, Supuran CT, Krasavin M. Multicomponent chemistry in the synthesis of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2016;31:185–99.
- Supuran CT, Scozzafava A, Jurca BC, Ilies MA. Carbonic anhydrase inhibitors. Part 49. Synthesis of substituted ureido- and thioureido derivatives of aromatic/heterocyclic sulfonamides with increased affinities for isozyme I. Eur J Med Chem 1998;33:83–93.
- Alkaya ZA, İlkimen H, Yenikaya C, et al. A novel proton transfer salt of 2-amino-6-sulfamoylbenzothiazole and its metal complexes: the evaluation of their inhibition effects on human cytosolic carbonic anhydrases. J Enzyme Inhib Med Chem 2017;32:231–9.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Yamali C, Gul HI, Sakagami H, Supuran CT. Synthesis and bioactivities of halogen bearing phenolic chalcones and their corresponding bis Mannich bases. J Enzyme Inhib Med Chem 2016;31:125–31.
- Mollica A, Locatelli M, Macedonio G, et al. Microwave-assisted extraction, HPLC analysis, and inhibitory effects on carbonic anhydrase I, II, VA, and VII isoforms of 14 blueberry Italian cultivars. J Enzyme Inhib Med Chem 2016;31:1–6.
- Margheri F, Ceruso M, Carta F, et al. Overexpression of the transmembrane carbonic anhydrase isoforms IX and XII in the inflamed synovium. J Enzyme Inhib Med Chem 2016;31:60–3.
- Mishra CB, Kumari S, Angeli A, et al. Design, synthesis and biological evaluation of N-(5-methyl-isoxazol-3-yl/1,3,4-thiadiazol-2-yl)-4-(3-substitutedphenylureido) benzenesulfonamides as human carbonic anhydrase isoenzymes I, II, VII and XII inhibitors. J Enzyme Inhib Med Chem 2016;31:174–9.
- Diaz JR, Fernández Baldo M, Echeverría G, et al. A substituted sulfonamide and its Co (II), Cu (II), and Zn (II) complexes as potential antifungal agents. J Enzyme Inhib Med Chem 2016;31:51–62.
- Supuran CT, Kalinin S, Tanç M, et al. Isoform-selective inhibitory profile of 2-imidazoline-substituted benzene sulfonamides against a panel of human carbonic anhydrases. J Enzyme Inhib Med Chem 2016;31:197–202.
- Federici C, Lugini L, Marino ML, et al. Lansoprazole and carbonic anhydrase IX inhibitors sinergize against human melanoma cells. J Enzyme Inhib Med Chem 2016;31:119–25.
- Chohan ZH, Scozzafava A, Supuran CT. Unsymmetrical 1,1′-disubstituted ferrocenes: synthesis of Co(ii), Cu(ii), Ni(ii) and Zn(ii) chelates of ferrocenyl -1-thiadiazolo-1′-tetrazole, -1-thiadiazolo-1′-triazole and -1-tetrazolo-1′-triazole with antimicrobial properties. J Enzyme Inhib Med Chem 2002;17:261–6.
- Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodiumfalciparum – the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96.
- Supuran CT, Scozzafava A, Mastrolorenzo A. Bacterial proteases: current therapeutic use and future prospects for the development of new antibiotics. Expert Opin Ther Pat 2001;11:221–59.
- Supuran CT, Barboiu M, Luca C, et al. Carbonic anhydrase activators. Part 14. Synthesis of mono- and bis- pyridinium salt derivatives of 2-amino-5-(2-aminoethyl)- and 2-amino-5-(3-aminopropyl)-1,3,4-thiadiazole, and their interaction with isozyme II. Eur J Med Chem 1996;31:597–606.
- Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors. Part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents with potential use as antiglaucoma drugs. Eur J Med Chem 1998;33:247–54.
- Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors: perfluoroalkyl/aryl-substituted derivatives of aromatic/heterocyclic sulfonamides as topical intraocular pressure-lowering agents with prolonged duration of action. J Med Chem 2000;43:4542–51.
- Supuran CT, Clare BW. Carbonic anhydrase inhibitors. Part 57. Quantum chemical QSAR of a group of 1,3,4-thiadiazole and 1,3,4-thiadiazoline disulfonamides with carbonic anhydrase inhibitory properties. Eur J Med Chem 1999;34:41–50.
- Puccetti L, Fasolis G, Vullo D, et al. Carbonic anhydrase inhibitors. Inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, IX, and XII with Schiff’s bases incorporating chromone and aromatic sulfonamide moieties, and their zinc complexes. Bioorg Med Chem Lett 2005;15:3096–101.
- Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8.
- Supuran CT. Carbonic anhydrase inhibitors: an Editorial. Expert Opin Ther Pat 2013;23:677–9.
- Sentürk M, Gülçin I, Beydemir S, et al. In Vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9.
- Carta F, Di Cesare Mannelli L, Pinard M, et al. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg Med Chem 2015;23:1828–40.
- Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother 2016;16:961–8.
- Chohan ZH, Arif M, Akhtar MA, et al. Metal-based antibacterial and antifungal agents: synthesis, characterization, and in vitro biological evaluation of Co(II), Cu(II), Ni(II), and Zn(II) complexes with amino acid-derived compounds. Bioinorg Chem Appl 2006;2006:83131.
- Nocentini A, Ferraroni M, Carta F, et al. Benzenesulfonamides incorporating flexible triazole moieties are highly effective carbonic anhydrase inhibitors: synthesis and kinetic, crystallographic, computational, and intraocular pressure lowering investigations. J Med Chem 2016;59:10692–704.
- Mete E, Comez B, Inci Gul H, et al. Synthesis and carbonic anhydrase inhibitory activities of new thienyl-substituted pyrazoline benzenesulfonamides. J Enzyme Inhib Med Chem 2016;31(sup2):1–5.