
Abstract
The β-carbonic anhydrases (CAs, EC 4.2.1.1) from the pathogenic bacterium Clostridium perfringens (CpeCA) was recently characterised kinetically and for its anion inhibition profile. In the search of effective CpeCA inhibitors, possibly useful to inhibit the growth/pathogenicity of this bacterium, we report here an inhibition study of this enzyme with a panel of aromatic, heterocyclic and sugar sulphonamides/sulphamates. Some sulphonamides, such as acetazolamide, ethoxzolamide, dichlorophenamide, dorzolamide, sulthiame and 4-(2-hydroxymethyl-4-nitrophenyl-sulphonamido)ethylbenzenesulphonamide were effective CpeCA inhibitors, with KIs in the range of 37.4–71.6 nM. Zonisamide and saccharin were the least effective such inhibitors, whereas many other aromatic and heterocyclic sulphonamides were moderate – weak inhibitors with KIs ranging between 113 and 8755 nM. Thus, this study provides the basis for developing better clostridial enzyme inhibitors with potential as antiinfectives with a new mechanism of action.
1. Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are metalloenzymes present in all three of life’s phylogenetic domains (Bacteria, Archaea and Eukarya) and various isoforms present in most organisms investigated so farCitation1–12. By converting metabolism-generated CO2 to soluble products, bicarbonate and protons, these enzymes are crucial in a multitude of physiologic processes connected among others with pH homoeostasis, biosynthetic reactions in which CO2/bicarbonate are involved, electrolyte secretion, photosynthesis, etc.Citation1–12. The α-class CAs present in vertebrates, including humansCitation1–6, are drug targets for obtaining antiglaucoma agentsCitation13,Citation14, anticonvulsantsCitation15, drugs for the treatment of idiopathic intracranial hypertension and other neurologic disordersCitation15,Citation16, antiobesity agentsCitation17 and diureticsCitation18–20. Most of these clinically used CA inhibitors (CAIs) belong to the sulphonamide class, as they possess the primary sulphonamide (or its isosteres, sulphamate and sulphamide moieties) as the zinc-binding functionCitation1–6,Citation21. Indeed, these compounds bind (in deprotonated, anionic form) to the CA active-site zinc ion, which is crucial for the catalytic activityCitation21. Representatives of this class of pharmacological agents have multiple therapeutic applications for decades, although many of these first-/second-generation agents do show side effects connected with the inhibition of off-target isoforms, due to the fact that in humans there are 15 CAs which do not differ significantly in their active site architectureCitation3–5,Citation22 and most of them show high affinity for this class of CAICitation1–6. However, in the last decade, a large number of different classes of CAIs and diverse inhibition mechanisms were reported, with a range of new chemotypes such as the coumarinsCitation23–26, sulphocoumarinsCitation27,Citation28, polyaminesCitation29, dithiocarbamatesCitation30,Citation31 and carboxylatesCitation32,Citation33 among others. Many of these novel types of CAIs show significant isoform-selective inhibition profiles, making this class of drugs much more attractive as candidates for the development of new generation pharmacological agentsCitation4,Citation23–33.
Bacteria encode CAs belonging to three classes, the α-, β- and γ-CAsCitation7,Citation34–40. These enzymes seem to be involved in crucial metabolisms, which probably explains both their wide distribution in Gram-negative and Gram-positive bacteria, as well as their generally very effective catalytic properties for the hydration of CO2 to bicarbonate and protonsCitation34–42. Thus, ultimately, inhibition of bacterial CAs has been proposed as an alternative approach for obtaining antibiotics with an alternative mechanism of action compared to the classical drugs that interfere with bacterial cell wall biosynthesis, DNA-gyrase or similar such targets, which led to an extensive drug resistance phenomenonCitation7,Citation12,Citation34,Citation36.
In previous work from our groups, we have reported the cloning and characterisation of a new β-CA from the bacterial pathogen Clostridium perfringens, CpeCA [Citation41] that has also been investigated for its interaction with anions and other small molecules known to interact with metalloenzymes such as CACitation42. We previously observed that most anions are millimolar CpeCA inhibitors, whereas sulphamate, sulphamide, phenylboronic acid and phenylarsonic acid are the most effective inhibitors, with KIs in the range of 7–75 μM. Thus, no highly effective CpeCA inhibitors were detected so far and this is the reason why we investigated the interaction of this enzyme with sulphonamides and sulphamates, the class of CAIs which usually leads to effective antimicrobial agents.
2. Materials and methods
2.1. Chemistry
Compounds 1–24 and AAZ-HCT were commercially, highest purity available derivatives from Sigma-Aldrich (Milan, Italy) and were used without further purification or were prepared as reported earlier by our groupCitation43–51.
2.2. Carbonic anhydrase assay
An applied photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity [Citation52]. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nM, with 20 mM TRIS (pH 8.3) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionised water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were pre-incubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation23–27 and represent the mean from at least three different determinations. All CA isofoms were recombinant ones obtained in-house as reported earlierCitation41,Citation42.
3. Results and discussion
We investigated the inhibition of CpeCA with a panel of sulphonamides of type 1–24, which include both aromatic and heterocyclic derivatives, employed extensively for the design of various classes of CAIs with interesting physicochemical propertiesCitation43–51 (). The clinically used agents acetazolamide AAZ, methazolamide MZA, ethoxzolamide EZA, dichorophenamide DCP, dorzolamide DZA, brinzolamide BRZ, benzolamide BZA, topiramate TPM, zonisamide ZNS, sulpiride SLP, indisulam IND, valdecoxib and VLX, celecoxib CLX, sulthiame SLT, saccharin SAC and hydrochlorothiazide HCT [Citation9] were also included in this study as they incorporate the sulphonamide/sulphamate zinc-binding function and act as potent CAIs against many β-CAs investigated earlier [Citation7]. The inhibition observed with these derivatives against CpeCA and the human (h) off-target α-class enzymes hCA I and II, are shown in .
Figure 1. Structure of sulphonamides investigated as CAIs in this work.
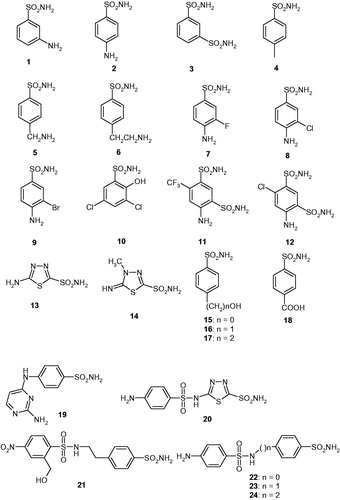
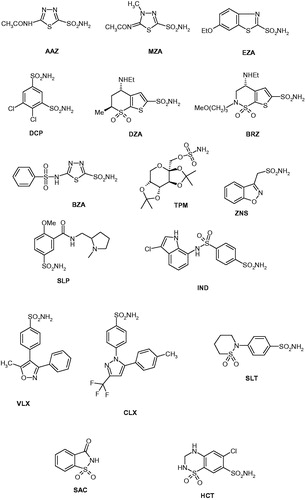
Table 1. Inhibition of human isoforms hCA I and hCA II (off-target enzymes), as well as the bacterial enzyme from C. perfringens (CpeCA) with sulphonamides 1–24 and the clinically used drugs AAZ–HCT, by a stopped-flow, CO2 hydrase assay [Citation52].
The following structure–activity relationship (SAR) can be drawn from the data of regarding CpeCA inhibition with these compounds
The least effective CpeCA inhibitors were zonisamide and saccharin, which did not affect the enzyme activity up to 100 µM (). ZNS is in fact the only aliphatic sulphonamide, whereas SAC the only secondary, acylated sulphonamide among the investigated compounds.
Moderate–weak inhibitory action, in the micromolar range, was observed for the following sulphonamides: 5, 6, 10–12 and 18, which had KIs in the range of 1.268–8.755 µM. These compounds belong to the aminoalkyl–benzenesulphonamide (5 and 6) and tetrasubstituted benzenesulphonamide/disulphonamide (10–12) series. Probably, the large number of substituents on the phenyl ring for the last type of derivatives is detrimental to their efficient binding to the enzyme.
More effective but moderate CpeCA inhibitors were the following compounds: 1–4, 7–9, 13, 14, 17, 19, 21–23, BZA, TPM, SLP–CLX and HCT, which had KIs in the range of 160–713 nM. It is obvious that these derivatives belong to a variety of different classes, with both aromatic, heterocyclic and sugar derivatives among them. Thus, a real SAR is difficult to draw, but it is important to note that many structural variations in the scaffold of aromatic/heterocyclic sulphonamides are tolerated without a significant loss of the CpeCA inhibitory action.
The most effective CpeCA inhibitors were 15, 16, 20, 24, AAZ, MZA, EZA, DCP, DZA, BRZ and SLT, which showed KIs in the range of 37.4–145 nM (). Again many different chemotypes led to quite effective CAIs, among which the most notable are dorzolamide, a rather bulky bicyclic sulphonamide (the best inhibitor with a KI of 37.4 nM), acetazolamide (the second best inhibitor with a KI of 49.1 nM) as well as the aromatic compound 4–(2-hydroxymethyl-4-nitrophenyl-sulphonamido)ethylbenzenesulphonamide 24, with a KI of 51.2 nM (). All of them are highly different structurally, which is of extreme importance for the possible design of even better CpeCA inhibitors belonging to the sulphonamide class.
The off-target isoforms hCA I and II have a very different inhibition profile with the compounds investigated here (), whereas hCA I has generally a lower affinity for most of these inhibitors, hCA II is highly inhibited by most of them, usually in the low nanomolar range, which makes it quite difficult to obtain CpeCA-selective inhibitors form this class of agents.
4. Conclusions
Species belonging to the genus Clostridium, such as Clostridium tetani, C. botulinum, C. barati, C. butirycum, C. hystolyticum and C. perfringens among others, are strictly anaerobic pathogens that provoke serious human disease, such as tetanus, botulism, gas gangrene, bacterial corneal keratitis and other infectionsCitation53,Citation54. Although some progress has been achieved ultimately for designing pharmacological agents effective against these diseases, such as for example protease inhibitors targeting various metalloproteases essential for the life cycle of these pathogens, there is a constant search for novel drug targets that may lead to new classes of such agents, considering the serious antibiotic drug resistance problems emerging worldwide with the clinically used drugsCitation53,Citation54. In the search of effective compounds interfering with the metabolism of these pathogens, in this paper, we investigated potential CpeCA inhibitors, possibly useful to inhibit the growth/pathogenicity of this bacterium. A panel of aromatic, heterocyclic and sugar sulphonamides/sulphamates were employed for the inhibition of this bacterial β-class enzyme. Some sulphonamides, such as acetazolamide, ethoxzolamide, dichlorophenamide, dorzolamide, sulthiame and 4-(2-hydroxymethyl-4-nitrophenyl-sulphonamido)ethylbenzenesulphonamide were effective CpeCA inhibitors, with KIs in the range of 37.4–71.6 nM. Zonisamide and saccharin were the least effective inhibitors, whereas many other aromatic and heterocyclic sulphonamides were moderate–weak inhibitors with KIs ranging between 113 and 8755 nM. This study thus provides the basis for developing better clostridial enzyme inhibitors with potential as antiinfectives with a new mechanism of action.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO(2) capture. J Enzyme Inhib Med Chem 2013;28:229–30.
- Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704.
- Lomelino CL, Supuran CT, McKenna R. Non-classical inhibition of carbonic anhydrase. Int J Mol Sci 2016;17:E1150.
- Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- Emameh RZ, Barker HR, Syrjänen L, et al. Identification and inhibition of carbonic anhydrases from nematodes. J Enzyme Inhib Med Chem 2016;31:176–84.
- Syrjänen L, Kuuslahti M, Tolvanen M, et al. The β-carbonic anhydrase from the malaria mosquito Anopheles gambiae is highly inhibited by sulfonamides. Bioorg Med Chem 2015;23:2303–9.
- Modak JK, Liu YC, Supuran CT, Roujeinikova A. Structure-activity relationship for sulfonamide inhibition of helicobacter pylori α-carbonic anhydrase. J Med Chem 2016;59:11098–109.
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Patents 2013;23:705–16.
- Nocentini A, Ferraroni M, Carta F, et al. Benzenesulfonamides incorporating flexible triazole moieties are highly effective carbonic anhydrase inhibitors: synthesis and kinetic, crystallographic, computational, and intraocular pressure lowering investigations. J Med Chem 2016;59:10692–704.
- Mishra CB, Kumari S, Angeli A, et al. Discovery of benzenesulfonamides with potent human carbonic anhydrase inhibitory and effective anticonvulsant action: design, synthesis, and pharmacological assessment. J Med Chem 2017;60:2456–69.
- Supuran CT. Acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6.
- Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Patents 2013;23:725–35.
- Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91.
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74.
- Supuran CT. Drug interaction considerations in the therapeutic use of carbonic anhydrase inhibitors. Expert Opin Drug Metab Toxicol 2016;12:423–31.
- Winum JY, Supuran CT. Recent advances in the discovery of zinc-binding motifs for the development of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:321–4.
- Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996;31:1001–10.
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62.
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44.
- Maresca A, Supuran CT. Coumarins incorporating hydroxy- and chloro-moieties selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II. Bioorg Med Chem Lett 2010;20:4511–14.
- Ferraroni M, Carta F, Scozzafava A, Supuran CT. Thioxocoumarins show an alternative carbonic anhydrase inhibition mechanism compared to coumarins. J Med Chem 2016;59:462–73.
- Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300.
- Grandane A, Tanc M, Zalubovskis R, Supuran CT. 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem Lett 2014;24:1256–60.
- Carta F, Temperini C, Innocenti A, et al. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 2010;53:5511–22.
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates strongly inhibit carbonic anhydrases and show antiglaucoma action in vivo. J Med Chem 2012;55:1721–30.
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun (Camb) 2012;48:1868–70.
- Langella E, D'Ambrosio K, D'Ascenzio M, et al. A combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes in the active site of carbonic anhydrases. Chemistry 2016;22:97–100.
- D'Ambrosio K, Carradori S, Monti SM, et al. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem Commun (Camb) 2015;51:302–5.
- Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs–antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzyme Inhib Med Chem 2014;29:379–87.
- Supuran CT, Capasso C. New light on bacterial carbonic anhydrases phylogeny based on the analysis of signal peptide sequences. J Enzyme Inhib Med Chem 2016;31:1254–60.
- Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32.
- Ferry JG. The gamma class of carbonic anhydrases. Biochim Biophys Acta 2010;1804:374–81.
- Smith KS, Ferry JG. Prokaryotic carbonic anhydrases. FEMS Microbiol Rev 2000;24:335–66.
- Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal beta-class (Cab) and gamma-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–8.
- Zimmerman S, Innocenti A, Casini A, et al. Carbonic anhydrase inhibitors. Inhibition of the prokariotic beta and gamma-class enzymes from Archaea with sulfonamides. Bioorg Med Chem Lett 2004;14:6001–6.
- Kumar RS, Hendrick W, Correll JB, et al. Biochemistry and physiology of the β class carbonic anhydrase (Cpb) from Clostridium perfringens strain 13. J Bacteriol 2013;195:2262–9.
- Vullo D, Sai Kumar RS, Scozzafava A, et al. Anion inhibition studies of a β-carbonic anhydrase from Clostridium perfringens. Bioorg Med Chem Lett 2013;23:6706–10.
- Supuran CT, Clare BW. Carbonic anhydrase inhibitors–Part 57: Quantum chemical QSAR of a group of 1, 3, 4-thiadiazole-and 1, 3, 4- thiadiazoline disulfonamides with carbonic anhydrase inhibitory properties. Eur J Med Chem 1999;34:41–50.
- Puccetti L, Fasolis G, Vullo D, et al. Carbonic anhydrase inhibitors. Inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, IX, and XII with Schiff’s bases incorporating chromone and aromatic sulfonamide moieties, and their zinc complexes. Bioorg Med Chem Lett 2005;15:3096–101.
- Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008–2013). Expert Opin Ther Pat 2013;23:737–49.
- Scozzafava A, Menabuoni L, Mincione F, Supuran CT. Carbonic anhydrase inhibitors. a general approach for the preparation of water-soluble sulfonamides incorporating polyamino − polycarboxylate tails and of their metal complexes possessing long-lasting, topical intraocular pressure-lowering properties. J Med Chem 2002;45:1466–76.
- Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8.
- Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8.
- Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3.
- Dubois L, Peeters S, Lieuwes NG, et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 2011;99:424–31.
- Carta F, Scozzafava A, Supuran CT. Sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2012;22:747–58.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Mastrolorenzo A, Supuran CT, Botulinum toxin, tetanus toxin and anthrax lethal factor inhibitors. In: Supuran CT, Winum J, eds. Drug design of zinc-enzyme inhibitors: functional, structural, and disease applications. Hoboken (NJ): Wiley; 2009:705–720.
- Supuran CT, Clostridium histolyticum collagenase inhibitors in the drug design. In: Supuran CT, Winum JY, eds. Drug design of zinc-enzyme inhibitors: functional, structural, and disease applications. Hoboken (NJ): Wiley; 2009:721–730.