
Abstract
The M2 isoform of pyruvate kinase (PKM2) is a potential antitumor therapeutic target. In this study, we designed and synthesised a series of 2, 3-didithiocarbamate substituted naphthoquinones as PKM2 inhibitors based on the lead compound 3k that we previously reported. Among them, compound 3f (IC50 = 1.05 ± 0.17 µM) and 3h (IC50 = 0.96 ± 0.18 µM) exhibited potent inhibition of PKM2, and their inhibitory activities are superior to compound 3k (IC50 = 2.95 ± 0.53 µM) and the known PKM2 inhibitor shikonin (IC50 = 8.82 ± 2.62 µM). In addition, we evaluated in vitro antiproliferative effects of target compounds using MTS assay. Most target compounds exhibited dose-dependent cytotoxicity with IC50 values in nanomolar concentrations against HCT116, MCF7, Hela, H1299 and B16 cells. These small molecule PKM2 inhibitors not only provide candidate compounds for cancer therapy, but also offer a tool to probe the biological effects of PKM2 inhibition on cancer cells.
Introduction
The metabolism in cancer cells substantially differs from that in healthy cellsCitation1–3. The tumour cells rely on glycolysis to produce energy and have a high rate of glucose uptake but low rates of oxidative phosphorylation (Warburg effect)Citation4,Citation5. Cancer cells were found to have very strong metabolic dependencies, which are not associated with normal cellsCitation6. The interventions on tumour glycolysis become a novel strategy for selective anti-cancer therapiesCitation7–9.
Pyruvate kinase (PK) is the last rate-limiting enzyme in the glycolytic pathway and catalyses the transfer of a phosphate group from phosphoenolpyruvate (PEP) to adenosine diphosphate (ADP) to obtain pyruvate and adenosine triphosphate (ATP)Citation10,Citation11. There are M1, M2, L and R isoforms of PK in mammalian cells: the M1 isoform (PKM1) is expressed in many differentiated tissues (skeletal muscle, heart and brain), PKM2 is expressed during embryonic development or over expressed in tumour tissues, PKL and PKR are expressed in liver and erythrocytes, respectivelyCitation12–14. PKM2 is important for cancer metabolism and tumour growthCitation15,Citation16. Tumourigenesis is associated with the re-expression of PKM2 together with a downregulation of the expression of PKM1 and other isozymesCitation17,Citation18. Therefore, targeting of PKM2 offers an opportunity to target cancer cell metabolism and reduce the side effects of cancer therapyCitation17,Citation19. However, an exact mechanistic understanding of PKM2 is still lacking. The identification of PKM2 inhibitors not only can provide candidate compounds for cancer therapy but also is helpful to deciphering other cellular functions of PKM2.
In 2010, Vander Heiden et al. screened a library of compounds to identify a small molecule PKM2 inhibitor compound A with IC50 value of 10 µM ()Citation20. Compound A was reported to presumably block the allosteric site of PKM2. In 2011, Chen et al. reported shikonin () and its enantiomeric isomer alkannin to inhibit PKM2 at low concentrationsCitation21,Citation22. Recently, we reported a new PKM2 inhibitor compound 3k () with IC50 value of 2.95 µM, which exhibits more potent PKM2 inhibitory activity than the known optimal PKM2 inhibitor shikonin (IC50 = 8.82 µM)Citation23.
Figure 1. Structures of compound 3, shikonin and compound 3k.
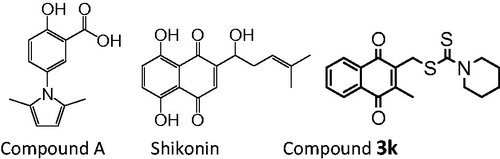
To further optimise this activity, in this study, some new analogues of 3k were designed and synthesised. The PKM2 inhibitor compound 3f and 3h have high PKM2 inhibition responses, which are more potent than shikonin and compound 3k, and they also exhibit high antiproliferative activity in several tumour cells.
Materials and methods
Chemistry
All chemicals, reagents and solvents were purchased from commercial sources. When necessary, they were purified and dried by standard methods. Reactions were checked by thin-layer chromatography (TLC) on pre-coated silica gel F254 plates. Column chromatography was carried out with Silica gel H (200–300 mesh or 500 mesh). Detection was by iodine vapour staining and UV light irradiation (UV lamp, model UV-IIB). Melting points were determined on an X4-type apparatus and are not corrected. 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE III-400 spectrometer, Chemical shifts δ in ppm with Me4Si as internal standard, coupling constants J in Hertz. High-resolution mass spectrum (HRMS) was recorded on a Thermo Scientific Orbitrap Elite MS.
Procedure for preparation of 2,3-bis-chloromethyl-[1,4]naphthoquinone (2)Citation24
The 1,4-naphthaquinone (1) (1 g, 6.3 mmol) in glacial acetic acid (20 ml) was taken in a 100-ml round-bottomed flask, and 36% aqueous formaldehyde (6 ml) was added. The reaction solution was cooled in ice water. Dry hydrogen chloride passed in for 2 h. The solution became red, then being kept at room temperature for 48 h. The reaction mixture was poured on ice and extracted with ethyl acetate. The combined organic fractions were washed with brine, dried (Na2SO4) and concentrated under reduced pressure. Purification of the crude residue by column chromatography (petroleum ether/ethyl acetate) afforded the compound 2 (yellow solid). The yield of this reaction was 68.9%. 1H NMR (400 MHz, CDCl3) δ 8.18–8.20 (m, 2H, ArH), 7.81–7.83 (m, 2H, ArH), 4.72 (s, 4H, 2CH2Cl).
General procedure for preparation of dithiocarbamic acid 3-thiocarbamoylsulphanylmethyl-1,4-dioxo-1,4-dihydro-naphthalen-2-ylmethyl ester (3a-3h)
Carbon disulphide (180 μL, 3 mmol) and amine (3 mmol) were added to CH3CN (5 ml) and the resulting solution was stirred for 30 min. 2, 3-Bis-chloromethyl-[1,4]naphthoquinone (2) (254 mg, 1 mmol) was added in portions at frequent intervals. Then, the reaction mixture was kept at room temperature for 48 h. The reaction mixture was concentrated in vacuo, diluted with H2O, and extracted with CH2Cl2. The combined organic fractions were washed with brine, dried (Na2SO4) and concentrated under reduced pressure. Purification of the crude residue by column chromatography (petroleum ether/CH2Cl2) afforded the title compound.
Data for dimorpholine-dithiocarbamic acid 3-dimorpholinethiocarbamoylsulphanylmethyl-1,4-dioxo-1,4-dihydro-naphthalen-2-ylmethyl ester (3a): yellow solid (78.4%); mp 153–154 °C. 1H NMR (400 MHz, CDCl3) δ 8.11–8.13 (m, 2H, ArH), 7.75–7.77 (m, 2H, ArH), 4.89 (s, 4H, 2CH2S), 3.99–4.28 (m, 8H, 4OCH2), 3.76 (s, 8H, 4NCH2). 13C NMR (100 MHz, CDCl3) δ 196.1, 183.9, 143.7, 134.0, 132.0, 126.7, 66.3, 66.2, 34.0. HR-MS (ESI+) m/z: 509.0697 [M + H]+. Found: 509.0686 [M + H]+.
Data for dimethyl-dithiocarbamic acid 3-dimethylthiocarbamoylsulphanylmethyl-1,4-dioxo-1,4-dihydro-naphthalen-2-ylmethyl ester (3b): yellow solid (92.0%); mp 157–158 °C. 1H NMR (400 MHz, CDCl3) δ 8.11–8.13 (m, 2H, ArH), 7.74–7.76 (m, 2H, ArH), 4.83 (s, 4H, 2CH2S), 3.56 (s, 3H, NCH3), 3.36 (s, 3H, NCH3). 13C NMR (100 MHz, CDCl3) δ 195.7, 183.9, 143.8, 133.9, 132.0, 126.7, 45.7, 41.5, 34.1. HR-MS (ESI+) m/z: 425.0486 [M + H]+,447.0305 [M + Na]+. Found: 425.0487 [M + H]+, 447.0304 [M + Na]+.
Data for diethyl-dithiocarbamic acid 3-diethylthiocarbamoylsulphanylmethyl-1,4-dioxo-1,4-dihydro-naphthalen-2-ylmethyl ester (3c): yellow solid (94.3%); mp 130–131 °C. 1H NMR (400 MHz, CDCl3) δ 8.12–8.14 (m, 2H, ArH), 7.74–7.76 (m, 2H, ArH), 4.83 (s, 4H, 2CH2S), 4.03 (q, 4H, 2NCH2), 3.72 (m, 4H, 2NCH2), 1.29 (m, 12H, 4CH3). 13C NMR (100 MHz, CDCl3) δ 194.2, 183.9, 144.0, 133.8, 132.1, 126.7, 49.9, 46.8, 34.0, 12.6, 11.6. HR-MS (ESI+) m/z: 481.1112 [M + H]+, 503.0931 [M + Na]+. Found: 481.1111 [M + H]+, 503.0934 [M + Na]+.
Data for dipropyl-dithiocarbamic acid 3-dipropylthiocarbamoylsulphanylmethyl-1,4-dioxo-1,4-dihydro-naphthalen-2-ylmethyl ester (3d): yellow solid (92.9%); mp 111–112 °C. 1H NMR (400 MHz, CDCl3) δ 8.12–8.14 (m, 2H, ArH), 7.73–7.76 (m, 2H, ArH), 4.80 (s, 4H, 2CH2S), 3.91 (q, 4H, 2NCH2), 3.60 (m, 4H, 2NCH2), 1.73–1.79 (m, 8H, 4CH2CH3), 0.93–0.95 (m, 12H, 4CH3). 13C NMR (100 MHz, CDCl3) δ 194.7, 183.8, 144.1, 133.8, 132.1, 126.7, 57.3, 54.5, 34.1, 20.8, 19.6, 11.2. HR-MS (ESI+) m/z: 537.1738 [M + H]+. Found: 537.1728 [M + H]+, 559.1567 [M + Na]+.
Data for diallyl-dithiocarbamic acid 3-diallylthiocarbamoylsulphanylmethyl-1,4-dioxo-1,4-dihydro-naphthalen-2-ylmethyl ester (3e): yellow liquid (83.3%); 1H NMR (400 MHz, CDCl3) δ 8.11–8.14 (m, 2H, ArH), 7.74–7.76 (m, 2H, ArH), 5.80–5.91 (m, 4H, 4CH=CH2), 5.27 (d, 2H, CH=CH2), 5.25 (d, 2H, CH=CH2), 5.24 (d, 2H, CH=CH2), 5.20 (d, 2H, CH=CH2), 4.83 (s, 4H, 4CH2S), 4.66 (d, 4H, 2NCH2), 4.30 (d, 4H, 2NCH2). 13C NMR (100 MHz, CDCl3) δ 196.6, 183.8, 143.9, 133.9, 132.0, 131.0, 130.3, 126.7, 118.9, 118.7, 56.9, 53.8, 34.4. HR-MS (ESI+) m/z: 529.1112 [M + H]+. Found: 529.1113 [M + H]+.
Data for dithiamorpholine-dithiocarbamic acid 3-dithiamorpholinethiocarbamoylsulphanylmethyl-1,4-dioxo-1,4-dihydro-naphthalen-2-ylmethyl ester (3f): yellow solid (90.7%); mp 146–147 °C. 1H NMR (400 MHz, CDCl3) δ 8.11–8.13 (m, 2H, ArH), 7.74–7.77 (m, 2H, ArH), 4.66 (s, 4H, 2CH2S), 4.57–4.58 (m, 4H, 2NCH2), 4.26–4.29 (m, 4H, 2NCH2), 2.76 (s, 8H, 4CH2S). 13C NMR (100 MHz, CDCl3) δ 195.3, 183.9, 143.8, 134.0, 131.9, 126.7, 34.7, 27.3. HR-MS (ESI+) m/z: 541.0240 [M + H]+, 563.0060 [M + Na]+. Found: 541.0343 [M + H]+,563.0178 [M + Na]+.
Data for dipyrrolidine-dithiocarbamic acid 3-dipyrrolidinethiocarbamoylsulphanylmethyl-1,4-dioxo-1,4-dihydro-naphthalen-2-ylmethyl ester (3g): yellow solid (88.2%); mp 150–151 °C. 1H NMR (400 MHz, CDCl3) δ 8.11–8.13 (m, 2H, ArH), 7.73–7.75 (m, 2H, ArH), 4.88 (s, 4H, 2CH2S), 3.95 (q, 4H, 2NCH2), 3.64 (q, 4H, 2NCH2), 1.96–2.09 (m, 8H, 2CH2CH2). 13C NMR (100 MHz, CDCl3) δ 191.3, 184.0, 143.9, 133.9, 132.0, 126.6, 55.3, 50.6, 33.5, 26.2, 24.3. HR-MS (ESI+) m/z: 477.0799 [M + H]+, 499.0618[M + Na]+. Found: 477.0791 [M + H]+, 499.0626[M + Na]+.
Data for dithiazolidine-dithiocarbamic acid 3-dithiazolidinethiocarbamoylsulphanylmethyl-1,4-dioxo-1,4-dihydro-naphthalen-2-ylme-thyl ester (3h): yellow solid (88.8%); mp 158–159 °C. 1H NMR (400 MHz, CDCl3) δ 8.11–8.14 (m, 2H, ArH), 7.75–7.77 (m, 2H, ArH), 4.86 (s, 4H, 2CH2S), 3.11–5.04 (m, 12H, 2NCH2CH2SCH2). 13C NMR (100 MHz, CDCl3) δ 192.5, 183.9, 143.6, 134.0, 131.9, 126.7, 56.6, 52.7, 34.2, 31.2, 29.1. HR-MS (ESI+) m/z: 512.9927 [M + H]+, 534.9747 [M + Na]+. Found: 512.9944 [M + H]+, 534.9720 [M + Na]+.
Biological activity
Purification of recombinant pyruvate kinase isoforms
Human cDNA for PKM2 was cloned into pET28a+ with a N-terminal His tag and purified from Escherichia coli strain BL21 (Invitrogen) using Ni-Agarose beads (Qiagen) as described previouslyCitation25.
PKM2 activity assay
Pyruvate kinase activity was measured with a fluorescent pyruvate kinase-lactate dehydrogenase coupled assay as previously describedCitation20.
Cell culture
Cell lines were grown with routine culture techniques in RPMI 1640 supplemented with 9% foetal bovine serum at 37 °C in 5% CO2.
MTS cell proliferation assay
Cells were plated in 96-well plates at a density of 5000 cells per well. Twelve hours after seeding, cells were treated with various concentrations of test compounds for 48 h. Cell viability was assessed with the MTS assay (Promega) according to the manufacturer's instruction.
Results and discussion
Chemistry
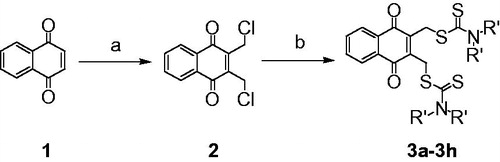
2, 3-Didithiocarbamate-substituted naphthoquinones (3a-3h) were prepared as target compounds as shown in Scheme 1. 1,4-Naphthoquinone 1 was reacted with formaldehyde in the presence of dry hydrogen chloride in a cold mixed solvent of H2O and acetic acid to provide 2,3-dichloromethyl-1,4-naphthoquinone 2. Compound 2 was treated with CS2 and various amines to obtain target compounds 3a-3h in moderate to high yields.
Scheme 1. Synthesis of 2,3-didithiocarbamate-substituted naphthoquinones. Reagents and conditions: (a) formaldehyde, HCl, HAc, H2O, 0 °C, 68.9%; (b) CS2, amine, CH3CN, rt, 78–94%.
Biological activity
PKM2 inhibition activity of compounds 3a-3h
We first tested the effect of compound 3a-3h on PKM2 activity using a fluorescent PK-LDH coupled assay according to a previously reported methodCitation20. Shikonin, which is a known inhibitor of PKM2, was used as the positive control. Most of the target compounds exhibited some degree of PKM2 inhibitory activity. Compound 3f (IC50 = 1.05 ± 0.17 µM) and 3h (IC50 = 0.96 ± 0.18 µM) displayed the higher inhibitory activity than the lead compound 3k (IC50 = 2.95 ± 0.53 µM) and shikonin (IC50 = 8.82 ± 2.62 µM) (). The preliminary SAR can be summarised as follows. The amine moiety of target compounds greatly influenced PKM2 inhibitory activity. Introduction of a short chain N, N-dimethylamine reduced the inhibitory activity (3b vs. 3c and 3d). When the n-propyl amine in 3d was replaced by allyl amine (3e), the inhibitory activity was lowered. The thiazolidinyl 3h (IC50 = 0.96 ± 0.18 µM) and thiamorpholinyl 3f (IC50 = 1.05 ± 0.17 µM) substituted compounds respectively demonstrated stronger activity than pyrrolidinyl 3g (IC50 = 2.33 ± 0.52 µM) and morpholinyl 3a (IC50 = 2.64 ± 0.98 µM) substituted compounds. Introduction of a sulphur atom therefore contributed to improvement of PKM2 inhibitory activity.
Table 1. PKM2 inhibitory activity of compounds 3a-3h.
Antiproliferative effects of target compounds 3a-3h
To determine the efficiency of 3a-3h as antitumour agents, we assessed the in vitro cytotoxicity of 3a-3h using several different tumour cell lines derived from human colon cancer (HCT116), breast cancer (MCF7), cervical cancer (Hela) and lung cancer (H1299) and mouse melanoma (B16). The results are presented in . Most target compounds reduced cancer cell viability at nanomolar concentrations in MTS reduction assays, showing higher cytotoxicity than shikonin. Specially, compound 3b exhibited an optimal dose-dependent cytotoxicity with IC50 values against HCT116, MCF7, Hela, H1299 and B16 cells from 69 nM to 122 nM. The preliminary SAR showed that introduction of a long-chain amine in target compounds lowered cytotoxicity (3b vs. 3c vs 3d), which was not consistent with the enzyme activity. This discrepancy may be due to the different properties of these compounds such as cell penetration that is important in the cellular assay. In addition, replacing the chain amines with various cyclic amines, morpholinyl (3a), thiamorpholinyl (3f), pyrrolidinyl (3g) and thiazolidinyl (3h) substitution compounds also demonstrated the great potency.
Table 2. In vitro cytotoxicity of target compounds
To further explore the selectivity of target compounds against cancer cells, we tested the cytotoxicity of representative compound 3f in BEAS-2B cells derived from normal human bronchial epithelial cells. The result showed that IC50 value of compound 3f was 11.49 ± 0.62 µM, which indicated the target compound had higher selectivity for cancer cells than normal cells.
Conclusions
We have described the identification and characterisation of a previously undescribed chemical class of PKM2 inhibitors. Most target compounds as PKM2 inhibitors, especially compound 3f and 3h, have high inhibition responses and are more potent than shikonin and compound 3k. These compounds are therefore optimal PKM2 inhibitors with the best characteristics reported to date. They may lock PKM2 into a low activity conformation and forces disruption of cancer cell metabolism in a manner that is less metabolically flexible than the normal state.
In addition, we have investigated in vitro cytotoxicity of these PKM2 inhibitors. Most target compounds show higher antitumour effects than shikonin in MTS assay. The compound 3b and 3c exhibited optimal dose-dependent cytotoxicity with IC50 values against HCT116, MCF7, Hela, H1299 and B16 cells, respectively, from 69 nM to 122 nM and from 84 nM to 251 nM.
However, there is absence of correlation between the PKM2 inhibitory activity and in vitro antitumor activity of the target compounds. This suggests that these compounds may have other mechanisms to influence the tumour cells. In future studies, we will focus on evaluation as yet unidentified mechanisms of these compounds.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013;4:e532.
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029–33.
- Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 2006;66:8927–30.
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008;7:11–20.
- Warburg O. On respiratory impairment in cancer cells. Science 1956;124:269–70.
- Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med 2008;49:(Suppl 2):24S–42S.
- Chen Z, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr 2007;39:267–74.
- Gatenby RA, Gillies RJ. Glycolysis in cancer: a potential target for therapy. Int J Biochem Cell Biol 2007;39:1358–66.
- Porporato PE, Dhup S, Dadhich RK, et al. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol 2011;2:49.
- Wong N, De Melo J, Tang D. PKM2, a Central Point of Regulation in Cancer Metabolism. Int J Cell Biol 2013;2013:242513
- Yang W, Lu Z. Regulation and function of pyruvate kinase M2 in cancer. Cancer Lett 2013;339:153–8.
- Gupta V, Bamezai RN. Human pyruvate kinase M2: a multifunctional protein. Protein Sci 2010;19:2031–44.
- Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol. Metab 2012;23:560–6.
- Noguchi T, Inoue H, Tanaka T. The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem 1986;261:13807–12.
- Yang W, Lu Z. Pyruvate kinase M2 at a glance. J Cell Sci 2015;128:1655–60.
- Chaneton B, Gottlieb E. Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends Biochem Sci 2012;37:309–16.
- Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008;452:230–3.
- Morgan HP, O'Reilly FJ, Wear MA, et al. M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation. Proc Natl Acad Sci USA 2013;110:5881–6.
- Dayton TL, Jacks T, Vander Heiden MG. PKM2, cancer metabolism, and the road ahead. EMBO Rep 2016;17:1721–30.
- Vander Heiden MG, Christofk HR, Schuman E, et al. Identification of small molecule inhibitors of pyruvate kinase M2. Biochem Pharmacol 2010;79:1118–24.
- Chen J, Xie J, Jiang Z, et al. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011;30:4297–306.
- Li W, Liu J, Zhao Y. PKM2 inhibitor shikonin suppresses TPA-induced mitochondrial malfunction and proliferation of skin epidermal JB6 cells. Mol Carcinog 2014;53:403–12.
- Ning X, Qi H, Li R, et al. Discovery of novel naphthoquinone derivatives as inhibitors of the tumor cell specific M2 isoform of pyruvate kinase. Eur J Med Chem 2017;138:343–52.
- Thomson RH. 240. Quinones. Part I. Chloroalkylation. J Chem Soc 1953;0:1196–9.
- Zhang Y, Liu B, Wu X, et al. New pyridin-3-ylmethyl carbamodithioic esters activate pyruvate kinase M2 and potential anticancer lead compounds. Bioorganic Med Chem 2015;23:4815–23.