
Abstract
Pyridinium containing sulfonamides have been largely investigated as carbonic anhydrase inhibitors (CAIs), showing interesting selectivity features. Nevertheless, only few structural studies are so far available on adducts that these compounds form with diverse CA isoforms. In this paper, we report the structural characterization of the adduct that a triphenylpyridinium derivative forms with hCA II, showing that the substitution of the pyridinium ring plays a key role in determining the conformation of the inhibitor in the active site and consequently the binding affinity to the enzyme. These findings open new perspectives on the basic structural requirements for designing sulfonamide CAIs with a selective inhibition profile.
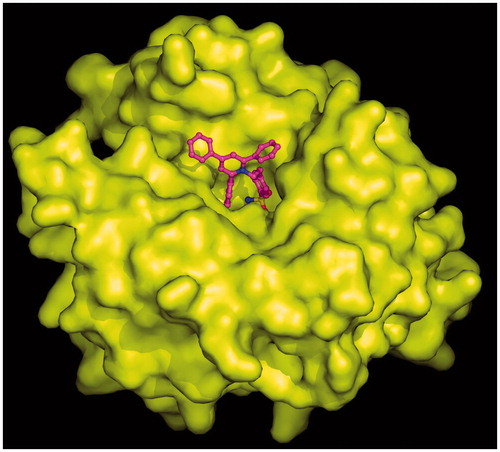
Graphical Abstract
Introduction
Heterocyclic/aromatic sulfonamides such as dorzolamide, brinzolamide, acetazolamide, methazolamide, ethoxzolamide, and dichlorophenamide () represent the first generations of clinically used inhibitors of the metalloenzyme carbonic anhydrase (CA) (EC 4.2.1.1)Citation1–3. They are very strong, typically low nanomolar inhibitors, of most of the 15 CA isoforms presently known in humansCitation2. Dorzolamide and brinzolamide are widely used, topically-acting antiglucoma agentsCitation4, ethoxzolamideCitation5 has fewer clinical applications, whereas acetazolamide, methazolamide, and dichlorophenamide are systemically used antiglaucoma drugs, still employed clinically, even if they were discovered decades agoCitation4. The latter compounds also show clinical benefits for the treatment of other conditions such as epilepsyCitation6–8, idiopathic intracranial hypertensionCitation9, obesityCitation10–12, and as diureticsCitation13, but their applications are severely limited due to a range of side effects connected with inhibition of CA isoforms not involved in those specific pathologiesCitation2. For this reason, the development of isoform-selective CA inhibitors (CAIs) represented an important drug design challenge for the last two decadesCitation2,Citation14, leading to interesting developments, with several classes of compounds identified so far, which show selective inhibition of the different CA isoforms. They belong to sulfonamideCitation15, coumarinCitation16–18, sulfocoumarinCitation19,Citation20, polyamineCitation21, dithiocarbamateCitation22,Citation23, carboxylateCitation24,Citation25 chemotypes, among others. Such isoform-selective compounds opened new scenarios for the applications of CAIs as antitumor drugsCitation26–29, anti-neuropathic pain agentsCitation30,Citation31, or even for the management of cerebral ischemiaCitation32, arthritisCitation33, or bacterial/fungal/protozoan infectionsCitation34,Citation35.
Figure 1. Chemical structures of clinically used CAIs and pyridinium containing sulfonamides 1 and 2.
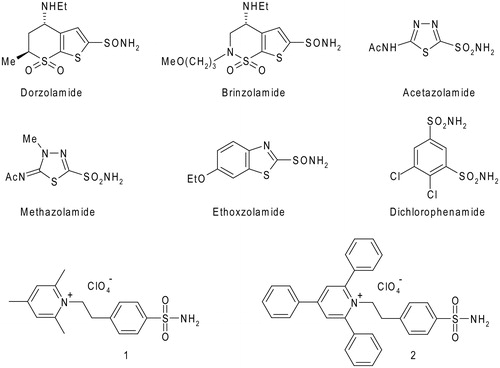
One of the most interesting class of isoform-selective sulfonamide CAIs is constituted by the pyridinium salts, obtained by reaction of amino-benzenesulfonamides with pyrylium saltsCitation36–38. These compounds represent the first class of CAIs, which were demonstrated to possess a high selectivity for inhibiting membrane-associated (CA IV, IX, XII, and XIV) over cytosolic or mitochondrial CA isoformsCitation36,Citation37,Citation39. Furthermore, due to their cationic nature, they are also membrane-impermeantCitation36–38, which makes them highly attractive for targeting extracellular CAsCitation26,Citation39,Citation40. Among the pyridinium containing sulfonamides, compounds 1 and 2, which incorporate 2,4,6-trisubstituted pyridinium moieties, were shown by our group to possess low nanomolar affinity for CA IX, a tumour-associated enzyme validated as an antitumor targetCitation26, and also to be less effective inhibitors of widespread, off target isoforms CA I, II, and IVCitation36,Citation37,Citation39.
Despite the interesting selectivity features of the pyridinium containing sulfonamides as CAIs, only few structural studies are so far available on the adducts that these compounds form with diverse CA isoforms, with the hCA II/1 complex being the only one characterized by X-ray diffraction studiesCitation41. In order to get more insights into the CA binding mechanism and the interesting selective inhibition profile of these molecules, we report here the crystal structure of the hCA II/2 adduct and compare it with the previously described structure of the hCA II/1 complexCitation41. The inhibition of the second tumour-associated isoform, hCA XII, with derivatives 2 is also reported here for the first time.
Materials and methods
CA inhibition
Inhibition constants reported in were previously determinedCitation1,Citation39 with the exception of KI of 2 against hCA XII which has been determined here. In detail, an applied photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activityCitation42. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. Six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rate was determined in the same manner and subtracted from the total observed rate. Stock solution of inhibitors (0.1 mM) were prepared in distilled–deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, to allow for the formation of the E–I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 (GraphPad Software Inc., San Diego, CA, USA) and the Cheng–Prusoff equation and represent the mean from at least three different determinations.
Table 1. Inhibition of isozymes hCA I, hCA II, hCA IV, hCA IX, and hCA XII with the pyridinium salts 1, 2, and the standard, clinically used sulfonamide CAIs.
X-ray crystallography
The hCA II/2 adduct was obtained using a procedure previously described for other hCA II/inhibitor complexesCitation43,Citation44. In detail, a 50-fold excess of the inhibitor was added to a 0.2 mg/mL enzyme solution in 20 mM Tris-HCl pH 8.0. After incubation overnight at 4 °C, the complex was concentrated to 10 mg/mL by using a 5-kDa cutoff ultrafiltration device (Vivaspin® 500; Sartorius, Göttingen, Germany). Crystals were obtained at 20 °C using the hanging drop vapour diffusion technique by equilibrating drops containing 1 µL of complex solution and an equal volume of precipitant solution consisting of 1.3 M sodium citrate, 0.1 M TRIS-HCl, pH 8.5, over a reservoir containing 0.5 mL of precipitant solution. Crystals appeared after 3 days. Diffraction data were collected to 1.65 Å resolution, in-house at −180 °C, using a Rigaku MicroMax-007 HF generator producing Cu Kα radiation and equipped with a Saturn 944 CCD detector. Cryoprotection of the crystals was achieved transferring the crystals into the precipitant solution with the addition of 10% (v/v) glycerol. Data were indexed, integrated, and scaled using HKL2000Citation45. Crystal parameters and data collection statistics are summarized in .
Table 2. Data collection and refinement statistics for the hCA II/2 complex.
A previously solved structure of hCA II (PDB code 5O07)Citation46, with inhibitor and non-protein atoms omitted, was used as starting model for rigid body refinement in CNSCitation47,Citation48. Initial refinement was continued in CNS using positional and slow cooling protocols followed by restrained B-value refinement. The inhibitor molecule was identified from peaks in |Fo| − |Fc| maps and gradually built into the model over several rounds of refinement. Composite simulated-annealing omit maps were used regularly during the building process to verify and correct the modelCitation47,Citation48. Crystallographic refinement was carried out against 95.1% of the measured data. The remaining 4.9% of the observed data, which was randomly selected, was used for R-free calculations to monitor the progress of refinement. Topology files of the inhibitor were generated using the PRODRG2 serverCitation49. Restraints on inhibitor bond angles and distances were taken from similar structures in the Cambridge Structural DatabaseCitation50, whereas standard restraints were used on protein bond angles and distances throughout refinement. The correctness of stereochemistry was finally checked using PROCHECKCitation51. The refinement statistics of final model are summarized in . Coordinates and structure factors were deposited in the Protein Data Bank (accession code 6EQU).
Results and discussion
Sulfonamides 1 and 2 were previously described by our groups, and were obtained by reaction of 4-aminoethyl-benzenesulfonamide with 2,4,6-trisubstituted pyrylium saltsCitation36,Citation38. In , the CA inhibitory action of these two positively-charged, membrane-impermeant sulfonamides, as well as those of the six clinically used drugs shown in , are presented. From the table it is evident that acetazolamide, methazolamide and ethoxzolamide are promiscuous CAIs, inhibiting effectively at least four of the five investigated isoforms, whereas dichlorophenamide, dorzolamide, and brinzolamide possess a more selective inhibition profile, as their activity against hCA I and hCA IV are modest, being however effective inhibitors of three isoforms, hCA II, IX, and XII. A different inhibition profile is observed for the two pyridinium-containing sulfonamides. Indeed, whereas the trimethylpyridinium derivative 1 is a low nanomolar inhibitor of hCA IX and XII, effectively inhibits hCA II and hCA IV, but it is less effective as hCA I inhibitor, the triphenylpyridinium 2 is a quite effective hCA XII inhibitor, it also inhibits hCA IX, but its affinity for the other isoforms is in the micromolar range (see ). Thus, sulfonamide 2 shows the most isoform-selective inhibition profile among the eight compounds considered here. Connected to the fact that it is a membrane-impermeant compoundCitation36,Citation37, and that the presence of the three phenyl moieties may induce also a better lipophilic character compared to 1, this compound constitutes an interesting lead for obtaining molecules to be investigated in detail for the selective inhibition of the tumour associated CA isoforms. The most interesting feature of 2 with respect to 1 is its reduced ability to inhibit the ubiquitous hCA II, although maintaining good inhibition constants against hCA IX and hCA XII. Indeed, due to the fact that hCA II is an ubiquitous, house-keeping isoform, its inhibition may be detrimental when the targeting of the tumour-associated isoforms CA IX and XII is envisaged, leading to many undesired side effectsCitation2. Thus, to get more insights into the molecular basis responsible for the reduced affinity for hCA II of compound 2 with respect to compound 1, the X-ray crystal structure of the hCA II/2 adduct was solved and compared with that previously reported of the hCA II/1 complexCitation41.
Crystals of the hCA II/2 complex were obtained as previously described for other sulfonamide CA inhibitorsCitation43,Citation44 and the structure was solved and refined using a previously reported procedureCitation46,Citation52–55. The final refined model had an R-work and R-free value of 17.5% and 21.0%, respectively, and was of high overall quality with 88.2% of the non-glycine residues located in the most favoured regions of the Ramachandran plot (). Since the initial stages of crystallographic refinement, electron density maps showed the presence of the inhibitor molecule bound within the enzyme active site. However, these maps were very well defined for the 4-ethylbenzenesulfonamide part of the inhibitor but poorly defined for the 2,4,6-triphenylpyridinium group, indicating that this region was flexible within the active site cavity (). Accordingly, B-factor values of this region (33.9 Å2) were higher with respect to those observed for the 4-ethylbenzenesulfonamide moiety (11.7 Å2). The binding of the inhibitor within the active site did not cause significant changes in the enzyme structure as demonstrated by the low value of the RMSD calculated by superposing the Cα atoms in the adduct and the non-inhibited enzyme (0.3 Å).
Figure 2. Active site region of the hCA II/2 adduct, showing σA-weighted |2Fo − Fc| simulated annealing omit map (contoured at 1.0σ) relative to the inhibitor molecule. Active site Zn2+ coordination (red continuous lines) and hydrogen bonds (red dotted lines) are also reported.
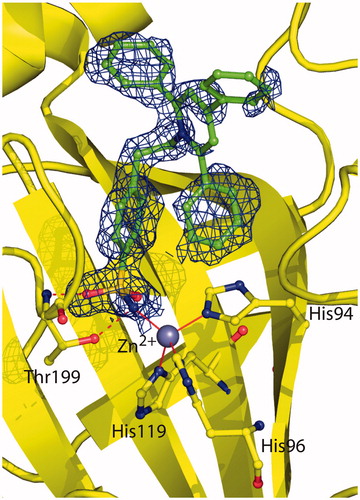
As expected for a benzenesulfonamide inhibitor, compound 2 was bound to the enzyme with its sulfonamide group coordinated to the zinc ion in a tetrahedral geometryCitation2. This group was also involved in two hydrogen bond interactions with residue Thr199, as already described for all hCA II/benzenesulfonamide adducts so far structurally characterized ()Citation2. No other polar interactions were observed between the enzyme and the inhibitor. Indeed, the ethylbenzene moiety was located in the middle of the active site establishing several hydrophobic interactions with Leu198, whereas the 2,4,6-triphenylpyridinium moiety did not establish strong interactions with enzyme (see above).
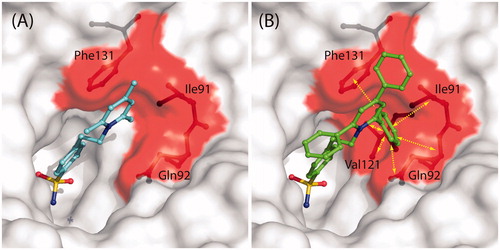
reports the structural superposition of compounds 2 and 1 when bound to the hCA II active site, showing that even if the benzenesulfonamide groups of the two inhibitors are quite perfectly superimposable, the two trisubstituted pyridinium moieties are oriented toward different regions of the active site. In particular, the 2,4,6-trimethylpyridinium moiety of 1, which was described as perfectly defined in the electron density mapsCitation41, fits perfectly into a hydrophobic pocket, defined by residues Ile91, Gln92, and Phe131, where it is involved in a strong face-to-face stacking interaction with the Phe131 aromatic ring (). On the contrary the 2,4,6-triphenylpyridinium moiety of 2, although oriented toward the hydrophilic part of the active site, is flexible and does not establish many stabilizing interactions with enzyme’s residues. Interestingly, the only differences between the two inhibitors are the substituents of the pyridinium ring, namely three methyl groups for compound 1 and three phenyl moieties for compound 2, which make it much more bulky and do not allow its accommodation into the hydrophobic pocket defined by residues Ile91, Gln92, and Phe131. Indeed, in this position the substituted ring would strongly clash with residues which delimit the pocket (). The impossibility for the pyridinium ring of 2 to be accommodated within the aforementioned hydrophobic pocket leads to the loss of the strong face-to-face interaction with Phe131 and is most likely responsible of its lower affinity for hCA II with respect to compound 1.
Figure 3. Structural superposition between 1 (cyan, PDB code 1ZE8)Citation41 and 2 (green) when bound to hCA II active site.
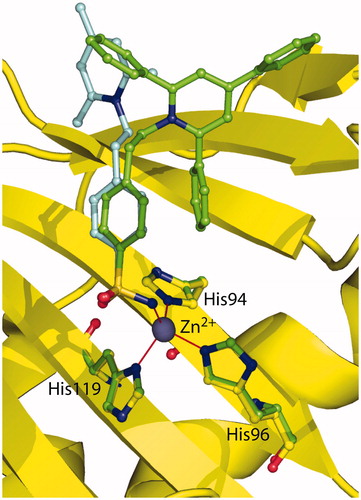
Figure 4. (A) Solvent accessible surface of hCA II/1 active siteCitation41. Residues delimiting the hydrophobic pocket, where the trimethyl-pyridinium ring is located, are highlighted in red (Ile91, Gln92, Phe131). Compound 1 and residues Ile91, Gln92, and Phe131 are represented as ball-and-stick. (B) Solvent accessible surface of hCA II active site. Colour code is as in (A). The hypothetical conformation that compound 2 would have had if its pyridinium ring was positioned in the same hydrophobic pocket of 1 is shown. Yellow arrows indicate protein residues which clashes with inhibitor (Ile91, Gln92, Val121, Phe131).
Conclusions
The X-ray data presented here explain why the triphenylpyridinium-substituted sulfonamide 2 is a much weaker hCA II inhibitor compared to its structural analogue 1 incorporating three methyl moieties at the pyridinium ring. Although there is a decreased affinity of 2 also towards the tumour-associated isoforms hCA IX and XII, compared to compound 1 (acting as a very potent inhibitor against both these isozymes), the triphenyl derivative 2 showed a selective inhibition profile for the tumour over cytosolic isoforms, which represents a valuable feature for compounds to be investigated as antitumor agents. Thus, understanding the detailed interactions between inhibitors belonging to similar structural classes, as 1 and 2 discussed here, may shed new light on the basic structural requirements for designing sulfonamide CAIs with a selective inhibitory profile.
Acknowledgements
We thank Maurizio Amendola and Giosuè Sorrentino for their skilful technical assistance with X-ray measurements.
Disclosure statement
The authors report no declaration of interest
Related Research Data
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nature Rev Drug Discov 2008;7:168–81.
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16.
- Di Fiore A, Pedone C, Antel J, et al. Carbonic anhydrase inhibitors: the X-ray crystal structure of ethoxzolamide complexed to human isoform II reveals the importance of thr200 and gln92 for obtaining tight-binding inhibitors. Bioorg Med Chem Lett 2008;18:2669–74.
- De Simone G, Scozzafava A, Supuran CT. Which carbonic anhydrases are targeted by the antiepileptic sulfonamides and sulfamates? Chem Biol Drug Des 2009;74:317–21.
- Bibi D, Mawasi H, Nocentini A, et al. Design and comparative evaluation of the anticonvulsant profile, carbonic-anhydrate inhibition and teratogenicity of novel carbamate derivatives of branched aliphatic carboxylic acids with 4-aminobenzensulfonamide. Neurochem Res 2017;42:1972–82.
- Mishra CB, Kumari S, Angeli A, et al. Discovery of benzenesulfonamides with potent human carbonic anhydrase inhibitory and effective anticonvulsant action: design, synthesis, and pharmacological assessment. J Med Chem 2017;60:2456–69.
- Supuran CT. Acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6.
- De Simone G, Di Fiore A, Supuran CT. Are carbonic anhydrase inhibitors suitable for obtaining antiobesity drugs? Curr Pharm Des 2008;14:655–60.
- Supuran CT, Di Fiore A, De Simone G. Carbonic anhydrase inhibitors as emerging drugs for the treatment of obesity. Expert Opin Emerg Drugs 2008;13:383–92.
- Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35.
- Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91.
- Supuran CT. Drug interaction considerations in the therapeutic use of carbonic anhydrase inhibitors. Expert Opin Drug Metab Toxicol 2016;12:423–31.
- Carta F, Supuran CT, Scozzafava A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med Chem 2014;6:1149–65.
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44.
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62.
- Maresca A, Supuran CT. Coumarins incorporating hydroxy- and chloro-moieties selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II. Bioorg Med Chem Lett 2010;20:4511–14.
- Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300.
- Grandane A, Tanc M, Zalubovskis R, Supuran CT. 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem Lett 2014;24:1256–60.
- Carta F, Temperini C, Innocenti A, et al. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 2010;53:5511–22.
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun 2012;48:1868–70.
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates strongly inhibit carbonic anhydrases and show antiglaucoma action in vivo. J Med Chem 2012;55:1721–30.
- Langella E, D’Ambrosio K, D’Ascenzio M, et al. A combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes in the active site of carbonic anhydrases. Chemistry 2016;22:97–100.
- D’Ambrosio K, Carradori S, Monti SM, et al. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem Commun 2015;51:302–5.
- Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008–2013). Expert Opin Ther Pat 2013;23:737–49.
- Monti SM, Supuran CT, De Simone G. Carbonic anhydrase IX as a target for designing novel anticancer drugs. Curr Med Chem 2012;19:821–30.
- Ondriskova E, Debreova M, Pastorekova S. Tumor-associated carbonic anhydrases IX and XII. In: Supuran CT, De Simone G, eds. Carbonic anhydrases as biocatalysts. From theory to medical and industrial applications. Amsterdam: Elsevier; 2015:169–205.
- Guler OO, De Simone G, Supuran CT. Drug design studies of the novel antitumor targets carbonic anhydrase IX and XII. Curr Med Chem 2010;17:1516–26.
- Carta F, Di Cesare Mannelli L, Pinard M, et al. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg Med Chem 2015;23:1828–40.
- Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother 2016;16:961–8.
- Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. J Enzyme Inhib Med Chem 2016;31:894–9.
- Bua S, Di Cesare Mannelli L, Vullo D, et al. Design and synthesis of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the treatment of rheumatoid arthritis. J Med Chem 2017;60:1159–70.
- Vermelho AB, Capaci GR, Rodrigues IA, et al. Carbonic anhydrases from Trypanosoma and Leishmania as anti-protozoan drug targets. Bioorg Med Chem 2017;25:1543–55.
- Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704.
- Scozzafava A, Briganti F, Ilies MA, Supuran CT. Carbonic anhydrase inhibitors: synthesis of membrane-impermeant low molecular weight sulfonamides possessing in vivo selectivity for the membrane-bound versus cytosolic isozymes. J Med Chem 2000;43:292–300.
- Supuran CT, Scozzafava A, Ilies MA, Briganti F. Carbonic anhydrase inhibitors: synthesis of sulfonamides incorporating 2,4,6-trisubstituted-pyridinium-ethylcarboxamido moieties possessing membrane-impermeability and in vivo selectivity for the membrane-bound (CA IV) versus the cytosolic (CA I and CA II) isozymes. J Enzyme Inhib 2000;15:381–401.
- Supuran CT, Scozzafava A, Ilies MA, et al. Carbonic anhydrase inhibitors – Part 53 – synthesis of substituted-pyridinium derivatives of aromatic sulfonamides: the first non-polymeric membrane-impermeable inhibitors with selectivity for isozyme IV. Eur J Med Chem 1998;33:577–94.
- Pastorekova S, Casini A, Scozzafava A, et al. Carbonic anhydrase inhibitors: the first selective, membrane-impermeant inhibitors targeting the tumor-associated isozyme IX. Bioorg Med Chem Lett 2004;14:869–73.
- Perut F, Carta F, Bonuccelli G, et al. Carbonic anhydrase IX inhibition is an effective strategy for osteosarcoma treatment. Expert Opin Ther Targets 2015;19:1593–605.
- Menchise V, De Simone G, Alterio V, et al. Carbonic anhydrase inhibitors: stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J Med Chem 2005;48:5721–7.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- La Regina G, Coluccia A, Famiglini V, et al. Discovery of 1,1′-biphenyl-4-sulfonamides as a new class of potent and selective carbonic anhydrase XIV inhibitors. J Med Chem 2015;58:8564–72.
- Vullo D, Supuran CT, Scozzafava A, et al. Kinetic and X-ray crystallographic investigations of substituted 2-thio-6-oxo-1,6-dihydropyrimidine-benzenesulfonamides acting as carbonic anhydrase inhibitors. Bioorg Med Chem 2016;24:3643–8.
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997;276:307–26.
- De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11.
- Brunger AT, Adams PD, Clore GM, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 1998;54:905–21.
- Brunger AT. Version 1.2 of the crystallography and NMR system. Nat Protoc 2007;2:2728–33.
- Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr D Biol Crystallogr 2004;60:1355–63.
- Allen FH. The Cambridge structural database: a quarter of a million crystal structures and rising. Acta Crystallogr. B 2002;58:380–8.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 1993;26:283–91.
- Rami M, Dubois L, Parvathaneni NK, et al. Hypoxia-targeting carbonic anhydrase IX inhibitors by a new series of nitroimidazole-sulfonamides/sulfamides/sulfamates. J Med Chem 2013;56:8512–20.
- Di Fiore A, Scozzafava A, Winum JY, et al. Carbonic anhydrase inhibitors: binding of an antiglaucoma glycosyl-sulfanilamide derivative to human isoform II and its consequences for the drug design of enzyme inhibitors incorporating sugar moieties. Bioorg Med Chem Lett 2007;17:1726–31.
- Bruno E, Buemi MR, Di Fiore A, et al. Probing molecular interactions between human carbonic anhydrases (hCAs) and a novel class of benzenesulfonamides. J Med Chem 2017;60:4316–26.
- Di Fiore A, Vergara A, Caterino M, et al. Hydroxylamine-O-sulfonamide is a versatile lead compound for the development of carbonic anhydrase inhibitors. Chem Commun 2015;51:11519–22.