
Abstract
A new series of 4,6-disubstituted 2-(4-(dimethylamino)styryl)quinoline 4a,b–9a,b was synthesized by the reaction of 2-(4-(dimethylamino)styryl)-6-substituted quinoline-4-carboxylic acids 3a,b with thiosemicarbazide, p-hydroxybenzaldehyde, ethylcyanoacetate, and 2,4-pentandione. In addition, the antitumour activity of all synthesized compounds 3a,b–9a,b was studied via MTT assay against two cancer cell lines (HepG2 and HCT116). Furthermore, epidermal growth factor receptor (EGFR) inhibition, using the most potent antitumour compounds, 3a, 3b, 4a, 4b, and 8a, was evaluated. The interpretation of the results showed clearly that the derivatives 3a, 4a, and 4b exhibited the highest antitumour activities against the tested cell lines HepG2 and HCT116 with IC50 range of 7.7–14.2 µg/ml, in comparison with the reference drugs 5-fluorouracil (IC50 = 7.9 and 5.3 µg/ml, respectively) and afatinib (IC50 = 5.4 and 11.4 µg/ml, respectively). In vitro EGFR screening showed that compounds 3a, 3b, 4a, 4b, and 8a exhibited moderate inhibition towards EGFR with IC50 values at micromolar levels (IC50 range of 16.01–1.11 µM) compared with the reference drugs sorafenib (IC50 = 1.14 µM) and erlotinib (IC50 = 0.1 µM). Molecular docking was performed to study the mode of interaction of compounds 3a and 4b with EGFR kinase.
Graphical Abstract
1. Introduction
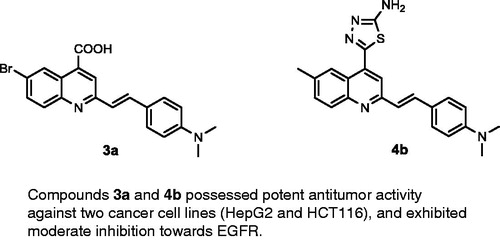
Development of a novel antitumour drug with potent activity remains critically important due to the majority of human deaths globally being attributable to cancerCitation1–13. Epidermal growth factor receptors (EGFR) are an important class of kinase enzymes used in cancer treatment, which are overexpressed in several tumours, such as brain, liver, colon, prostate, breast, and non-small-cell lung cancersCitation14–18. The inhibition of EGFR is affected by blocking tyrosine kinase at ATP-binding sites with small molecules, such as quinazoline derivativesCitation19–22. Recently, afatinib, gefitinib, and erlotinib (), quinazoline derivatives designed to inhibit EGFR kinase, have been approved by the FDA for the treatment of non-small cell lung and breast cancersCitation23–29. Moreover, it was reported that the nitrogen atom at position-3 of the quinazoline core formed water-mediated hydrogen bond with Thr766 (gatekeeper residue) within the EGFR pocketCitation29,Citation30. On the contrary, bioisosteric replacement of the quinazoline ring system with quinoline, through conversion of the nitrogen atom at position-3 by carbon or carbonitrile fragments, yielded quinoline derivatives such as neratinib and pelitinib (), which are potent EGFR kinase inhibitorsCitation31–38. This bioisosteric replacementCitation39 did not require a water molecule to mediate the binding with the amino acid residue Thr766. 2-Styrylquinoline (SQ) derivatives have been reported as promising antitumour compounds against several tumour cell linesCitation40–43. Recently, a series of 2-styryl-4-aminoquinoline (SQ-I, ) was developed which possessed potent in vitro antiproliferative activity against lung, colon, and liver cancer cell lines, comparable to gefitinibCitation44.
Figure 1. Reported EGFR inhibitors and antitumour agents, and design of the newly synthesized 2-styrylquinolines.
The above-mentioned results encouraged us in designing and synthesizing a series of SQs, which are anticipated to be as potent as structurally related quinazoline bioisosteres. The antitumour activity of the target compounds was evaluated using two tumour cell lines, namely, human hepato-cellular carcinoma cell line (HepG2) and human colorectal carcinoma cell line (HCT116)Citation45–48. In addition, some compounds were evaluated for their inhibitory activity against the EGFR tyrosine kinase enzyme. Moreover, a molecular docking method was used to study the putative binding mode of the target molecules into the receptor pocket of EGFR kinaseCitation1,Citation8–9,Citation12,Citation30.
2. Experimental
2.1. Chemistry
Melting points (°C) were recorded using Stuart melting point apparatus and were uncorrected. IR spectra were recorded on a Mattson 5000 FT-IR spectrometer (in cm−1) (Mattson Instruments, Cambridge, UK) using KBr disk at the Faculty of Pharmacy, Mansoura University. 1 H-NMR and 13C NMR spectra were recorded on a Bruker Avance spectrometer (400 MHz) in DMSO-d6 at Georgia State University, Atlanta, GA. The chemical shifts in ppm are expressed in δ units, using tetramethylsilane as an internal standard, and coupling constants in Hz. Mass spectrum analyses were performed on Thermo Fisher Scientific (Waltham, MA, USA) SID GC GC/MS, DSQ II in the Faculty of Science, Mansoura University. Reaction times were determined using a TLC technique on silica gel plates 60 F245E. Merck, and the spots were visualised by U.V. (366, 245 nm). Biological screening was conducted at the Pharmacognosy Department, Faculty of Pharmacy, Mansoura University. Compound 3a was prepared according to its previous reportCitation49.
2.1.1. Synthesis of compound 3b
A mixture of the appropriate 5-substituted isatin (5 mmol), 4-(4-(dimethylamino)phenyl)but-3-en-2-one (1) (0.945 g, 5 mmol) and potassium hydroxide (1.28 g, 23 mmol) in 50% aqueous ethanol (20 ml) was heated under reflux for 24 h. The reaction mixture was then diluted with 30% aqueous ethanol solution (20 ml) and neutralised with 50% acetic acid. The precipitated solid was filtered, dried, and crystallised from ethanol.
2.1.1.1. 2–(4-(Dimethylamino)styryl)-6-methylquinoline-4-carboxylic acid (3b)
Light yellow crystals; yield: 78%; m.p. 275 °C. IR (KBr) γ/cm−1: 3400–3448 (br, OH), 1681 (C = O acid). 1 H-NMR (400 MHz-DMSO-d6): δ 2.28 (s, 3 H, Ar-CH3), 3.00 (s, 6 H, 2CH3), 6.50 (d, J = 8 Hz, 1 H, styryl-H), 6.62 (d, J = 4 Hz, 1 H, styryl-H), 6.80 (d, 2 H, Ar-H), 7.45 (d, 2 H, Ar-H), 7.57 (d, J = 8 Hz, 1 H, Ar-H), 7.92 (s, 1 H, Ar-H), 8.03 (d, J = 8 Hz, 1 H, Ar-H), 8.35 (s, 1 H, Ar-H), 11.69 (s, 1 H, COOH, D2O exchangeable). 13C NMR (100 MHz-DMSO-d6): δ 21.5, 40.2, 111.1, 115.2, 124.5, 125.3, 126.8, 127.3, 127.5, 128.8, 129.9, 130.5, 132.1, 134.2, 144.9, 150.2, 166.8. MS (m/z%): 333 (M+ + 1, 10.55), 332 (M+, 42.60), 204 (100). Anal. Calcd for C21H20N2O2 (332.40): C, 75.88; H, 6.06; N, 8.43. Found: C, 75.91; H, 6.08; N, 8.47.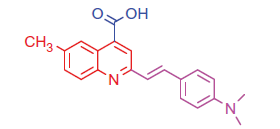
2.1.2. General method for synthesis of compounds 4a,b
Phosphorus oxychloride (4.6 g, 34 mmol) was added dropwise to an ice-cold mixture of compounds 3a,b and thiosemicarbazide (0.91 gm, 10 mmol) and the reaction mixture was heated under reflux for 8 h. The reaction mixture was cooled, poured into ice-cold water, and neutralised with 10% sodium carbonate solution. The precipitated solid was filtered, washed with water, dried, and crystallised from ethanol.
2.1.2.1. 5-(6-Bromo-2–(4-(dimethylamino)styryl)quinolin-4-yl)-1,3,4-thiadiazol-2-amine (4a)
White crystals; yield: 82%; m.p. 223 °C. IR (KBr) γ/cm−1: 3380 (NH2), 685 (C-S-C). 1 H-NMR (400 MHz-DMSO-d6): δ 3.20 (s, 6 H, 2CH3), 6.53 (d, J = 8 Hz, 1 H, styryl-H), 6.61 (d, J = 4 Hz, 1 H, styryl-H), 6.86 (d, 2 H, Ar-H), 7.53 (d, 2 H, Ar-H), 7.54 (d, J = 8 Hz, 1 H, Ar-H), 8.00 (s, 1 H, Ar-H), 8.13 (d, J = 8 Hz, 1 H, Ar-H), 8.29 (s, 1 H, Ar-H), 8.97 (s, 2 H, NH2, D2O exchangeable). MS (m/z%): 453 (M+ + 1, 35.14), 451 (M+ –1, 62.03), 324 (100). Anal. Calcd for C21H18BrN5S (452.37): C, 55.76; H, 4.01; N, 15.48. Found: C, 55.80; H, 4.06; N, 15.51.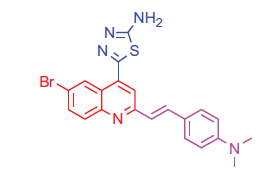
2.1.2.2. 5-(2-(4-(Dimethylamino)styryl)-6-methylquinolin-4-yl)-1,3,4-thiadiazol-2-amine (4b)
White crystals; yield: 85%; m.p. 226 °C. IR (KBr) γ/cm−1: 3384 (NH2), 690 (C-S-C). 1 H-NMR (400 MHz-DMSO-d6): δ 2.20 (s, 3 H, Ar-CH3), 3.14 (s, 6 H, 2CH3), 6.48 (d, J = 8 Hz, 1 H, styryl-H), 6.60 (d, J = 4 Hz, 1 H, styryl-H), 6.78 (d, 2 H, Ar-H), 7.45 (d, 2 H, Ar-H), 7.61 (d, J = 8 Hz, 1 H, Ar-H), 8.10 (s, 1 H, Ar-H), 8.17 (d, J = 8 Hz, 1 H, Ar-H), 8.33 (s, 1 H, Ar-H), 8.91 (s, 2 H, NH2, D2O exchangeable). 13C NMR (100 MHz-DMSO-d6): δ 19.8, 40.9, 110.5, 116.4, 121.2, 122.8, 126.3, 127.1, 128.2, 130.0, 131.2, 132.5, 133.3, 142.6, 143.4, 151.0, 157.1, 162.0, 173.7. MS (m/z%): 389 (M+ + 2, 8.85), 388 (M+ + 1, 42.21), 387 (M+, 53.20), 259 (100). Anal. Calcd for C22H21N5S (387.50): C, 68.19; H, 5.46; N, 18.07; S, 8.27. Found: C, 68.22; H, 5.50; N, 18.13; S, 8.29.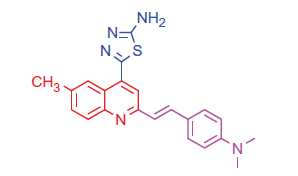
2.1.3. General method for synthesis of 2-(4-(dimethylamino)styryl)-6-substituted quinoline-4-carbonyl chlorides (5a,b)
Thionyl chloride (4.17 g, 30 mmol) was added to quinolone carboxylic acid 3 (10 mmol). The reaction mixture was heated under reflux for 3 h. After cooling, it was evaporated under reduced pressure and the obtained solid was used directly in the next step without further purification.
2.1.4. General method for synthesis of compounds 6a,b and 7a,b
A mixture of the acid chloride 5 (10 mmol), 4-hydroxybenzaldehyde (1.22 g, 10 mmol) or phenyl hydrazine (1.08 g, 10 mmol) and potassium carbonate (1.38 g, 10 mmol) in dimethylformamide (25 ml) was heated at 90 °C for 24 h. After cooling, the reaction mixture was poured into ice water. The obtained solid was filtered, washed with water, dried, and crystallised from ethanol to give pure products.
2.1.4.1. 4-Formylphenyl 6-bromo-2-(4-(dimethylamino)styryl)quinoline-4-carboxylate (6a)
Buff crystals; yield: 90%; m.p. 242 °C. IR (KBr) γ/cm−1: 1715 (C = O aldehyde), 1690 (C = O ester). 1 H-NMR (400 MHz-DMSO-d6): δ 2.96 (s, 6 H, 2CH3), 6.37 (d, J = 8 Hz, 1 H, styryl-H), 6.42 (d, J = 4 Hz, 1 H, styryl-H), 6.65 (d, 2 H, Ar-H), 6.82 (d, 2 H, Ar-H), 7.48 (d, J = 8 Hz, 1 H, Ar-H), 7.56 (d, 2 H, Ar-H), 7.61 (d, 2 H, Ar-H), 8.07 (s, 1 H, Ar-H), 8.13 (d, J = 8 Hz, 1 H, Ar-H), 8.35 (s, 1 H, Ar-H), 9.07 (s, 1 H, CHO). MS (m/z%): 502 (M+ + 1, 6.73), 501 (M+, 42.52), 500 (M+–1, 60.21), 373 (100). Anal. Calcd for C27H21BrN2O3 (501.38): C, 64.68; H, 4.22; N, 5.59. Found: C, 64.72; H, 4.26; N, 5.63.
2.1.4.2. 4-Formylphenyl 2-(4-(dimethylamino)styryl)-6-methylquinoline-4-carboxylate (6 b)
Pale brown crystals; yield: 92%; m.p. 248 °C. IR (KBr) γ/cm−1: 1710 (C = O aldehyde), 1695 (C = O ester). 1 H-NMR (400 MHz-DMSO-d6): δ 2.30 (s, 3 H, Ar-CH3), 3.12 (s, 6 H, 2CH3), 6.39 (d, J = 8 Hz, 1 H, styryl-H), 6.40 (d, J = 4 Hz, 1 H, styryl-H), 6.85 (d, 2 H, Ar-H), 7.47 (d, J = 8 Hz, 1 H, Ar-H), 7.56 (d, 2 H, Ar-H), 7.62 (d, 2 H, Ar-H), 7.84 (d, 2 H, Ar-H), 8.11 (s, 1 H, Ar-H), 8.18 (d, J = 8 Hz, 1 H, Ar-H), 8.32 (s, 1 H, Ar-H), 9.13 (s, 1 H, CHO). MS (m/z%): 437 (M+ + 1, 22), 436 (M+, 37), 308 (100). Anal. Calcd for C28H24N2O3 (436.51): C, 77.04; H, 5.54; N, 6.42. Found: C, 77.08; H, 5.57; N, 6.46.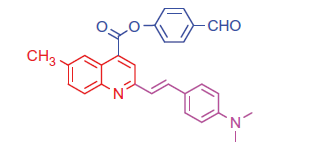
2.1.4.3. 6-Bromo-2-(4-(dimethylamino)styryl)-N'-phenylquinoline-4-carbohydrazide (7a)
Brown crystals; yield: 89%; m.p. 232 °C. IR (KBr) γ/cm−1: 3362 (NH), 1650 (C = O amide). 1 H-NMR (400 MHz-DMSO-d6): δ 3.12 (s, 6 H, 2CH3), 4.92 (s, 1 H, NH, D2O exchangeable), 5.20 (s, 1 H, NH, D2O exchangeable), 6.39 (d, J = 8 Hz, 1 H, styryl-H), 6.40 (d, J = 4 Hz, 1 H, styryl-H), 6.80 (d, 2 H, Ar-H), 7.47 (d, J = 8 Hz, 1 H, Ar-H), 7.56 (d, 2 H, Ar-H), 8.04 (s, 1 H, Ar-H), 8.18 (d, J = 8 Hz, 1 H, Ar-H), 8.21–8.29 (m, 3 H, Ar-H), 8.32 (s, 1 H, Ar-H), 8.41–8.48 (m, 2 H, Ar-H). 13C NMR (100 MHz-DMSO-d6): δ 42.1, 111.3, 114.0, 114.9, 118.5, 121.8, 123.7, 125.3, 126.9, 127.2, 129.1, 130.2, 131.7, 132.2, 138.9, 142.4, 143.8, 148.6, 150.0, 155.4, 163.9. MS (m/z%): 488 (M+ + 1, 18.52), 487 (M+, 56.12), 486 (M+–1, 68.03), 359 (100). Anal. Calcd for C26H23BrN4O (487.40): C, 64.07; H, 4.76; N, 11.50. Found: C, 64.11; H, 4.79; N, 11.54.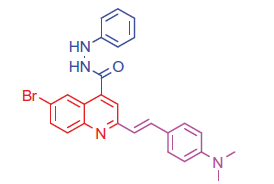
2.1.4.4. 2-(4-(Dimethylamino)styryl)-6-methyl-N'-phenylquinoline-4-carbohydrazide (7 b)
Brown crystals; yield: 86%; m.p. 228 °C. IR (KBr) γ/cm−1: 3365 (NH), 1655 (C = O amide). 1 H-NMR (400 MHz-DMSO-d6): δ 2.41 (s, 3 H, Ar-CH3), 3.33 (s, 6 H, 2CH3), 4.85 (s, 1 H, NH, D2O exchangeable), 5.21 (s, 1 H, NH, D2O exchangeable), 6.28 (d, J = 8 Hz, 1 H, styryl-H), 6.39 (d, J = 4 Hz, 1 H, styryl-H), 6.84 (d, 2 H, Ar-H), 7.55 (d, 2 H, Ar-H), 7.60 (d, J = 8 Hz, 1 H, Ar-H), 8.02 (s, 1 H, Ar-H), 8.10 (d, J = 8 Hz, 1 H, Ar-H), 8.19–8.26 (m, 3 H, Ar-H), 8.32 (s, 1 H, Ar-H), 8.39–8.47 (m, 2 H, Ar-H). MS (m/z%): 423 (M+ + 1, 32.31), 422 (M+, 55.17), 294 (100). Anal. Calcd for C27H26N4O (422.53): C, 76.75; H, 6.20; N, 13.26. Found: C, 76.79; H, 6.28; N, 13.30.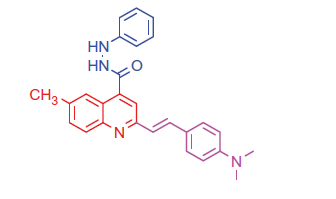
2.1.5. General method for synthesis of compounds 8a,b and 9a,b
To the acid chloride 5 (10 mmol), sodium salt of ethyl 2-cyanoacetate or pentane-2,4-dione [prepared by using sodium ethoxide and ketone derivatives in ethanol (20 ml)] was added and stirred at room temperature overnight. The reaction mixture was filtered, washed with water, dried, and crystallised from DMF.
2.1.5.1. Ethyl 3-(6-bromo-2-(4-(dimethylamino)styryl)quinolin-4-yl)-2-cyano-3-oxopropanoate (8a)
White crystals; yield: 82%; m.p. > 300 °C. 1 H-NMR (400 MHz-DMSO-d6): δ 1.22 (t, J = 8 Hz, 3 H, CH3), 3.14 (s, 6 H, 2CH3), 4.20 (q, J = 8 Hz, 2 H, CH2), 4.60 (s, 1 H, CH-CN), 6.58 (d, J = 8 Hz, 1 H, styryl-H), 6.66 (d, J = 4 Hz, 1 H, styryl-H), 6.82 (d, 2 H, Ar-H), 7.47 (d, 2 H, Ar-H), 7.52 (d, J = 8 Hz, 1 H, Ar-H), 8.00 (s, 1 H, Ar-H), 8.23 (d, J = 8 Hz, 1 H, Ar-H), 8.41 (s, 1 H, Ar-H). MS (m/z%): 493 (M+ + 1, 42.11), 491 (M+–1, 34.17), 363 (100). Anal. Calcd for C25H22BrN3O3 (492.37): C, 60.99; H, 4.50; N, 8.53. Found: C, 60.95; H, 4.48; N, 8.50.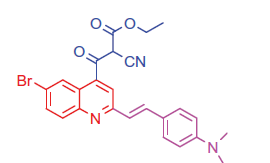
2.1.5.2. Ethyl 2-cyano-3-(2-(4-(dimethylamino)styryl)-6-methylquinolin-4-yl)-3-oxopropanoate (8 b)
White crystals; yield: 80%; m.p. > 300 °C. IR (KBr) γ/cm−1: 2210 (C≡N), 1705 (C = O ester), 1628 (C = O ketone). 1 H-NMR (400 MHz-DMSO-d6): δ 1.24 (t, J = 8 Hz, 3 H, CH3), 3.00 (s, 3 H, Ar-CH3), 3.10 (s, 6 H, 2CH3), 4.18 (q, J = 8 Hz, 2 H, CH2), 4.53 (s, 1 H, CH-CN), 6.59 (d, J = 8 Hz, 1 H, styryl-H), 6.60 (d, J = 4 Hz, 1 H, styryl-H), 6.80 (d, 2 H, Ar-H), 7.44 (d, J = 8 Hz, 1 H, Ar-H), 7.52 (d, 2 H, Ar-H), 8.05 (s, 1 H, Ar-H), 8.12 (d, J = 8 Hz, 1 H, Ar-H), 8.38 (s, 1 H, Ar-H). MS (m/z%): 428 (M+ + 1, 22.39), 427 (M+, 50.11), 299 (100). Anal. Calcd for C26H25N3O3 (427.50): C, 73.05; H, 5.89; N, 9.83. Found: C, 73.09; H, 5.92; N, 9.87.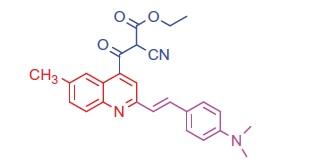
2.1.5.3. 3-(6-Bromo-2-(4-(dimethylamino)styryl)quinoline-4-carbonyl)pentane-2,4-dione (9a)
White crystals; yield: 77%; m.p. > 300 °C. IR (KBr) γ/cm−1: 1620 (C = O ketone). 1 H-NMR (400 MHz-DMSO-d6): δ 3.00 (s, 6 H, 2CH3), 3.37 (s, 6 H, 2COCH3), 4.64 (s, 1 H, CH-CO), 6.50 (d, J = 8 Hz, 1 H, styryl-H), 6.62 (d, J = 4 Hz, 1 H, styryl-H), 6.85 (d, 2 H, Ar-H), 7.49 (d, 2 H, Ar-H), 7.57 (d, J = 8 Hz, 1 H, Ar-H), 7.95 (s, 1 H, Ar-H), 8.03 (d, J = 8 Hz, 1 H, Ar-H), 8.35 (s, 1 H, Ar-H). 13C NMR (100 MHz-DMSO-d6): δ 27.2, 41.5, 80.2, 112.4, 114.9, 123.8, 125.0, 126.2, 128.0, 128.2, 129.1, 129.8, 131.0, 132.4, 134.8, 144.5, 154.8, 194.0, 199.7. MS (m/z%): 480 (M+ + 1, 20.44), 478 (M+–1, 31.00), 351 (100). Anal. Calcd for C25H23BrN2O3 (479.37): C, 62.64; H, 4.84; N, 5.84. Found: C, 62.60; H, 4.81; N, 5.80.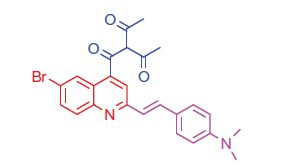
2.1.5.4. 3-(2-(4-(Dimethylamino)styryl)-6-methylquinoline-4-carbonyl)pentane-2,4-dione (9 b)
White crystals; yield: 79%; m.p. > 300 °C. IR (KBr) γ/cm−1: 1625 (C = O ketone). 1 H-NMR (400 MHz-DMSO-d6): δ 2.93 (s, 6 H, 2CH3), 3.11 (s, 3 H, Ar-CH3), 3.34 (s, 6 H, 2COCH3), 4.60 (s, 1 H, CH-CO), 6.44 (d, J = 8 Hz, 1 H, styryl-H), 6.60 (d, J = 4 Hz, 1 H, styryl-H), 6.80 (d, 2 H, Ar-H), 7.42 (d, J = 8 Hz, 1 H, Ar-H), 7.52 (d, 2 H, Ar-H), 7.92 (d, J = 8 Hz, 1 H, Ar-H), 8.04 (s, 1 H, Ar-H), 8.21 (s, 1 H, Ar-H). MS (m/z%): 415 (M+ + 1, 31.26), 414 (M+, 50.37), 286 (100). Anal. Calcd for C26H26N2O3 (414.51): C, 75.34; H, 6.32; N, 6.76. Found: C, 75.37; H, 6.37; N, 6.79.
2.2. Biological assay
2.2.1. Antitumour activity using MTT assay
The designed compounds were evaluated for their in vitro antitumour effect using the standard MTT method against two human tumour cell lines, namely, HepG2 and HCT116Citation45–48. The quantitative evaluation of the cytotoxicity was performed using tetrazolium salt MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2 H-tetrazolium bromide) assay. The cytotoxic activity was expressed as the concentration of the compound that caused 50% growth inhibition (IC50, mean ± SEM) compared with the growth of untreated cells.
2.2.2. EGFR kinase inhibition assay
EGFR kinase activity was determined via enzyme-linked immunosorbent assay (ELISA) in 96-well platesCitation50. The EGFR kinase activity for each compound were expressed as IC50 values. Data were represented as mean ± SD from three independent experiments, and differences between groups were considered statistically significant at p < .05.
2.3. Docking methodology
All modelling experiments were conducted with MOE programs running on PC computer (MOE 2008.10, Chemical Computing Group, Inc., Montreal, QC, Canada)Citation51–54. Starting coordinates of the X-ray crystal structure of the EGFR enzyme in complex with erlotinib (PDB code 1M17) were obtained from the RCSB Protein Data BankCitation29.
3. Results and discussion
3.1. Chemistry
The target compounds were prepared as outlined in Schemes 1 and 2. The structures of the target compounds were established based on elemental analysis, IR, 1 H NMR, 13C NMR, and MS data. The starting compounds, 2-(4-(dimethylamino)styryl)-6-substituted quinoline-4-carboxylic acids 3a,b, were prepared through the Pfitzinger reaction, which offers a very convenient synthetic entry to the quinoline-4-carboxylic acid derivatives 3a,b by heating 4–(4-(dimethylamino)phenyl)but-3-en-2-one (1) and 5-substituted isatins 2a,b in an aqueous/alcoholic KOH solution (Scheme 1)Citation55–57.
Scheme 1. Synthesis of the designed 2-styryl-4-quinoline carboxylic acids, and 1,3,4-thiadiazoles 3a,b and 4a,b.
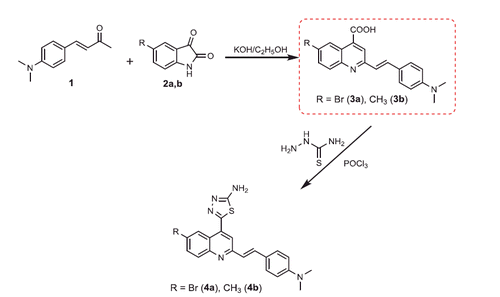
Scheme 2. Synthesis of compounds 5a,b–9a,b.
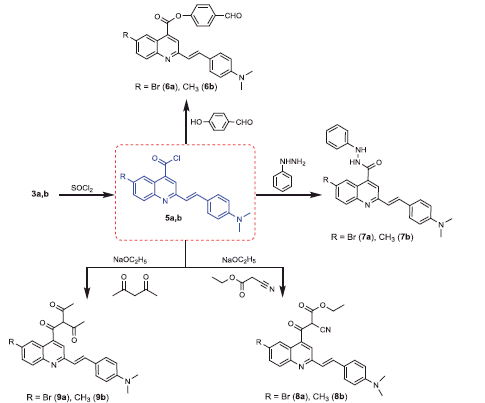
The condensation of quinoline-4-carboxylic acids 3a,b with thiosemicarbazide in the presence of phosphorus oxychlorideCitation58 afforded the 2-amino-1,3,4-thiadiazoles 4a,b (Scheme 1). The IR spectra of the compounds 4a,b showed disappearance of peak at 3400–3448 cm−1 (OH group) and peak at 1681 cm−1 (C = O group) in compounds 3a,b, while peaks in the ranges 3380–3384 cm−1 and 685–690 cm−1, attributed to (NH2) and (C–S–C) groups, respectively, were observed. In addition, 1 H NMR spectra of compounds 4a,b showed signals in the range of 8.91–8.97 ppm characteristic for the –NH2 group with disappearance of –COOH group signal of compounds 3a,b at 11.69 ppm.
Generation of the acid chlorides 5a,b from their corresponding carboxylic acids 3a,b was achieved through heating under reflux with thionyl chloride (Scheme 2). These acid chlorides 5a,b were subjected to reaction with 4-hydroxybenzaldehyde and phenylhydrazine in dimethylformamide containing potassium carbonate to yield 4-formylphenyl 2-(4-(dimethylamino)styryl)-6-substituted quinoline-4-carboxylates 6a,b and 2-(4-(dimethylamino)styryl)-6-substituted N′-phenylquinoline-4-carbohydrazides 7a,b, respectively (Scheme 2). The IR spectra of compounds 6a,b and 7a,b showed absorption bands at 1644–1657 cm−1 and 3362–3365 cm−1 attributed to formyl and amino groups, respectively. In addition, 1 H NMR spectra of compounds 6a,b showed signals in the range of 9.07–9.13, characteristic of the formyl moieties. The acid chlorides 5a,b were further condensed with either ethyl cyanoacetate or acetyl acetone, to afford compounds 8a,b and 9a,b (Scheme 2). 1 H NMR spectra of compounds 8a,b showed characteristic triplet and quartet signals for the ethyl ester groups at 1.22–1.24 ppm and 4.18–4.20 ppm, respectively. Furthermore, compounds 9a,b showed singlet signals for the methyl ketone groups (O = C–CH3) at approximately 3.34–3.37 ppm in addition to a singlet peak at 4.60–4.64 ppm characteristic of the –CH– groups (–CH–CO–CH3).
3.2. Biological evaluation
3.2.1. Antitumour activity
The antitumour activity of the designed compounds 3a,b–9a,b and both reference drugs 5-fluorouracil (5-FU) and afatinib against HepG2 and HCT116 cell lines is shown in and Citation45–48. It is clear that compounds 3a and 4a,b exhibited the highest antitumour activities against the tested cell lines, with an IC50 range of 7.7–14.0 µg/ml, in comparison with the IC50 values of the reference drugs 5-FU (IC50 range of 5.3–7.9 µg/ml) and afatinib (IC50 range of 5.4–11.4 µg/ml). Moreover, 2–(4-(dimethylamino)styryl)-6-methylquinoline-4-carboxylic acid (3b) exhibited strong cytotoxic effects against HepG2 and HCT116 cell lines with IC50 values of 17.2 and 14.8 µg/ml, respectively. In addition, ethyl 3-(6-bromo-2-(4-(dimethylamino)styryl)quinolin-4-yl)-2-cyano-3-oxopropanoate (8a) showed strong activity against the HCT116 cell line (IC50 = 16.0 µg/ml) and moderate activity against the HepG2 cell line (IC50 = 26.2 µg/ml). On the contrary, compounds 7a and 8 b showed moderate efficacy against HepG2 and HCT116 cell lines with an IC50 range of 43.7–52.6 µg/ml. Finally, 4-formylphenyl 2-(4-(dimethylamino)styryl)quinoline-4-carboxylates 6a,b and 4-carbonylpentane-2,4-diones 3-(2-(4-(dimethylamino)styryl)quinoline-4-carbonyl)pentane-2,4-diones 9a,b exhibited weak to very weak cytotoxic activity, with an IC50 range of 57.3–100 µg/ml.
Figure 2. Relative viability of cells (%) against concentration of the newly synthesized compounds.
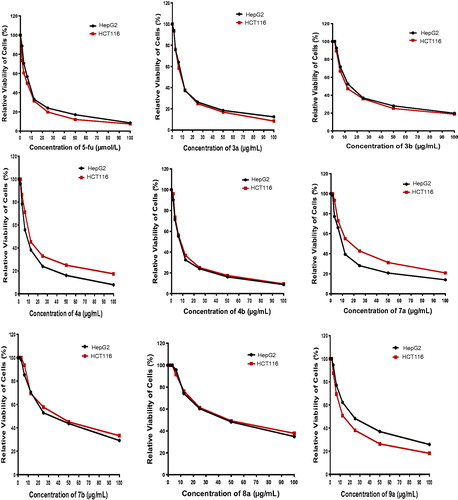
Table 1. In vitro antitumour activity of the tested compounds.
3.2.2. EGFR inhibitory activity
The mechanism of antitumour activity of the target compounds was studied using ELISA-based EGFR-TK assayCitation50. Five compounds with the highest antitumour activities were evaluated against EGFR kinase activity assays with sorafenib and erlotinib as the reference drugs. IC50 values of the tested compounds were calculated and are listed in , where compounds 3a (IC50 = 2.23 µM) and 4b (IC50 = 1.11 µM) showed the highest inhibitory activity against EGFR, compared to the other tested compounds. The activities of compounds 3b (IC50 = 8.01 µM), 4a (IC50 = 8.78 µM), and 8a (IC50 = 16.01 µM) against EGFR were found to be weakly active comparable to those of sorafenib (IC50 = 1.14 µM) and erlotinib (IC50 = 0.1 µM). Based on these results, we can conclude that EGFR-TK inhibitory activity of the target compounds is correlated to their antitumour activities against HepG2 and HCT116.
Table 2. IC50 values of the designed compounds toward EGFR kinase and docking interaction energy.
3.3. Molecular docking study
The inhibitory activities of compounds 3a and 4b on EGFR kinase prompted us to carry out molecular docking into the putative binding site of EGFR kinase. Both compounds 3a and 4b were flexibly docked into the active site of EGFR kinase along with the reference inhibitor erlotinib (PDB code: 1M17)Citation29. All docking calculations were performed using MOE 2008.10 softwareCitation7b,Citation51–54.
The interaction energies of compounds 3a and 4b and erlotinib, docked into the active site of EGFR, were –18.54, –20.89, and –29.01 kcal/mol, respectively ( and ). The molecular docking results of the most active compound 4b demonstrated a hydrophobic interaction of the quinoline ring with surrounding amino acids, such as Val702, Leu694, and Leu820. The substituent group at C-4 of the quinoline ring is the main moiety affecting the binding mode of compound 4b in both activation and catalytic loops, where a 2-aminothiadiazole ring uniquely formed trifurcated hydrogen bonds with the distinctive residue Met769, Gln767, and Thr766. Moreover, the 2-styryl fragment of compound 4b was firmly extended to the backbone, similar to the 6,7-dialkoxy moiety of erlotinib, augmenting the recognition and the overall inhibitory activity (, lower left panel).
Figure 3. Three-dimensional interactions of erlotinib (upper left panel), compounds 4b (lower left panel), and 3a (lower right panel) with the receptor pocket of EGFR kinase. Hydrogen bonds are shown as green line. Upper right panel shows superimposition of compounds 4b (green coloured) and 3a (yellow coloured) on erlotinib (red coloured) inside the pockets of the active site.
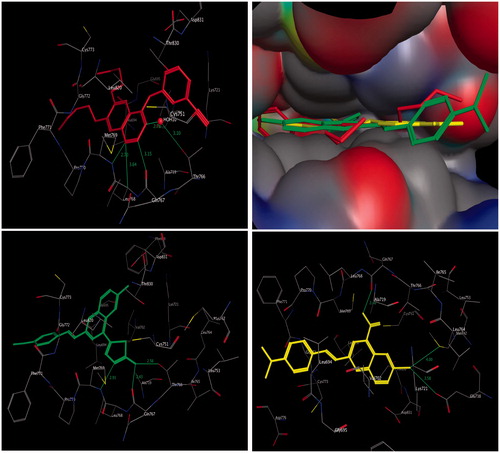
Compound 3a binds in a similar manner to compound 4b, where hydrophobic interaction is clearly observed among amino acid residues Val702, Leu694, and Leu820 and quinoline core. It was found that the carboxylic group at position-4 of the quinoline core was clearly recognised, with hydrogen bonding to the amino acid residue Gln767, while this carboxylic group was shifted away from the distinctive amino acid residue Met769. Moreover, the bromine atom at position-6 formed bifurcated hydrogen bonds with the amino acid residue Met742 and Glu738 (, lower right panel). It is clear that the results of the molecular docking can be used to design novel quinoline derivatives with potential antitumour activity and binding to EGFR kinase.
4. Conclusions
Novel 4,6-disubstituted 2-SQ derivatives 3a,b–9a,b have been synthesized, and their antitumour activity and EGFR inhibition have been evaluated. Among the tested compounds, 3a and 4a,b (IC50 ≅ 7.7–9.8 µg/ml) were identified as the most potent antitumour agents against HepG2 and HCT116 cancer cell lines, with activity comparable to that of 5-FU (IC50 ≅ 5.37–7.9 µg/ml) and afatinib (IC50 ≅ 5.4 –11.4 µg/ml). Moreover, compound 3b exhibited strong antitumour activities against HepG2 and HCT116 cancer cell lines with IC50 values of 17.2 and 14.8 µg/ml, respectively. Compounds 3a and 4b have moderate inhibitory activity on EGFR with IC50 values of 2.23 and 1.22 µM, respectively. Accordingly, both compounds 3a and 4b are expected to exert their antitumour activity through inhibition of EGFR. A molecular docking study was conducted for compounds 3a and 4b and the putative binding site of EGFR kinase, which revealed a binding mode similar to that of the reference inhibitor erlotinib.
Acknowledgements
Authors deeply thank Dr. Abdelbasset A. Farahat, Georgia State University, United States for his great help in carrying out NMR. The authors extend their appreciation to the Holding Company for Biological Products and Vaccines (VACSERA), Cairo, Egypt, for performing the biological screening.
Additional information
Funding
Related Research Data
References
- Alanazi AM, Abdel-Aziz AA-M, Shawer TZ, et al. Synthesis, antitumor and antimicrobial activity of some new 6-methyl-3-phenyl-4(3H)-quinazolinone analogues: in silico studies. J Enzyme Inhib Med Chem 2016;31:721–35.
- Mohamed MA, Ayyad RR, Shawer TZ, et al. Synthesis and antitumor evaluation of trimethoxyanilides based on 4(3H)-quinazolinone scaffolds. Eur J Med Chem 2016;112:106–13.
- Abdel-Aziz AA-M, El-Azab AS, El-Subbagh HI, et al. Design, synthesis, single-crystal and preliminary antitumor activity of novel arenesulfonylimidazolidin-2-ones. Bioorg Med Chem Lett 2012;22:2008–14.
- Avendańo C, Menéndez J. Medicinal chemistry of anticancer agents. Amsterdam: Elsevier; 2008.
- Varmus H. The new era in cancer research. Science 2006;312:1162–5.
- Eckhardt S. Recent progress in the development of anticancer agents. Curr Med Chem Anticancer Agents 2002;2:419–39.
- (a) Abdel-Aziz AA-M, El-Azab AS, Alanazi AM, et al. Synthesis and potential antitumor activity of 7-(4-substituted piperazin-1-yl)-4-oxoquinolines based on ciprofloxacin and norfloxacin scaffolds: in silico studies. J Enzyme Inhib Med Chem 2016;31:796–809. (b) Abdel-Aziz AA-M, El-Azab AS, Alanazi AM, et al. Design, synthesis and antibacterial activity of fluoroquinolones containing bulky arenesulfonyl fragment: 2D-QSAR and docking study. Eur J Med Chem 2011;46:5487–97.
- El-Azab AS, Al-Omar MA, Abdel-Aziz AA-M, et al. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: molecular docking study. Eur J Med Chem 2010;45:4188–98.
- Al-Suwaidan IA, Abdel-Aziz AA-M, Shawer TZ, et al. Synthesis, antitumor activity and molecular docking study of some novel 3-benzyl-4 (3H) quinazolinone analogues. J Enzyme Inhib Med Chem 2016;31:78–89.
- Al-Obaid A, Abdel-Hamide S, El-Kashef H, et al. Synthesis, in vitro antitumor activity and molecular modeling study of certain 2-thieno-4 (3H)-quinazolinone analogs. Eur J Med Chem 2009;44:2379–91.
- Alanazi AM, Al-Suwaidan IA, Abdel-Aziz AA-M, et al. Design, synthesis and biological evaluation of some novel substituted 2-mercapto-3-phenethylquinazolines as antitumor agents. Med Chem Res 2013;22:5566–77.
- Al-Suwaidan IA, Alanazi AM, Abdel-Aziz AA-M, et al. Design, synthesis and biological evaluation of 2-mercapto-3-phenethylquinazoline bearing anilide fragments as potential antitumor agents: molecular docking study. Bioorg Med Chem Lett 2013;23:3935–41.
- Alanazi AM, Abdel-Aziz AA-M, Al-Suwaidan IA, et al. Design, synthesis and biological evaluation of some novel substituted quinazolines as antitumor agents. Eur J Med Chem 2014;79:446–54.
- Bazley LA, Gullick WJ. The epidermal growth factor receptor family. Endocr Relat Cancer 2005;12(Suppl 1):S17–S27.
- Hirsch FR, Varella-Garcia M, Bunn PA Jr, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol 2003;21:3798–807.
- Bishayee S. Role of conformational alteration in the epidermal growth factor receptor (EGFR) function. Biochem Pharmacol 2000;60:1217–23.
- Ogiso H, Ishitani R, Nureki O, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002;110:775–87.
- Umekita Y, Ohi Y, Sagara Y, Yoshida H. Co-expression of epidermal growth factor receptor and transforming growth factor-alpha predicts worse prognosis in breast-cancer patients. Int J Cancer 2000;89:484–7.
- Zhang F, Zhang H, Wang F. EGFR inhibition studies by hybrid scaffolds for their activity against ovarian cancer. J Buon 2016;21:1482–90.
- Sheikh IA, Hassan HM. In silico identification of novel erlotinib analogues against epidermal growth factor receptor. Anticancer Res 2016;36:6125–32.
- Mowafy S, Galanis A, Doctor ZM, et al. Toward discovery of mutant EGFR inhibitors; Design, synthesis and in vitro biological evaluation of potent 4-arylamino-6-ureido and thioureido-quinazoline derivatives. Bioorg Med Chem 2016;24:3501–12.
- Sun M, Zhao J, Chen X, et al. Synthesis and biological evaluation of novel tricyclic oxazine and oxazepine fused quinazolines. Part 2: gefitinib analogs. Bioorg Med Chem Lett 2016;26:4842–5.
- Minkovsky N, Berezov A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr Opin Investig Drugs 2008;9:1336–46.
- Hirsh V. Afatinib (BIBW 2992) development in non-small-cell lung cancer. Future Oncol 2011;7:817–25.
- Barlesi F, Tchouhadjian C, Doddoli C, et al. Gefitinib (ZD1839, Iressa) in non-small-cell lung cancer: a review of clinical trials from a daily practice perspective. Fundam Clin Pharmacol 2005;19:385–93.
- Arteaga CL, Johnson DH. Tyrosine kinase inhibitors-ZD1839 (Iressa). Curr Opin Oncol 2001;13:491–8.
- Barker AJ, Gibson KH, Grundy W, et al. Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg Med Chem Lett 2001;11:1911–14.
- Ganjoo KN, Wakelee H. Review of erlotinib in the treatment of advanced non-small cell lung cancer. Biologics 2007;1:335–46.
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem 2002;277:46265–72.
- Wissner A, Berger DM, Boschelli DH, et al. 4-Anilino-6,7-dialkoxyquinoline-3-carbonitrile inhibitors of epidermal growth factor receptor kinase and their bioisosteric relationship to the 4-anilino-6,7-dialkoxyquinazoline inhibitors. J Med Chem 2000;43:3244–56.
- Minami Y, Shimamura T, Shah K, et al. The major lung cancer-derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI-272. Oncogene 2007;26:5023–7.
- Wissner A, Mansour TS. The development of HKI-272 and related compounds for the treatment of cancer. Arch Pharm (Weinheim) 2008;341:465–77.
- Rabindran SK, Discafani CM, Rosfjord EC, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res 2004;64:3958–65.
- Kiesel BF, Parise RA, Wong A, et al. LC-MS/MS assay for the quantitation of the tyrosine kinase inhibitor neratinib in human plasma. J Pharm Biomed Anal 2017;134:130–6.
- Laheru D, Croghan G, Bukowski R, et al. A phase I study of EKB-569 in combination with capecitabine in patients with advanced colorectal cancer. Clin Cancer Res 2008;14:5602–9.
- Luethi D, Durmus S, Schinkel AH, et al. Liquid chromatography-tandem mass spectrometry assay for the EGFR inhibitor pelitinib in plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2013;934:22–5.
- Pisaneschi F, Nguyen QD, Shamsaei E, et al. Development of a new epidermal growth factor receptor positron emission tomography imaging agent based on the 3-cyanoquinoline core: synthesis and biological evaluation. Bioorg Med Chem 2010;18:6634–45.
- Lu S, Zheng W, Ji L, et al. Synthesis, characterization, screening and docking analysis of 4-anilinoquinazoline derivatives as tyrosine kinase inhibitors. Eur J Med Chem 2013;61:84–94.
- Pawar VG, Sos ML, Rode HB, et al. Synthesis and biological evaluation of 4-anilinoquinolines as potent inhibitors of epidermal growth factor receptor. J Med Chem 2010;53:2892–901.
- Mrozek-Wilczkiewicz A, Spaczynska E, Malarz K, et al. Design, synthesis and in vitro activity of anticancer styrylquinolines. The p53 independent mechanism of action. PLoS One 2015;10:e0142678.
- Chang FS, Chen W, Wang C, et al. Synthesis and antiproliferative evaluations of certain 2-phenylvinylquinoline (2-styrylquinoline) and 2-furanylvinylquinoline derivatives. Bioorg Med Chem 2010;18:124–33.
- Musiol R, Jampilek J, Kralova K, et al. Investigating biological activity spectrum for novel quinoline analogues. Bioorg Med Chem 2007;15:1280–8.
- Podeszwa B, Niedbala H, Polanski J, et al. Investigating the antiproliferative activity of quinoline-5,8-diones and styrylquinolinecarboxylic acids on tumor cell lines. Bioorg Med Chem Lett 2007;17:6138–41.
- Jiang N, Zhai X, Li T, et al. Design, synthesis and antiproliferative activity of novel 2-substituted-4-amino-6-halogenquinolines. Molecules 2012;17:5870–81.
- Vega-Avila E, Pugsley MK. An overview of colorimetric assay methods used to assess survival or proliferation of mammalian cells. Proc West Pharmacol Soc 2011;54:10–14.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986;89:271–7.
- Mauceri HJ, Hanna NN, Beckett MA, et al. Combined effects of angiostatin and ionizing radiation in antitumour therapy. Nature 1998;394:287–91.
- John H. Chinolinderivate, XXXVIII. Synthese 2-phenylierter-3-Oxy-chinolin-4-carbonsäuren. Adv Synth Catal 1932;133:259–72.
- Zhong L, Guo XN, Zhang XH, et al. Expression and purification of the catalytic domain of human vascular endothelial growth factor receptor 2 for inhibitor screening. Biochim Biophys Acta 2005;1722:254–61.
- MOE. 2008.10 of Chemical Computing Group. Inc. Available from: http://www.chemcomp.com [last accessed 1 Sep 2017].
- (a) Al-Suwaidan IA, Alanazi AM, El-Azab AS, et al. Molecular design, synthesis and biological evaluation of cyclic imides bearing benzenesulfonamide fragment as potential COX-2 inhibitors. Part 2. Bioorg Med Chem Lett 2013;23:2601–5. (b) Goda FE, Abdel-Aziz AA-M, Ghoneim HA. Synthesis and biological evaluation of novel 6-nitro-5-substituted aminoquinolines as local anesthetic and anti-arrhythmic agents: molecular modeling study. Bioorg Med Chem 2005;13:3175–83.
- Alanazi AM, El-Azab AS, Al-Suwaidan IA, et al. Structure-based design of phthalimide derivatives as potential cyclooxygenase-2 (COX-2) inhibitors: anti-inflammatory and analgesic activities. Eur J Med Chem 2015;92:115–23.
- El-Gamal MI, Bayomi SM, El-Ashry SM, et al. Synthesis and anti-inflammatory activity of novel (substituted)benzylidene acetone oxime ether derivatives: molecular modeling study. Eur J Med Chem 2010;45:1403–14.
- Gomes R, Diniz AM, Jesus A, et al. The synthesis and reaction network of 2-styryl-1-benzopyrylium salts: an unexploited class of potential colorants. Dyes Pigm 2009;81:69–79.
- Sandmeyer T. Ueber Isonitrosoacetanilide und deren Kondensation zu Isatinen. Helv Chim Acta 1919;2:234–42.
- Massoud MA, El Bialy SA, Bayoumi WA, El Husseiny WM. Synthesis of new 2-and 3-hydroxyquinoline-4-carboxylic acid derivatives as potential antioxidants. Heterocycl Commun 2014;20:81–8.
- Holla BS, Poojary KN, Poojary B, Bhat KS, et al. Synthesis, characterization and antibacterial activity studies on some fluorine containing quinoline-4-carboxylic acids and their derivatives. Indian J Chem 2005;44B:2114–19.