
Abstract
Two sets of benzimidazole derivatives were synthesised and tested in vitro for activity against promastigotes of Leishmania tropica and L. infantum. Most of the tested compounds resulted active against both Leishmania species, with IC50 values in the low micromolar/sub-micromolar range. Among the set of 2-(long chain)alkyl benzimidazoles, whose heterocyclic head was quaternised, compound 8 resulted about 100-/200-fold more potent than miltefosine, even if the selectivity index (SI) versus HMEC-1 cells was only moderately improved. In the set of 2-benzyl and 2-phenyl benzimidazoles, bearing a basic side chain in position 1, compound 28 (2-(4-chlorobenzyl)-1-lupinyl-5-trifluoromethylbenzimidazole) was 12-/7-fold more potent than miltefosine, but exhibited a further improved SI. Therefore, compounds 8 and 28 represent interesting hit compounds, susceptible of structural modification to improve their safety profiles.
Graphical Abstract
Introduction
After malaria, leishmaniasis is the second most prevalent parasite infection worldwide for mortality in humansCitation1. It is transmitted by the bite of a sand-fly infected by a flagellate protozoan of the genus Leishmania. Three different forms of the disease are described: visceral, cutaneous and muco-cutaneous leishmaniasis. The disease is endemic in many tropical and subtropical Countries, leading annually to an estimated 700,000–1 million new cases and 20,000–30,000 deathsCitation1, mostly due to the visceral form caused by Leishmania donovani. The parasite exists in the ovoid non-flagellate form (amastigote) and in the flagellate promastigote, found in the sand-fly.
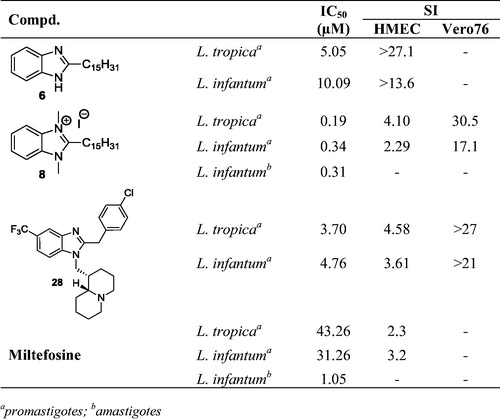
The therapy of leishmaniasis is still based on pentavalent antimonials (sodium stibogluconate and meglumine antimoniate) as first choice drugCitation2a,Citation2b, whereas amphotericin B, miltefosine, paromomycin and pentamidine are considered second-line drugsCitation3a–c. Some other drugs as edelfosine, sitamaquine, fexinidazole, tamoxifene, imiquimod and pentoxyphylline are reported to give variable cure rates when used either alone or, better, in association with antimonials to overcome resistanceCitation4 ().
Figure 1. First and second line or synergistic agents to treat leishmaniasis: (a) meglumine antimoniate (predominant species in aqueous solution); (b) sodium stibogluconate (predominant species in aqueous solution); (c) amphotericin B; (d) miltefosine; (e) edelfosine; (f) paromomycin; (g) pentamidine; (h) sitamaquine; (i) imiquimod; (j) tamoxifene; (k) fexinidazole; (l) pentoxyphylline.
All these drugs may cause several side effects and most of them are also expensive, and thus out of reach for the poor people living in tropical and sub-tropical countries, where the disease is endemic. The cited drugs exhibit very different chemical structures and hit a variety of biological targets, but in several cases the mechanism of action is still undefined or only partially known.
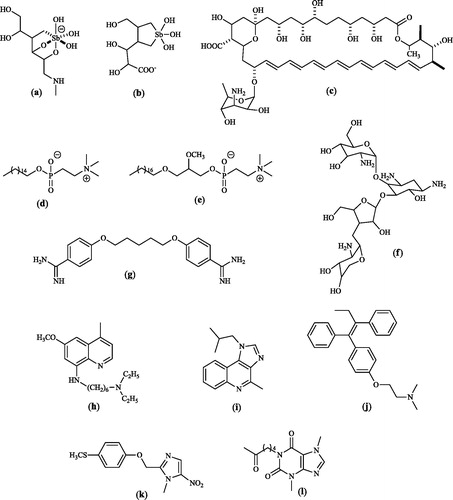
To meet the need of novel more efficacious, safe and unexpensive drugs to treat leishmaniasis, a number of studies are on-going, exploring a wide chemical space from several classes of natural productsCitation5 and or their semi-synthetic derivatives (sterolsCitation5, mono-, sequi-, di- and tri-terpensCitation6, alkaloidsCitation7, flavonoidsCitation8, etc.) to the most diversified synthetic compounds, from the simple chloroacetoanilidesCitation9, to organometallicsCitation10a (as auranofinCitation10b), aryldiselenidesCitation11, adamantylidene alkyl phosphocolineCitation12 and a variety of heterocyclesCitation13, particularly indoleCitation14, indazoleCitation15, benzotriazoleCitation16 and benzimidazoleCitation17–20 derivatives. Examples of these compounds are depicted in and .
Figure 2. Examples of investigational anti-leishmanial agentsCitation5–16.
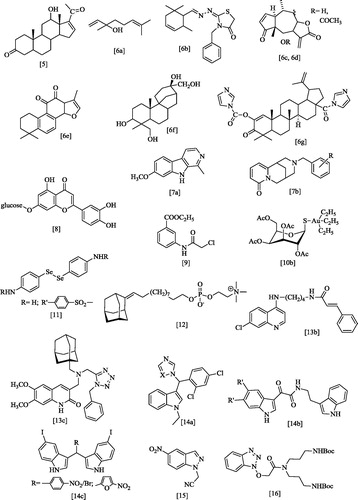
Figure 3. Benzimidazole derivatives previously tested as anti-leishmanial agentsCitation17–20.
Among the benzazolic derivatives, an important position is held by the 2-trifluoromethyl-Citation17 and 2-arylbenzimidazoleCitation18 derivatives that, besides activity versus several other protozoa, display antileishmanial action with potency in the low micromolar range. Interestingly, some bis-benzimidazolesCitation19,Citation20 exhibit sub-micromolar IC50, resulting 7- to 26-fold more potent than pentamidine.
Since many years we are interested in the chemistry and biological properties of benzimidazole derivatives, pursuing varied pharmacological aims, from analgesic-anti-inflammatory actionCitation21, conditioned avoidance response (CAR) inhibitionCitation22, choleretic activity and gastric protectionCitation23, antiviralCitation24 and antitumoralCitation25 activities. In order to further explore the biocidal potential of benzimidazole derivatives, we deemed interesting to evaluate the antileishmanial activity of a set of 2-alkyl/2-benzyl benzimidazoles whose heterocyclic head was quaternised to mimic the ammonium head of miltefosine and edelfosine. Additionally, we selected, among our in house library of benzimidazoles, a second set of 2-arylbenzimidazoles 1-substituted with basic side chains that might be loosely related to sitamaquine. As the anti-leishmanial activity of sitamaquine analogues is mainly related to the length and structure of their basic side chainsCitation4a, in this subset of benzimidazoles a variety of basic chains, featured by different sizes, steric hindrance and lipophilicity, have been included. The bicyclic quinolizidine (lupinyl) moiety is of particular relevance, having been shown to produce analogous or superior activity against Leishmania promastigotes in comparison to sitamaquineCitation4c when replacing the diethylaminohexyl side chain of the latter (our unpublished results). On the whole 38 compounds ( and ) were tested against the promastigotes of Leishmania tropica, responsible for cutaneous leishmaniasis (CL), and 33 of them (depending on availability) were also tested against L. infantum, the causative agent of visceral leishmaniasis (VL). The two best compounds were also assayed against L. infantum amastigotes.
Figure 4. Investigated benzimidazole derivatives without basic side chain.
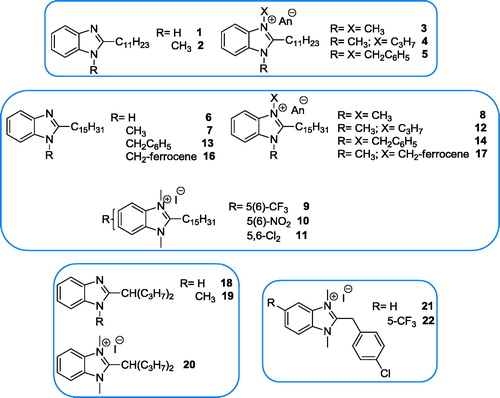
Figure 5. Investigated benzimidazole derivatives with basic side chain.
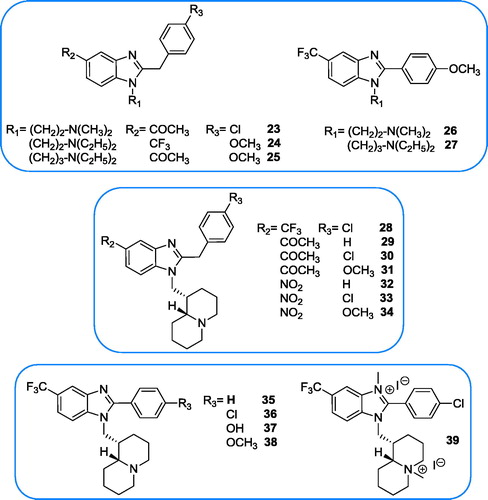
Materials and methods
General
Chemicals, solvents and reagents used for the syntheses were purchased from Sigma-Aldrich, Fluka or Alfa Aesar, and were used without any further purification unless otherwise stated. CC = flash column chromatography. Melting points (uncorrected) were determined with a Büchi apparatus. 1H NMR and 13C NMR spectra were recorded with a Varian Mercury 300VX or Varian Gemini-200 spectrometers in CDCl3 or acetone-d6; the chemical shifts were expressed in ppm (δ), coupling constants (J) in Hertz (Hz). High-resolution mass spectra (HRMS) were performed on a FT-Orbitrap mass spectrometer in positive electrospray ionisation (ESI). Elemental analyses were performed on a Carlo Erba EA-1110 CHNS instrument in the Microanalysis Laboratory of the Department of Pharmacy of Genoa University. Compounds were generally characterised by 1H and 13C NMR spectra and elemental analysis or HRMS; a few intermediates were characterised by elemental analysis and 1H NMR.
General procedure for the synthesis of 1 H-benzimidazoles 1, 6
Benzene-1,2-diamine (500 mg, 4.62 mmol) and the appropriate acid (5.55 mmol) were stirred at 145 °C for 24 h under inert atmosphere. The resulting residue was purified by CC (silica gel; eluent as indicated for each compound). These compounds were already obtained through different procedureCitation26,Citation27.
2-Undecyl-1 H-benzimidazole (1): CC (silica gel; cyclohexane/EtOAc; in gradient up to 92:8). The solid residue was rinsed with petroleum ether and the title compound was obtained as a white solid. Yield: 32%. m.p. 108.1–109.3 °C (lit.Citation26, 107.5 °C). 1H NMR (300 MHz, CDCl3): 9.41 (s, 1H, NH). 7.56 (dd, 2H, J = 5.9 and 3.1 Hz), 7.22 (dd, 2H, J = 5.9 and 3.1 Hz), 2.94 (t, 2H, J = 7.7 Hz), 1.91–1.81 (m, 2H), 1.39–1.23 (m, 16H), 0.88 (t, 3H, J = 6.6 Hz). 13C NMR (50 MHz, CDCl3): 154.5, 137.3, 121.1, 113.5, 30.9, 28.6, 28.4, 28.35, 28.3, 27.4, 21.6, 13.1. Anal. Calcd for C18H28N2: C, 79.36; H, 10.36; N, 10.28. Found: C, 79.29; H, 10.53; N, 10.06.
2-Pentadecyl-1 H-benzimidazole (6): CC (silica gel; CH2Cl2/MeOH; in gradient up to 99.4:0.6). The solid residue was rinsed with ethyl ether and the title compound was obtained as a white solid. Yield: 63%. m.p. 88.8–94.2 °C (lit.Citation26, 96.5–97; litCitation27, 98–100 °C). 1H NMR (300 MHz, CDCl3): 9.36 (s, 1H, NH), 7.56 (dd, 2H, J = 5.8 and 3.1 Hz), 7.22 (dd, 2H, J = 5.8 and 3.1 Hz), 2.29 (t, 2H, J = 7.7 Hz), 1.90–1.80 (m, 2H), 1.39–1.24 (m, 24H), 0.88 (t, 3H, J = 6.5 Hz). 13C NMR (50 MHz, CDCl3): 154.1, 136.5, 121.1, 113.6, 30.9, 28.6, 28.4, 28.3, 27.2, 21.6, 13.0. Anal. Calcd for C22H36N2: C, 80.43; H, 11.04; N, 8.53. Found: C, 80.50; H, 11.39; N, 8.42.
General procedure for the synthesis of 2-alkyl-1-methyl-1 H-benzimidazoles (2, 7) and 2-alkyl-1,3-dimethyl-1 H-benzimidazol-3-ium iodides (3, 8)
To a solution of the appropriate 1H-benzimidazole (1 or 6, 0.37 mmol) in anhydrous THF (2 ml), K2CO3 (50.7 mg, 0.37 mmol) and methyl iodide (102 μL, 1.65 mmol) were added. The mixture was stirred at 40 °C for 76 h under inert atmosphere. After cooling at room temperature, inorganic salts were filtered and the solution was evaporated under reduced pressure. The resulting residue was treated with ethyl ether and rinsed with the same solvent giving compound 3 or 8 as a white-cream solid. The ethereal solution was then purified by CC (silica gel; eluent as indicated for each compound). Compounds 2, 3 and 8 were already described in the literatureCitation27–29, obtained by different methods.
1-Methyl-2-undecyl-1H-benzimidazole (2): CC (CH2Cl2; isocratic). The solid residue was rinsed with ethyl ether and the final product was obtained as a white-cream solid. Yield: 45%. m.p. 40.8–43.4 °C (lit.Citation28, yellow oil). 1H NMR (300 MHz, CDCl3): 7.75–7.72 (m, 1 H), 7.41–7.23 (m, 3H), 3.74 (s, 3H), 2.90 (t, 2H, J = 7.7 Hz), 1.87–1.84 (m, 2H), 1.45–0.85 (m, 19H); conforming to the previously described spectrumCitation28.
1,3-Dimethyl-2-undecyl-1 H-benzimidazol-3-ium iodide (3): Yield: 22%. m.p. 157.3–161.3 °C (lit.Citation29 167–168 °C). 1H NMR (300 MHz, CDCl3): 7.68–7.62 (m, 4H), 4.11 (s, 6H), 3.55 (t, 2H, J = 7.2 Hz), 1.74–1.73 (m, 2H), 1.60–1.59 (m, 2H), 1.48–1.47 (m, 2H), 1.25–1.24 (m, 12H), 0.87–0.86 (m, 3H).13C NMR (75 MHz, CDCl3): 154.0, 131.4, 126.7, 112.5, 33.2, 31.6, 29.3, 29.1, 29.0, 28.9, 27.1, 26.1, 22.4, 13.8. HRMS (ESI) m/z Calcd for C20H33N2+ [M]+: 301.2638; found: 301.2637.
1-Methyl-2-pentadecyl-1H-benzimidazole (7): CC (CH2Cl2; isocratic). The solid residue was rinsed with petroleum ether and the final product was obtained as a white-cream solid. Yield: 17%. m.p. 64.2–65.6 °C. 1H NMR (300 MHz, CDCl3): 7.74–7.71 (m, 1H), 7.32–7.23 (m, 3H), 3.74 (s, 3H), 2.88 (t, 2H, J = 7.7 Hz), 1.92–1.82 (m, 2H), 1.45–1.25 (m, 24H), 0.88 (t, 3H, J = 6.6 Hz). Anal. Calcd for C23H38N2: C, 80.64; H, 11.18; N, 8.18. Found: C, 80.44; H, 11.20; N, 7.90.
1,3-Dimethyl-2-pentadecyl-1 H-benzimidazol-3-ium iodide (8): Yield: 43%. m.p. 169.0–172.0 °C (lit.Citation27 187–188 °C). 1H NMR (300 MHz, CDCl3): 7.72–7.70 (m, 2H), 7.69–7.59 (m, 2H), 4.11 (s, 6H), 3.55 (t, 2H, J = 7.7 Hz), 1.77–1.60 (m, 2H), 1.50–1.18 (m, 24H), 0.87 (t, 3H, J = 6.6 Hz).13C NMR (75 MHz, CDCl3): 154.2, 131.6, 126.9, 112.7, 33.3, 31.8, 29.6, 29.5, 29.3, 29.2, 27.3, 26.3, 22.6, 14.1. HRMS (ESI) m/z Calcd for C24H41N2+ [M]+: 357.3264; found: 357.3263.
General procedure for the synthesis of 1-methyl-3-propyl-1 H-benzimidazol-3-ium iodides 4 and 12
To a solution of the appropriate 1-methyl-1H-benzimidazole (2 or 7, 0.16 mmol) in anhydrous THF (1 mL), 1-iodopropane (160 μL, 1.640 mmol) was added. The mixture was stirred at reflux for 24–60 h under nitrogen. After cooling at room temperature, ethyl ether was added to the reaction and the formed solid was then filtered and rinsed with the same solvent giving compound 4 or 12 as a white solid.
1-Methyl-3-propyl-2-undecyl-1 H-benzimidazol-3-ium iodide (4): Yield: 57%. m.p. 140.2–145.0 °C. 1H NMR (300 MHz, CDCl3): 7.71–7.61 (m, 4H), 4.40 (t, 2H, J = 7.2 Hz), 4.17 (s, 3H), 3.54 (t, 2H, J = 6.9 Hz), 2.05–2.03 (m, 2H), 1.74–1.73 (m, 2H), 1.55–1.52 (m, 3H), 1.25–1.24 (m, 13H), 1.10 (t, 3H, J = 7.4 Hz), 0.87–0.86 (m, 3H).13C NMR (75 MHz, CDCl3): 153.7, 131.8, 130.9, 126.8, 112.9, 112.7, 48.1, 33.7, 31.8, 29.5, 29.4, 29.3, 29.2, 29.1, 29.0, 27.1, 26.1, 23.1, 22.6, 14.1, 11.5. HRMS (ESI) m/z Calcd for C22H37N2+ [M]+: 329.2951; found: 329.2949.
1-Methyl-2-pentadecyl-3-propyl-1H-benzimidazol-3-ium iodide (12): Yield: 28%. m.p. 144.7–147.4 °C. 1H NMR (300 MHz, CDCl3): 7.73–7.59 (m, 4H), 4.40 (t, 2H, J = 7.5 Hz), 4.18 (s, 3H), 3.56 (t, 2H, J = 7.7 Hz), 2.08–2.01 (m, 2H), 1.77–1.70 (m, 2H), 1.59–1.53 (m, 3H), 1.40–1.25 (m, 21H), 1.12 (t, 3H, J = 7.4 Hz), 0.87 (t, 3H, J = 6.0 Hz). 13C NMR (75 MHz, CDCl3): 153.7, 131.8, 130.9, 126.8, 112.9, 112.7, 48.1, 33.7, 31.8, 29.6, 29.5, 29.4, 29.3, 29.1, 27.8, 26.1, 23.1, 22.6, 14.1, 11.5. HRMS (ESI) m/z Calcd for C26H45N2+ [M]+: 385.3577; found: 385.3580.
General procedure for the synthesis of 2-alkyl-1,3-dibenzyl-1 H-benzimidazol-3-ium chlorides 5, 15 and 1-benzyl-2-pentadecyl-1 H-benzimidazole 13
To a mixture of K2CO3 (70 mg, 0.50 mmol) and the appropriate 1 H-benzo[d]imidazole (1 or 6, 0.30 mmol) in anhydrous THF (2.5 ml), benzyl chloride (183 μL, 1.52 mmol) was added, then stirred at reflux for 60 h under inert atmosphere. After cooling at room temperature, inorganic salts were filtered and the solution was evaporated under reduced pressure. The resulting residue was treated with THF and rinsed with the same solvent giving compound 5 or 15 as a white-cream solid. The solution was then purified by CC (silica gel; eluent as indicated for each compound).
1,3-Dibenzyl-2-undecyl-1H-benzimidazol-3-ium chloride (5): Yield: 77%. m.p. 223.3–225.3 °C. 1H NMR (300 MHz, CDCl3): 7.60–7.51 (m, 4H), 7.37–7.26 (m, 10H), 5.90 (s, 4H), 3.64 (t, 2H, J = 7.7 Hz), 1.20–0.99 (m, 18H), 0.86 (t, 3H, J = 6.7 Hz). 13C NMR (75 MHz, CDCl3): 155.9, 133.4, 131.7, 129.4, 128.8, 126.9, 126.7, 113.3, 49.8, 31.8, 29.6, 29.4, 29.3, 29.2, 29.1, 28.8, 27.3, 26.1, 22.6, 14.1. HRMS (ESI) m/z Calcd for C32H41N2+ [M]+: 453.3264; found: 453.3257.
1-Benzyl-2-pentadecyl-1H-benzimidazole (13): CC (CH2Cl2; isocratic). The solid residue was rinsed with cold MeOH and the final product was obtained as a white-cream solid. Yield: 17%. m.p. 60.7–61.8 °C. 1HNMR (300 MHz, CDCl3): 7.78–7.75 (d, 1H, J = 7.5 Hz), 7.30–7.18 (m, 6H), 7.05–7.03 (m, 2H), 5.34 (s, 2H), 2.82 (t, 2H, J = 7.6 Hz), 1.87–1.77 (m, 2H), 1.34–1.25 (m, 24H), 0.88 (t, 3H, J = 6.5 Hz). Anal. calcd for C29H42N2: C, 83.20; H, 10.11; N, 6.69. Found: C, 83.16; H, 10.41; N, 6.86.
1,3-Dibenzyl-2-pentadecyl-1 H-benzimidazol-3-ium chloride (15): CC (silica gel; CH2Cl2/MeOH; in gradient up to 99.5:0.5). The solid residue was rinsed with THF and the final product was obtained as a white-cream solid. Yield: 40%. m.p. 214.9–216.3 °C. 1H NMR (300 MHz, CDCl3): 7.56–7.54 (m, 4H), 7.36–7.29 (m, 10H), 5.90 (s, 4H), 3.67–3.65 (m, 2H), 1.25–0.87 (m, 29H). 13C NMR (75 MHz, CDCl3): 156.0, 133.3, 131.6, 129.4, 128.8, 126.9, 126.7, 113.2, 49.8, 31.8, 29.7, 29.6, 29.5, 29.4, 29.3, 29.0, 28.8, 27.3, 26.1, 22.6, 14.1. HRMS (ESI) m/z Calcd for C36H49N2+ [M]+: 509.3890; found: 509.3878.
3-Benzyl-1-methyl-2-pentadecyl-1 H-benzimidazol-3-ium chloride (14)
Benzyl chloride (168 μL, 1.43 mmol) was added to a solution of 1-methyl-2-pentadecyl-1H-benzimidazole (compound 7, 0.15 mmol) in anhydrous THF (1 ml). The reaction was stirred at reflux for 80 h under inert atmosphere. After cooling at room temperature, the formed solid was filtered and rinsed first with THF and then with ethyl ether, providing compound 14 as a white solid. Yield: 18%. m.p. 215.7–219.3 °C. 1H NMR (300 MHz, CDCl3): 7.77–7.26 (m, 9H), 5.84 (s, 2H), 6.87 (s, 3H), 3.67 (s, 2H), 1.34–1.14 (m, 26H), 0.88 (t, 3H, J = 6.1 Hz). 13C NMR (75 MHz, CDCl3): 155.5, 133.5, 131.8, 131.5, 129.4, 128.9, 126.9, 126.8, 112.9, 112.8, 77.0, 49.7, 32.9, 31.9, 29.7, 29.6, 29.5, 29.4, 29.3, 29.2, 29.1, 27.2, 25.8, 22.7, 14.1. HRMS (ESI) m/z Calcd for C30H45N2+ [M]+: 433.3577; found: 433.3576.
1-(Ferrocenylmethyl)-2-pentadecyl-1 H-benzimidazole (16)
To a mixture of K2CO3 (63 mg, 0.45 mmol) and 2-pentadecyl-1H-benzimidazole (compound 6, 0.30 mmol) in anhydrous THF/CH3CN (1:3.5 mL), ferrocenylmethyl trimethylammonium iodide (117 mg, 0.46 mmol) was added. The reaction was stirred at room temperature for 18 h under inert atmosphere. After the completion of reaction, the solution was evaporated under reduced pressure. The resulting residue was taken up with CH2Cl2 and washed several times with H2O. The organic layer was dried with anhydrous Na2SO4, filtered and evaporated to dryness to obtain a pale orange oil, which was purified by CC (silica gel; CH2Cl2; isocratic). The title compound was obtained as a pale yellow solid. Yield: 75%. m.p. 77.2–78.7 °C. 1H NMR (300 MHz, CDCl3): 7.79 (d, 1H, J = 6.3 Hz), 7.38 (s, 1H), 7.23–7.22 (m, 2H), 5.05 (s, 2H), 4.42–4.11 (m, 9H), 2.90 (t, 2H, J = 7.1 Hz), 1.89–1.88 (m, 2H), 1.64–1.25 (m, 24H), 0.88–0.85 (m, 3H). Hydrochloride: m.p. 149.8–150.2 °C. Anal. calcd for C33H47ClFeN2: C, 70.40; H, 8.41; N, 4.98. Found: C, 70.42; H, 8.91; N, 5.02.
1-(Ferrocenylmethyl)-3-methyl-2-pentadecyl-1 H-benzimidazol-3-ium iodide (17)
Methyl iodide (200 μL, 3.24 mmol) was added to a solution of 1-ferrocenyl-3-methyl-2-pentadecyl-1 H-benzo[d]imidazole (compound 16, 0.09 mmol) in anhydrous ethyl ether (1.5 mL). The reaction was stirred at 40 °C for 80 h under inert atmosphere. After cooling at room temperature, the formed solid was filtered and rinsed with ethyl ether giving compound 17 as a white solid. Yield: 52%. m.p. 162.2–165.7 °C. 1H NMR (300 MHz, CDCl3): 7.74–7.73 (m, 1H), 7.60–7.59 (m, 3H), 5.55 (s, 2H), 4.36–4.21 (m, 9H), 4.05 (s, 3H), 3.53–3.52 (m, 2H), 1.52–1.51 (m, 4H), 1.26–1.24 (m, 22H), 0.88–0.87 (m, 3H). 13C NMR (75 MHz, CDCl3): 153.8, 131.6, 130.9, 126.7, 113.0, 112.6, 79.3, 69.4, 69.3, 47.1, 33.1, 31.9, 29.7, 29.6, 29.5, 29.3, 29.2, 27.5, 26.6, 22.6, 14.1. HRMS (ESI) m/z Calcd for C34H49N2Fe+ [M]+: 541.3240; found: 541.3234.
2-(Heptan-4-yl)-1 H-benzimidazole (18)
2-Propylpentanoyl chloride (338 mg, 2.08 mmol) was added at 0 °C to a solution of benzene-1,2-diamine (225 mg, 2.08 mmol) in anhydrous 1,4-dioxane (1 mL) and the reaction mixture was stirred at room temperature for 15 h under nitrogen. After that time, BF3.Et2O (263 μL) was added and the mixture was stirred at reflux for other 12 h. The solvent was then stripped off and the obtained residue was diluted with EtOAc, washed with a cold solution of 5% HCl and with 2 M NaOH. The organic layer was dried with anhydrous Na2SO4, filtered and evaporated to dryness to obtain a residue that was purified by CC (silica gel; CH2Cl2/cyclohexane; in gradient up to 80:20). The fractions containing the purified product were gathered up and rinsed with diethyl ether to provide a white solid. Yield: 18%. m.p. 224.7–225.8 °C. 1H NMR (300 MHz, acetone-d6): 7.49–7.46 (m, 2H), 7.39 (s, 1H), 7.13–7.11 (m, 2H), 3.03–2.95 (m, 1H), 1.91–1.81 (m, 2H), 1.79–1.64 (m, 2H), 1.33–1.20 (m, 4H), 0.89–0.84 (m, 6H). 13C NMR (50 MHz, CDCl3): 157.8, 136.8, 121.2, 113.6, 39.4, 36.1, 19.2, 12.9. Anal. calcd for C14H20N2: C, 77.73; H, 9.32; N, 12.95. Found: C, 77.71; H, 9.66; N, 12.83.
General procedure for the synthesis of 2-(heptan-4-yl)-1-methyl-1 H-benzimidazole 19 and 1,3-dimethyl-2-(heptan-4-yl)-1 H-benzimidazol-3-ium iodide 20
To a mixture of K2CO3 (33.0 mg, 0.24 mmol) and 2-(heptan-4-yl)-1H-benzimidazole (compound 18, 0.24 mmol) in anhydrous THF (1 mL), methyl iodide (814 μL, 13.14 mmol) was added. The reaction was stirred at 40 °C for 26 h under inert atmosphere. After cooling at room temperature, inorganic salts were filtered and the solution was evaporated under reduced pressure. The resulting residue was treated with ethyl ether and rinsed with the same solvent giving compound 20 as a white solid. The ethereal solution was then purified by CC (silica gel; eluent as indicated for each compound).
2-(Heptan-4-yl)-1-methyl-1 H-benzimidazole (19): CC (CH2Cl2; isocratic). The title compound was obtained as a pale grey oil. Yield: 47%. 1H NMR (300 MHz, CDCl3): 7.80–7.77 (m, 1H), 7.33–7.24 (m, 3H), 3.76 (s, 3H), 3.03–2.97 (m, 1H), 2.01–1.89 (m, 2H), 1.81–1.70 (m, 2H), 1.34–1.17 (m, 4H), 0.87 (t, 6H, J = 7.4 Hz). 13C NMR (50 MHz, CDCl3): 155.6, 130.7, 129.9, 125.4, 124.9, 114.9, 110.0, 36.6, 34.9, 30.5, 19.9, 12.7. Hydrochloride, m.p. 169.2–173.2 (EtOH/Et2O). Anal. calcd for C15H23ClN2: C 67.51, H 8.69, N 10.50, found: C 67.71, H 8.88, N 10.40.
1,3-Dimethyl-2-(heptan-4-yl)-1 H-benzimidazol-3-ium iodide (20): White powder. Yield: 25%. m.p. 192.2–192.9 °C. 1H NMR (300 MHz, CDCl3): 7.88–7.86 (m, 2H), 7.68–7.65 (m, 2H), 4.25 (m, 6H), 3.78–3.73 (m, 1H), 2.06–1.98 (m, 2H), 1.60–1.43 (m, 4H), 1.22–1.18 (m, 2H), 0.98–0.93 (t, 6H, J = 7.1 Hz). 13C NMR (75 MHz, CDCl3): 154.9, 133.1, 131.6, 127.4, 37.3, 34.5, 21.4, 13.9. HRMS (ESI) m/z Calcd for C16H25N2+ [M]+: 245.2012; found: 245.2011.
General procedure for the synthesis of N-(2-aminophenyl)palmitamide derivatives 40–42
To a solution of the proper 4- or 4,5-substituted 1,2-phenylendiamine (2.5 mmol) in THF (8 mL) in presence of Hunig base (5 mmol), a solution of palmitoyl chloride (2.5 mmol) in 5 ml of THF was added dropwise. The mixture was reacted at r.t. for 24 h with stirring. After removing the solvent, the residue was taken up with water, alkalinised with 2 N NaOH and exhaustively extracted with CHCl3. The dried organic layer (Na2SO4) was concentrated to dryness leaving a residue that was thoroughly washed with dry Et2O/hexane (1:1).
N-[2-Amino-4(5)-trifluoromethylphenyl]palmitamide (40): White powder. Yield: 42%. m.p. 71–73.5 °C (hexane/Et2O an.). 1H NMR (200 MHz, CDCl3): 7.63 (s, 1H, NHCO, collapses with D2O), 7.39 (s, 1H), 7.26 (d, 1H, J = 8.8 Hz), 6.75 (d, 1H, J = 8.8 Hz), 3.92 (s, 2H, NH2, collapse with D2O), 2.41 (t, 2H, J = 7.8 Hz), 1.87–1.56 (m, 2H), 1.29 (pseudo s, 24H), 0.91 (t, 3H, J = 6.8 Hz). Anal. calcd for C23H37F3N2O: C, 66.64; H, 9.00; N, 6.76. Found: C, 66.72; H, 9.09; N, 6.85.
N-[2-Amino-4(5)-nitrophenyl]palmitamide (41): Yellowish powder. Yield: 29%. m.p. 142–143 °C (hexane/Et2O an.). 1H NMR (200 MHz, CDCl3): 8.06 (br. s, 1H and 1H, NHCO, collapses with D2O, superimposed), 7.42 (m, 1H), 6.82 (d, 1H, J = 8.8 Hz), 3.40 (s, 2H, NH2, collapse with D2O), 2.49 (pseudo s, 2H), 1.96–1.70 (m, 2H), 1.30 (pseudo s, 24 H), 0.92 (pseudo s, 3H). Anal. calcd for C22H37N3O3: C, 67.49; H, 9.53; N, 10.73. Found: C, 67.74; H, 9.57; N, 10.93.
N-(2-Amino-4,5-dichlorophenyl)palmitamide (42): White powder. Yield: 35%. m.p. 96–98 °C (hexane/Et2O an.). 1H NMR (200 MHz, CDCl3): 7.40 (s, 1H, NHCO, collapses with D2O), 7.36 (s, 1H), 6.95 (s, 1H), 3.38 (s, 2H, NH2, collapse with D2O), 2.42 (t, 2H, J = 7.8 Hz), 1.82–1.54 (m, 2H), 1.26 (pseudo s, 24H), 0.91 (t, 3H, J = 7.0 Hz). Anal. calcd for C22H36Cl2N2O: C, 63.60; H, 8.73; N, 6.74. Found: C, 63.51; H, 8.73; N, 7.00.
General procedure for the synthesis of 2-pentadecyl-5/6-1H-benzimidazole derivatives 43–45
The N-(2-aminophenyl)palmitamides (0.50 mmol) in 4 N HCl (10 mL) were refluxed at 120 °C for 4 h. After cooling, the acidic solution was basified with 2 N NaOH and shaken with CH2Cl2. The organic layer was dried (Na2SO4) and evaporated to afford the benzimidazole that was thoroughly washed with dry Et2O/hexane (1:1). 2-Pentadecyl-5-trifluoromethyl-1H-benzimidazole was yield as an oil and was used as such for the preparation of the corresponding 1-methylbenzimidazole derivative.
2-Pentadecyl-5-trifluoromethyl-1 H-benzimidazole (43): Yield: 76%. Oil. 1H NMR (200 MHz, CDCl3): 9.24 (s, 1H, NH, collapses with D2O), 8.21 (s, 1H), 8.08 (d, 1H, J= 8.6 Hz), 7.80 (d, 1H, J = 8.6 Hz), 2.45 (t, 2H, J = 7.0 Hz), 1.84–1.1.59 (m, 2H), 1.29 (pseudo s, 24H), 0.91 (t, 3H, J = 6.8 Hz). Anal. calcd for C23H35F3N2: C, 69.67; H, 8.90; N, 7.06. Found: C, 69.45; H, 9.00; N, 8.75.
5-Nitro-2-pentadecyl-1 H-benzimidazole (44): Yield: 45%. m.p. 86–88 °C (hexane/Et2O an.). 1H NMR (200 MHz, CDCl3): 9.54 (s, 1H, NH, collapses with D2O), 8.52 (s, 1H), 8.21 (d, 1H, J = 9.0 Hz), 7.64 (d, 1H, J = 8.8 Hz), 3.04 (t, 2H, J = 8.0 Hz), 2.03–1.80 (m, 2H), 1.26 (pseudo s, 24H), 0.90 (t, 3H, J = 6.8 Hz). Anal. calcd for C22H35N3O2: C, 70.74; H, 9.44; N, 11.25. Found: C, 70.60; H, 9.59; N, 11.55.
5,6-Dichloro-2-pentadecyl-1 H-benzimidazole (45): Yield: 44%. m.p. 74–76 °C (hexane/Et2O an.). 1H NMR (200 MHz, CDCl3): 9.38 (s, 1H, NH, collapses with D2O), 7.60 (s, 1H), 7.26 (s, 1H), 3.01 (t, 2H, J = 8.0 Hz), 2.01–1.80 (m, 2H), 1.23 (pseudo s, 24H), 0.90 (t, 3H, J = 6.6 Hz). Anal. calcd for C22H34Cl2N2: C, 66.49; H, 8.62; N, 7.05. Found: C, 66.70; H, 8.95; N, 7.35.
General procedure for the synthesis of N-methyl-1 H-benzimidazole derivatives 46–48, 50 and 51
In a sealed tube, to a solution of the proper benzimidazole (0.10 mmol) in 5 mL of THF were added, in the order, Cs2CO3 (0.30 mmol) and iodomethane (0.15 mmol). The mixture was heated at 60 °C for 6–8 h with stirring. The solvent was evaporated and the residue was taken up with water, alkalinised with 2 N NaOH and extracted with CH2Cl2. After drying, the solvent was removed obtaining an oily residue that was washed with hexane.
N-Methyl-2-pentadecyl-5(6)-trifluoromethyl-1 H-benzimidazole (46): White powder. Yield: 90%. m.p. 65.8–67.9 °C (hexane). 1H NMR (200 MHz, CDCl3): 8.03 (s, 1H), 7.81 (d, 1H, J= 9.6 Hz), 7.51 (d, 1H, J = 9.6 Hz), 3.79 (s, 3H, NCH3), 2.90 (t, 2H, J = 8.0 Hz), 2.01–1.80 (m, 2H), 1.28 (pseudo s, 24H), 0.89 (t, 3H, J = 6.4 Hz). Anal. calcd for C24H37F3N2: C, 70.21; H, 9.08; N, 6.82. Found: C, 69.72; H, 9.23; N, 6.00.
N-Methyl-5(6)-nitro-2-pentadecyl-1 H-benzimidazole (47): Yellowish powder. Yield: 45%. m.p. 69.7–71.4 °C (hexane). 1H NMR (200 MHz, CDCl3): 8.55 (s, 1H), 8.38 (d, 1H, J= 9.8 Hz), 7.68 (d, 1H, J = 9.8 Hz), 4.09 (s, 3H, NCH3), 3.28 (t, 2H, J = 7.8 Hz), 2.09–1.85 (m, 2H), 1.28 (pseudo s, 24H), 0.90 (t, 3H, J = 6.2 Hz). Anal. calcd for C23H37N3O2: C, 71.28; H, 9.62; N, 10.84. Found: C, 71.28; H, 9.67; N, 11.18.
N-Methyl-5,6-dichloro-2-pentadecyl-1H-benzimidazole (48): White powder. Yield: 42%. m.p. 64.8–67.3 °C (hexane). 1H NMR (200 MHz, CDCl3): 8.15 (s, 1H), 7.69 (s, 1H), 3.97 (s, 3H, NCH3), 3.28 (t, 2H, J = 8.4 Hz), 2.06–1.87 (m, 2H), 1.28 (pseudo s, 24H), 0.91 (t, 3H, J = 6.4 Hz). Anal. calcd for C23H36Cl2N2: C, 67.14; H, 8.82; N, 6.81. Found: C, 67.17; H, 8.80; N, 7.15.
N-Methyl-2–(4-chlorobenzyl)-1 H-benzimidazole (50): White powder. Yield: 23%. m.p. 117–119 °C (hexane) conforming to the literatureCitation30.
N-Methyl-2-(4-chlorobenzyl)-5-trifluoromethyl-1 H-benzimidazole (51): White powder. Yield: 100%. Oil. 1H NMR (200 MHz, CDCl3): 7.80–7.04 (m, 7H), 4.36 (s, 2H), 3.95 (s, 3H, NCH3). Anal. calcd for C16H12ClF3N2: C, 59.18; H, 3.72; N, 8.63. Found: C, 59.30; H, 3.65; N, 8.49.
General procedure for the synthesis of benzimidazole quaternary ammonium salts 9–11, 21, 22 and 39
The suitable N-methylbenzimidazole derivative or N-lupinyl-5-trifluoromethyl-2-(4-chlorophenyl)benzimidazole (0.20 mmol) was reacted with iodomethane (0.5 mL, 8 mmol) at r.t. for 24 h with stirring. The reaction mixture was washed with dry Et2O affording the title quaternary ammonium salt.
1,3-Dimethyl-2-pentadecyl-5-trifluoromethyl-1 H-benzimidazol-3-ium iodide (9): Yield: 83%. m.p. 116–118 °C (Et2O an.). 1H NMR (200 MHz, CDCl3): 8.10–7.70 (m, 3H), 4.15 (s, 6H), 3.52 (t, 2H, J = 7.05 Hz), 1.93–1.64 (m, 2H), 1.57–0.80 (m, 27H). 13C NMR (50 MHz, CDCl3): 156.2, 132.7, 130.3, 122.7, 113.2, 109.6, 33.0, 32.6, 30.9, 28.6, 28.5, 28.4, 28.3, 28.1, 26.2, 26.0, 21.6, 13.1. Anal. calcd for C25H40F3IN2: C, 54.35; H, 7.30; N, 5.07. Found: C, 54.33; H, 6.92; N, 5.21.
1,3-Dimethyl-5-nitro-2-pentadecyl-1 H-benzimidazol-3-ium iodide (10): Yield: 51%. m.p. 154–156 °C (Et2O an.). 1H NMR (200 MHz, CDCl3): 8.63 (s, 1H), 8.29–8.17 (m, 1H), 7.88 (d, 1H, J = 8.10 Hz), 4.21 (s, 6H), 2.94 (t, 2H, J = 7.15 Hz), 2.00–0.80 (m, 29H). 13C NMR (50 MHz, CDCl3): 157.0, 133.0, 129.7, 117.0, 114.8, 107.7, 30.9, 29.2, 28.6, 28.4, 28.3, 26.7, 26.3, 21.7, 13.1; Anal. calcd for C24H40IN3O2: C, 54.44; H, 7.61; N, 7.94. Found: C, 54.37; H, 7.63; N, 7.98.
5,6-Dichloro-1,3-dimethyl-2-pentadecyl-1 H-benzimidazol-3-ium iodide (11): Yield: 100%. m.p. 200–203 °C (Et2O an.). 1H NMR (200 MHz, CDCl3): 7.96 (s, 2H), 4.14 (s, 6H), 3.51 (t, 2H, J = 7.15 Hz), 1.84–0.75 (m, 29H). 13C NMR (50 MHz, CDCl3): 155.5, 130.8, 129.7, 113.4, 32.4, 30.9, 28.6, 28.3, 26.2, 21.7, 13.1. Anal. calcd for C24H39Cl2IN2: C, 52.09; H, 7.10; N, 5.06. Found: C, 52.18; H, 7.17; N, 5.41.
2-(4-Chlorobenzyl)-1,3-dimethyl-1 H-benzimidazol-3-ium iodide (21): Yield: 35%. m.p. 94–98 °C (Et2O an.). 1H NMR (200 MHz, CDCl3): 8.10–7.00 (m, 8H), 4.32 (s, 3H), 4.17 (s, 3H), 3.45 (s, 2H). 13C NMR (50 MHz, CDCl3): 128.6, 126.6, 126.0, 120.2, 111.9, 106.3, 64.0, 32.5, 26.1. Anal. calcd for C16H16ClIN2: C, 48.20; H, 4.05; N, 7.03. Found: C, 42.45; H, 4.90; N, 6.16.
2-(4-Chlorobenzyl)-1,3-dimethyl-5-trifluoromethyl-1 H-benzimidazol-3-ium iodide (22): Yield: 41%. m.p. 60 °C (Et2O an.). 1H NMR (200 MHz, CDCl3): 8.16–7.08 (m, 7H), 4.25 (s, 3H), 4.18 (s, 3H), 3.58 (s, 2H). 13C NMR (50 MHz, CDCl3): 153.4, 132.7, 130.4, 128.7, 126.1, 123.2, 113.4, 110.0, 64.1, 33.4, 33.1, 30.9, 28.9. Anal. calcd for C17H15ClF3IN2: C, 43.75; H, 3.24; N, 6.00. Found: C, 43.92; H, 3.49; N, 6.00.
2-(4-Chlorophenyl)-3-methyl-1-{[5-methylammonio-(1 S,9aR)-octahydroquinolizin-1-yl]- methyl}-5-trifluoromethyl-1 H-benzimidazol-3-ium diiodide (39): Yield: 35%. m.p. 165–169 °C (Et2O an.). 1H NMR (200 MHz, DMSO): 8.75 (s, 1 arom. H), 8.56 (d, J = 9.0, 1H), 8.07 (d, J = 8.5, 2H), 7.91 (d, J = 8.5, 2H), 7.26 (s, 1H), 3.93 (s, 3H), 3.16 (s, 3H), 3.11–2.88 (m, 2H), 2.78–2.63 (m, 2H), 2.17–1.05 (m, 14H). 13C NMR (DMSO): 151.5, 138.2, 133.1, 132.3, 131.5, 130.0, 127.2, 123.0, 119.0, 115.1, 66.0, 64.5, 50.3, 49.1, 47.2, 33.4, 33.0, 20.6, 19.1, 18.7, 18.1. Anal. calcd for C26H31ClF3I2N3: C, 40.52; H, 4.97; N 5.47. Found: C, 40.67; H, 4.59; N 5.47.
Evaluation of anti-leishmanial activity
Promastigote stage of L. infantum strain MHOM/TN/80/IPT1 (kindly provided by Dr M. Gramiccia, ISS, Roma) and L. tropica (MHOM/IT/2012/ISS3130) were cultured in RPMI 1640 medium (EuroClone) supplemented with 10% heat-inactivated fetal calf serum (EuroClone), 20 mM Hepes, and 2 mM L-glutamine at 24 °C.
(b) In vitro intracellular amastigote susceptibility assays. THP-1 cells (human acute monocytic leukaemia cell line) were maintained in RPMI supplemented with 10% FBS, 50 μM 2-mercaptoethanol, 20 mM Hepes, 2 mM glutamine, at 37 °C in 5% CO2. For Leishmania infections, THP-1 cells were plated at 5 × 105 cells/mL in 16-chamber Lab-Tek culture slides (Nunc) and treated with 0.1 μM phorbol myristate acetate (PMA, Sigma) for 48 h to achieve differentiation into macrophages. Cells were washed and infected with metacyclic L. infantum promastigotes at a macrophage/promastigote ratio of 1/10 for 24 h. Cell monolayers were then washed and incubated with compounds for 72 h. Slides were fixed with methanol and stained with Giemsa. The percentage of infected macrophages in treated and non-treated cells was determined by light microscopy.
Cell cytotoxicity assays
(a) The long-term human microvascular endothelial cell line (HMEC-1) was maintained in MCDB 131 medium (Invitrogen, Milan, Italy) supplemented with 10% fetal calf serum (HyClone, Celbio, Milan, Italy), 10 ng/mL of epidermal growth factor (Chemicon), 1 µg/mL of hydrocortisone, 2 mM glutamine, 100 U/mL of penicillin, 100 l g/mL of streptomycin and 20 mM Hepes buffer (EuroClone). Unless stated otherwise, all reagents were from Sigma Italia, Milan, Italy. For the cytotoxicity assays, cells were treated with serial dilutions of test compounds and cell proliferation evaluated using the MTT assay already describedCitation33. The results are expressed as IC50, which is the dose of compound necessary to inhibit cell growth by 50%.
(b) Vero-76 cells were seeded at an initial density of 4 × 105 cells/mL in 24-well plates, in culture medium (Dulbecco’s Modified Eagle Medium (D-MEM) with l-glutamine, supplemented with foetal bovine serum (FBS), 0.025 g/L kanamycin). Cell cultures were then incubated at 37 °C in a humidified, 5% CO2 atmosphere in the absence or presence of serial dilutions of test compounds. Cell viability was determined after 48–96 h at 37 °C by the Crystal violet staining method.
The results are expressed as CC50, which is the concentration of compound necessary to inhibit cell growth by 50%. Each CC50 value is the mean and standard deviation of at least three separate experiments performed in duplicate.
Results and discussion
Synthesis
The 1-unsubstituted 2-alkylbenzimidazoles were prepared either by dry heating at 145 °C of a mixture of 1,2-phenylenediamine with the suitable acid (1 and 6), or by treating the diamine with valproyl chloride, in dioxane solution, followed by the action of ethereal boron trifluoride (18) (Schemes 1 and 4). The last method, in contrast with the indication of Tandon and KumarCitation34, gave only a modest yield of benzimidazole, being prevailing the formation of the N,N’-divalproyl-1,2-phenylendiamine (49). Compounds 1 and 6 were already described (see Materials and methods).
The treatment of the 1-unsubstituted benzimidazoles with excess of methyl iodide, in the presence of anhydrous K2CO3, gave place to mixtures of 1-methyl-2-substituted benzimidazoles (2, 7 and 19) and 1,3-dimethyl-2-substituted benzimidazolium iodides (3, 8 and 20) (Schemes 1 and 4). The dimethylated compounds were easily isolated being insoluble in dry ether, while the monomethylated compounds were separated from the N-unsubstituted benzimidazoles by CC on silica, eluting with CH2Cl2. Similarly, by treating compounds 1 and 6 with an excess of benzyl chloride the 1,3-dibenzyl benzimidazolium chlorides 5 and 15 were obtained, but the mono-benzylated compound (13) was isolated only in the case of 6 (Scheme 1). Compounds 2, 3 and 8 were already described (see Materials and methods).
Scheme 1. Reagents and conditions: (a) 145 °C, N2, 24 h; (b) CH3I, THF, K2CO3, 40 °C, 76 h; (c) C3H7I, THF, 24–60 h; (d) C6H5–CH2–Cl, THF, K2CO3, N2, reflux, 60 h; (e) C6H5–CH2–Cl, THF, N2, reflux, 80 h.
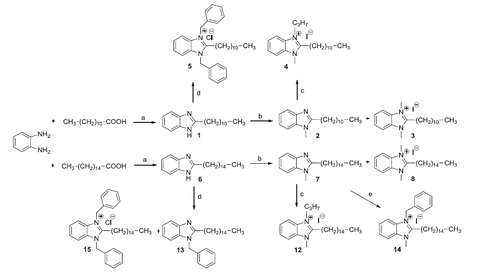
An attempt to improve the yield of 1-methyl-2-pentadecyl benzimidazole (7) by reacting directly the palmitic acid with N-methyl-1,2-phenylendiamine gave disappointing result (yield 17%).
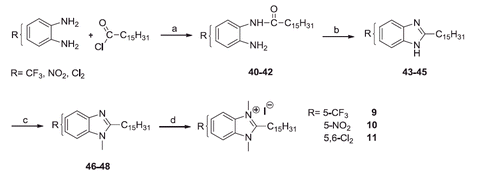
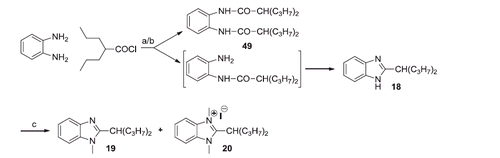
To obtain the 5-substituted compounds 9–11, the 4-substituted or 4,5-disubstituted-1,2-phenylenediamines were mono-acylated with palmitoyl chloride and the monoamides 40–42 were cyclised by the action of 4 N HCl. The benzimidazoles 43–45 were methylated with methyl iodide in the presence of Cs2CO3 (46–48) and, finally, quaternised at r.t. with excess of methyl iodide (Scheme 2). The intermediates 40, 41, 46 and 47 (Scheme 2) could be a mixture of two regioisomers, however we did not succeed in separating them, but it is not important for the structures of the final compounds 9–11.
Scheme 2. Reagents and conditions: (a) THF, N2, Hünig base (2 equiv), r.t., 24 h; (b) HCl 4 N, reflux, 4 h; (c) CH3I, THF, Cs2CO3, 60 °C, 6–8 h; (d) CH3I excess, r.t., 24 h.
The mono-methylated benzimidazoles 2 and 7 were converted into the quaternary salts 4, 12 and 14 (Scheme 1), by heating them with propyl iodide or with benzyl chloride for the latter. As suggested by Howarth and HanlonCitation35 for analogous compounds, by treating the 2-pentadecyl benzimidazole with (ferrocenylmethyl)trimethyl ammonium iodide at r.t., the 1-ferrocenylmethylbenzimidazole 16 was obtained in high yield, the latter was then quaternised with methyl iodide to 17 (Scheme 3).
Scheme 3. Reagents and conditions: (a) CH3CN/THF, K2CO3, N2, r.t., 18 h; (b) CH3I, dry Et2O, 40 °C, 80 h.
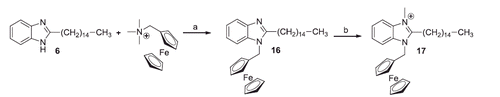
Scheme 4. Reagents and conditions: (a) dioxane, N2, r.t., 15 h; (b) BF3*Et2O, reflux, 12 h; (c) CH3I, THF, K2CO3, 50 °C, 26 h.
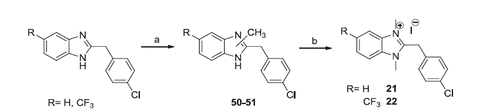
Finally, by treating the 2-(4-chlorobenzyl)benzimidazoleCitation36 and 2-(4-chlorobenzyl)-5-trifluoromethylbenzimidazoleCitation37 with methyl iodide in the presence of Cs2CO3, the corresponding 1-methylbenzimidazoles were obtained, that with excess of methyl iodide gave the quaternary salts 21 and 22 (Scheme 5).
Scheme 5. Reagents and conditions: (a) CH3I, THF, Cs2CO3, 60 °C, 6 h; (b) CH3I excess, r.t., 24 h.
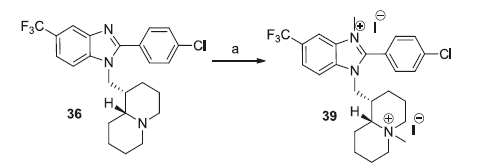
All but one (39) of the benzimidazole derivatives bearing a basic side chain were already described by some of us: 24, 28 and 32–34Citation21a; 23, 25 and 29–31Citation21b; 26 and 27Citation24c; 35, 36 and 38Citation21c; 37Citation25b. The novel bisquaternary salt 39 was obtained by treating with methyl iodide the previously described benzimidazole derivative 36Citation21c (Scheme 6). Attempts of selective quaternisation of quinolizidine nitrogen were unsuccessful.
Scheme 6. Reagents and conditions: (a) CH3I excess, r.t., 24 h.
Antileishmanial activity
With the exception of compound 2, all the (38) compounds of and were tested in vitro against promastigotes of L. tropica, while 33 of them were also tested against L. infantum, using the MTT assayCitation31,Citation32. Results are expressed as IC50 ± SD (µM) and reported in , together with the corresponding selectivity indexes (ratio of IC50 versus human microvascular endothelial cell line (HMEC-1), or monkey kidney cells (Vero76), and IC50 of compounds versus the two Leishmania species.
Table 1. In vitro data on antileishmanial activity against L. tropica and L. infantum promastigotes and cytotoxicity on the human endothelial cell line (HMEC-1) and/or monkey kidney cell (Vero-76) of benzimidazole derivatives 1 and 3–39.
The results collected in show that most of the tested compounds were active against L. tropica (30 over 38) and L. infantum (25 over 33). Among the compounds considered inactive (1, 7, 13, 16, 20, 21, 25 and 39), two (1 and 13) were tested only at concentrations up to 16 and 12 μM, respectively, and it is not excluded that they could exhibit some activity at higher concentrations. The active compounds resulted less potent than the reference drug amphotericin B, reaching, at the best, the 43% of its potency versus L. tropica (cpd 8) and the 58% versus L. infantum (cpd 4), respectively. However, comparing the tested compounds with miltefosine, another commonly used drug, they frequently resulted many fold (up to 228-fold) more potent. It is worth noting that in our experimental conditions miltefosine displayed an IC50 value versus the promastigote stage of L. infantum (31.26 μM) quite higher than the corresponding values found in the literature (15.0 μMCitation11a; 16.7 μMCitation6c; 19.6 μMCitation13b), while no data are available in the literature for miltefosine activity versus L. tropica to compare with our results (43 μM). Indeed, substantial variability has been observed for miltefosine susceptibility of several other Leishmania speciesCitation38. Anyhow, even taking into account the lowest IC50 value (15 μM) aforementioned, most of the tested compounds remain many-fold (up to 55- and 43-fold for compounds 4 and 8, respectively) more potent than miltefosine against L. infantum.
L. infantum was commonly (with the exception of compounds 4, 5, 10–12 and 22) less sensitive than L. tropica, which in two cases (compounds 23 and 26) was the only affected species.
Activity was largely present in both subsets of compounds, but the higher potencies (IC50≤ 5 µM) were mainly found among the 2-undecyl- and 2-pentadecylbenzimidazole derivatives, in which subset the activity was particularly high (IC50 minor or around 1 µM) when the benzimidazole ring was quaternised (compounds 3–5, 8–12 and 14).
Considering the quite different structural features that characterise the two subsets of compounds, the structure–activity relationships will be discussed separately for each subset. The distribution of activity among the two subsets is illustrated in .
Figure 6. Number of compounds inhibiting the growth of L. tropica and L. infantum promastigotes and range of their IC50 (µM).
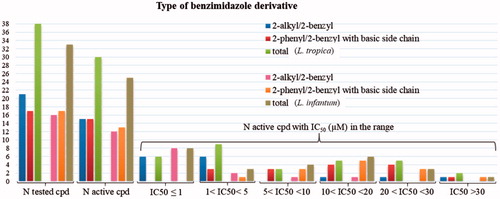
Regarding the subset of benzimidazole derivatives bearing in position 2 an aliphatic chain, it is observed that the 1-unsubstituted-2-alkylbenzimidazoles (1, 6 and 18) were not only either inactive or only moderately active, but also the least toxic versus HMEC-1 cells. The introduction in position 1 of a methyl, benzyl and ferrocenylmethyl residue abolished (7, 13 and 16) or reduced (19) the activity. However generating a fixed positive charge on the benzimidazole ring of the aforementioned compounds, by treating them with methyl or propyl iodide or benzyl chloride, a striking increase of activity was observed, obtaining compounds with IC50 in submicromolar (4, 5, 8, 12 and 14) or low micromolar range (3, 9–11, 15 and 17). Somewhat unexpected was the lack of activity observed for the quaternised compound 20 (1,3-dimethyl-2-(4-heptyl)benzimidazolium iodide), which was inactive even at a concentration up to 73 µM. Commonly, the quaternisation increased both the activity and the cytotoxicity, while quaternising compound 19 to 20, its activity was abolished leaving unchanged the low cytotoxicity.
In this subset of benzimidazole derivatives, compounds 4 and 8 were the most potent versus L. infantum and L. tropica, respectively, with IC50= 0.27 and 0.19 µM corresponding to the 58 and 28% of amphotericin B potency with respect to the two Leishmania species. In comparison to miltefosine, compound 4 was 116-fold more effective versus L. infantum, whilst compound 8 was 228 more potent versus L. tropica. The introduction of electron-withdrawing substituents on the benzimidazole ring reduced the activity (compare 8 with 9–11), but the activity-lowering effect was stronger versus L. tropica than versus L. infantum. However, comparing the couple of compounds 8–9 to 21–22, where the pentadecyl chain is replaced by a 4-chlorobenzyl moiety, it is observed that the introduction of a 5-trifluoromethyl group had a positive effect on the activity. Also in this case the activity on L. infantum was higher than on L. tropica. The toxicity of 4 and 8 (and similar compounds) versus the HMEC cells was not negligible, with selectivity index (SI) in the range 2.3–7.4, that, however, were better than the corresponding SI of miltefosine (2.0 and 3.2). Indeed, the HMEC cells are particularly sensitive to most kinds of chemicals, thus the best compounds 4 and 8 were also tested for toxicity against Monkey kidney Vero76 cells, sharing a quite more valuable SI value. Interestingly, the 1-unsubstituted benzimidazole 6, even displaying a moderate activity, exhibited a very valuable SI versus the sensitive HMEC cells (SI> 37 and >13). Thus, compounds 4, 6 and 8 represent interesting hit compounds for developing better anti-leishmania agents by increasing activity or reducing toxicity through further chemical manipulation (chain length, chain branching and unsaturation, number and nature of substituents on the benzimidazole and eventual benzyl group).
Compound 8 and its analogues (3–5, 9–12, 14, 15 and 17) may display their activity (as well as their toxicity) acting as cationic surfactants able to modify, like miltefosineCitation38, the cell membrane permeability; moreover, once inside the cell, they may activate several stress pathways, inhibit fatty acids and sterol biosynthesis, and/or cytochrome-C oxidase and other targets. Moreover, it is known that quaternary ammonium compounds are able to impair the uptake of cholineCitation39, required for the synthesis of parasite membrane phospholipids, but also to inhibit the 3-fold methylation of phosphatidyl ethanolamine that represents the primary route to the Leishmania phosphatidyl cholineCitation40. It is worth noting that sodium 2-pentadecylbenzimidazole-5-carboxylate (M&B35347B) besides acting as anionic surfactant, is an inhibitor of acetyl-CoA carboxylase able to derange fatty acid and cholesterol biosynthetic pathwaysCitation41.
Concerning the subset of 2-phenyl- and 2-benzylbenzimidazoles, the compounds bearing an open-chain basic head were only moderately active (24, 26 and 27) or inactive (23 and 25), while those bearing a lupinyl residue were all, but one (39), endowed with valuable activity. Among the 1-lupinylbenzimidazole the activity was influenced by the substituents in 2 and 5 positions. The 5-acetyl derivatives were less potent than the corresponding 5-trifluoromethyl- and 5-nitro derivatives (compare 28–34, particularly 28, 30 and 33). The negative effect of the 5-acetyl group was also evident among the 1-dialkylaminoalkyl derivatives 23–27.
The higher potency of compound 28 in comparison to 36 suggests that the 2-benzylbenzimidazoles may be more potent than the corresponding 2-phenyl analogues, and indeed, excluding from comparison compounds 29–31 for the presence of the acetyl group (negatively affecting the activity), the 2-benzyl-1-lupinylbenzimidazole were, on average, more potent than the 2-phenyl-1-lupinyl derivatives.
With this kind of compounds we did not succeed to quaternise the lupinyl moiety without affecting also the benzimidazole ring and it was observed that the double quaternisation produced the loss of activity (compare compounds 36 and 39).
In this subset of 2-arylbenzimidazoles, compound 28 appears as the most interesting because resulted 12-/7-fold more potent than miltefosine and did not manifest any discernible cytotoxicity on Vero cells (CC50> 100 µM and SI >27 and >21 versus L. tropica and L. infantum, respectively), while the toxicity on HMEC-1 cells was only moderate (SI= 4.58 and 3.56 versus the two Leishmania species). It is worth noting that compound 28 was already shown to possess antiproliferative activity, with GI50< 5 µM, against 24 human cancer cell lines, among which the renal cancer cell line UO31 was particularly sensitive (GI50= 0.019 µMCitation25b). Moreover, the same compound displayed moderate antiviral activity against Coxsackie virus B5 (CVB-5) and respiratory syncytial virus (RSV) with EC50 13 and 15 µM, respectivelyCitation24c. Also compounds 33 and 34 displayed good level of antileishmanial activity associate with modest toxicity on Vero76 cells and represent, together with 28, interesting hit compounds.
Possessing a basic side chain, compounds 23–39 might, like sitamaquineCitation3b,Citation4d, anchor to the anionic phospholipidic components of Leishmania cell membrane, disrupting its function.
Eventually, they could permeate the cell and accumulate into cytosolic acidic compartments. Once inside the cells, the benzimidazole derivatives might inhibit some of the enzymes that are essential for Leishmania survival and proliferation and are absent from their mammalian hostCitation42, like those involved in the biosynthesis of membrane ergosterol and the 24-alkylsterolsCitation3b,Citation43 or the zinc metalloprotease (leishmanolysin)Citation18,Citation44, playing crucial roles in the Leishmania parasite physiology and in host-parasite interaction.
Some benzimidazole derivatives bearing a basic side chain have, already, been shown to somewhat affect sterol biosynthesis, like 2-[(4-diethylaminoethoxy)phenyl]benzimidazole that blocks the reduction of 7-dehydrocholesterol to cholesterolCitation45 and 2-(4-chlorobenzyl)-1-(3-diethylaminopropyl)-5-trifluoromethylbenzimidazoleCitation21 (structurally close to compounds 23–25) that, at 50 mg/kg p.os, reduced significantly (>15%) the serum cholesterol concentration in hypercholesterolemic mice. The mechanism of action of these two kinds of benzimidazole derivatives was not further investigated, and the possibility of their interference in parasite ergosterol biosynthesis may be only conjectural.
On the other hand, some 2-aryl-5-substituted benzimidazoles, devoid of basic chain, have been shown to inhibit the stearoyl coenzyme A desaturase (SCD1), blocking the formation of oleic and palmitoleic triglycerides, cholesterol esters and phospholipidsCitation46. The SCD1, besides being investigated for the treatment of dislipidemic diseases and body weight control, has been found to participate, together with other desaturase enzymes, in the de novo synthesis of mono- and poly-unsaturated fatty acids (C18–C22 PUFA) of parasitic membrane. These biosynthetic pathways play a crucial role for parasitic viability at different life cycle stagesCitation47. Some other 2-arylbenzimidazoles, still lacking basic side chain (, central row), have been shown to exhibit leishmanicidal effect and to dock successfully in the binding pocket of the promastigote surface protease (leishmanolysin, GP63 protein), which contributes to parasite virulenceCitation18. Of course, for the discussed compounds, other, even multiple, mechanisms of action, not yet identified, may take place.
Finally, for a better insight of the real value of the studied compounds as antileishmanial agents, compounds 8 and 28, representative of the two subsets of benzimidazole derivatives that display the highest activity against the promastigote stage, were tested against the intramacrophagic amastigote stage of L. infantum. Compound 8 exhibited an IC50= 0.313 μM, with a 3.35-fold increased potency with respect to miltefosine, while compound 28, at 2 μM concentration (42% of its IC50 versus promastigotes) reduced the amastigote infection of THP-1 cells by 33.2% (human acute monocytic leukaemia cell line; IC50> 2 μM).
Conclusions
Two sets of benzimidazole derivatives (38 compounds) were tested in vitro for activity against promastigotes of L. tropica and L. infantum. A first set was formed by 2-(long chain)-alkyl/benzyl benzimidazoles (1–22), whose heterocyclic head was, in most cases, quaternised to mimic the ammonium head of miltefosine and related analogous anti-leishmanial drugs. The second set was composed of 2-benzyl and 2-phenyl benzimidazoles (23–39) bearing in position 1 a basic side chain (dialkylaminoalkyl- or lupinyl-).
Most of the tested compounds of both sets resulted active against L. tropica (30 over 38) and L. infantum (25 over 33) (). The IC50 values for the quaternised 2-alkylbenzimidazoles were in the low micromolar/submicromolar range. Compound 8 (IC50= 0.19 µM and 0.34 µM versus L. tropica and L. infantum, respectively) resulted 228- and 93-fold more potent than miltefosine, with SI in the range 4.1–2.3 versus HMEC cells, but displaying SI= 30 and 17 versus Vero76 cells. Among the compounds bearing a basic side chain, the 1-lupinyl derivatives were commonly more active than dialkylaminoalkyl ones, and compound 28 [2-(4-chlorobenzyl)-1-lupinyl-5-trifluoromethylbenzimidazole] displayed the highest potency (IC50= 3.70 µM and 4.76 µM for the two Leishmania species). This compound was just a little less toxic than 8 on HMEC cells (SI= 4.6 and 3.6 versus L. tropica and L. infantum, respectively), but did not manifest any discernible cytotoxicity against Vero76 cells (CC50> 100 µM and SI= 27 and 21 versus the two Leishmania species). Therefore, several compounds and particularly the benzimidazoles 8 and 28, whose activity was confirmed on intramacrophagic amastigote stage of L. infantum, represent interesting hit compounds, whose structure can be further variate in order to improve their safety profiles (toxicity/activity ratios).
Based on the chemical features of the relevant compounds, their interaction with the acidic components (mainly the phospholipids) of cell membrane, with consequent disruption of its function, may explain the observed anti-leishmanial activity. The internalisation of compounds and their interaction with different targets inside the cell might also have an important role, but its investigation is beyond the aim of the present preliminary study.
Acknowledgments
This work was in part supported by Ministero dell?Istruzione, dell’Università e della Ricerca (PRIN) Projects 2010C2LKKJ_006; 20154JRJPP_004.
Disclosure statement
All authors declare no conflicts of interest.
Related Research Data
References
- World Health Organization (WHO). Available from: http://www.who.int/neglected_diseases/diseases/en [last accessed 11 Jul 2017].
- (a) Frézard F, Demicheli C, Ribeiro RR. Pentavalent antimonials: new perspectives for old drugs. Molecules 2009;1:2317–36. (b) Frézard F, Martins PS, Barbosa MC, et al. New insights into the chemical structure and composition of the pentavalent antimonial drugs, meglumine antimonate and sodium stibogluconate. J Inorg Biochem 2008;102:656–65.
- (a) Murray HW, Berman JD, Davies CR, et al. Advances in leishmaniasis. Lancet 2005;3:1561–77. (b) Singh N, Kumar M, Singh RK, Leishmaniasis: current status of available drugs and new potential drug targets. J Trop Med 2012;5:485–97. (c) Ameen M. Cutaneous leishmaniasis: advances in disease pathogenesis, diagnostics and therapeutics. Clin Exp Dermatol 2010;35:699–705.
- (a) Berman JD, Lee LS. Activity of 8-aminoquinolines against Leishmania tropica within human macrophages in vitro. Am J Trop Med Hyg 1983;32:753–9. (b) Singh S, Sivakumar R. Challenges and new discoveries in the treatment of leishmaniasis. J Infect Chemother 2004;10:307–15. (c) Garnier T, Brown MB, Lawrence MJ, Croft SL. In-vitro and in-vivo studies on a topical formulation of sitamaquine dihydrochloride for cutaneous leishmaniasis. J Pharm Pharmacol 2006;58:1043–54. (d) Loiseau PM, Cojean S, Schrével J. Sitamaquine as a putative antileishmanial drug candidate: from the mechanism of action to the risk of drug resistance. Parasite 2011;18:115–19. (e) Almeida OL, Santos JB. Advances in the treatment of cutaneous leishmaniasis in the new world in the last ten years: a systematic literature review. An Bras Dermatol 2011;86:497–506.
- (a) Singh N, Mishra BB, Bajpai S. Natural product based leads to fight against leishmaniasis. Bioorg Med Chem 2014;22:18–45. (b) Cheuka PM, Mayoka G, Mutai P, et al. The role of natural products in drug discovery and development against neglected tropical diseases. Molecules 2017;22:E58.
- (a) do Socorro S, Rosa M, Mendonça-Filho RR, Bizzo HR, et al. Antileishmanial activity of a linalool-rich essential oil from Croton cajucara. Antimicrob Agents Chemother 2003;47:1895–901. (b) De Monte C, Bizzarri B, Gidaro MC, et al. Bioactive compounds of Crocus sativus L. and their semi-synthetic derivatives as promising anti-Helicobacter pylori, anti-malarial and anti-leishmanial agents. J Enzyme Inhib Med Chem 2015;30:1027–33. (c) Wulsten IF, Costa-Silva TA, Mesquita JT, et al. Investigation of the anti-Leishmania (Leishmania) infantum activity of some natural sesquiterpene lactones. Molecules 2017;22:e685. (d) Barrera PA, Jimenez-Ortiz V, Tonn C, et al. Natural sesquiterpene lactones are active against Leishmania mexicana. J Parasitol 2008;5:1143–9. (e) Sairafianpour M, Christensen J, Staerk D, et al. Leishmanicidal, antiplasmodial, and cytotoxic activity of novel diterpenoid 1,2-quinones from Perovskia abrotanoides: new source of tanshinones. J Nat Prod 2001;64:1398–403. (f) Kayser O, Kiderlen AF, Bertels S, et al. Antileishmanial activities of aphidicolin and its semisynthetic derivatives. Antimicrob Agents Chemother 2001;45:288–92. (g) Sousa MC, Varandas R, Santos RC, et al. Antileishmanial activity of semisynthetic lupane triterpenoids betulin and betulinic acid derivatives: synergistic effects with miltefosine. PLoS One 2014;9:e89939.
- (a) Di Giorgio C, Delmas F, Ollivier E, et al. In vitro activity of the beta-carboline alkaloids harmane, harmine, and harmaline toward parasites of the species Leishmania infantum. Exp Parasitol 2004;1:67–74. (b) Turabekova MA, Vinogradova VI, Werbovetz KA, et al. Structure-activity relationship investigations of leishmanicidal N-benzylcytisine derivatives. Chem Biol Drug Des 2011;78:183–9.
- Kirmizibekmez H, Calis I, Perozzo R, et al. Inhibiting activities of the secondary metabolites of Phlomis brunneogaleata against parasitic protozoa and plasmodial enoyl-ACP Reductase, a crucial enzyme in fatty acid biosynthesis. Planta Med 2004;70:711–17.
- Hiam A, Sebastien D, George B, et al. Microtubule target for new antileishmanial drugs based on ethyl 3-haloacetamidobenzoates. J Enzyme Inhib Med Chem 2006;21:305–12.
- (a) Sánchez-Delgado RA, Anzellotti A. Metal complexes as chemotherapeutic agents against tropical diseases: trypanosomiasis, malaria and leishmaniasis. Mini Rev Med Chem 2004;4:23–30. (b) Ilari A, Baiocco P, Messori L, et al. A gold-containing drug against parasitic polyamine metabolism: the X-ray structure of trypanothione reductase from Leishmania infantum in complex with auranofin reveals a dual mechanism of enzyme inhibition. Amino Acids 2012;42:803–11.
- (a) Plano D, Baquedano Y, Moreno-Mateos D, et al. Selenocyanates and diselenides: a new class of potent antileishmanial agents. Eur J Med Chem 2011;4:3315–23. (b) Baquedano Y, Moreno E, Espuelas S, et al. Novel hybrid selenosulfonamides as potent antileishmanial agents. Eur J Med Chem 2014;74:116–23.
- Papanastasiou I, Prousis KC, Georgikopoulou K, et al. Design and synthesis of new adamantyl-substituted antileishmanial ether phospholipids. Bioorg Med Chem Lett 2010;20:5484–7.
- (a) Pathak D, Yadav M, Siddiqui N, et al. Antileishmanial agents: an updated review. Pharm Chem 2011;3:239–49. (b) Vale-Costa S, Costa-Gouveia J, Pérez B, et al. N-cinnamoylated aminoquinolines as promising antileishmanial agents. Antimicrob Agents Chemother 2013;5:5112–15. (c) Brindisi M, Brogi S, Relitti N, et al. Structure-based discovery of the first non-covalent inhibitors of Leishmania major tryparedoxin peroxidase by high throughput docking. Sci Rep 2015;5:9705. (d) Barteselli A, Casagrande M, Basilico N, et al. Clofazimine analogs with antileishmanial and antiplasmodial activity. Bioorg Med Chem 2015;23:55–65.
- (a) Pagniez F, Abdala-Valencia H, Marchand P, et al. Antileishmanial activities and mechanisms of action of indole-based azoles. J Enzyme Inhib Med Chem 2006;21:277–83. (b) Gupta L, Talwar A, Nishi, et al. Synthesis of marine alkaloid: 8,9-dihydrocoscinamide B and its analogues as novel class of antileishmanial agents. Bioorg Med Chem Lett 2007;17:4075–9. (c) Bharate SB, Bharate JB, Khan SI, et al. Discovery of 3,3′-diindolylmethanes as potent antileishmanial agents. Eur J Med Chem 2013;63:435–43. (d) Roy A, Chowdhury S, Sengupta S, et al. Development of derivatives of 3, 3'-diindolylmethane as potent Leishmania donovani bi-subunit topoisomerase IB poisons. PLoS One 2011;6:e28493.
- Danan A, Charon D, Kirkiacharian S, et al. Synthesis and antiparasitic activities of amidinic azolated derivatives. Farmaco 1997;52:227–9.
- Jagu E, Pomel S, Diez-Martinez A, et al. Synthesis and in vitro antikinetoplastid activity of polyamine-hydroxybenzotriazole conjugates. Bioorg Med Chem 2017;25:84–90.
- Hernández-Luis F, Hernández-Campos A, Castillo R, et al. Synthesis and biological activity of 2-(trifluoromethyl)-1H-benzimidazole derivatives against some protozoa and Trichinella spiralis. Eur J Med Chem 2010;45:3135–41.
- Shaukat A, Mirza HM, Ansari AH, et al. Benzimidazole derivatives: synthesis, leishmanicidal effectiveness, and molecular docking studies. Med Chem Res 2013;22:3606–20.
- (a) Mayence A, Vanden Eynde JJ, LeCour L, Jr, et al. Piperazine-linked bisbenzamidines: a novel class of antileishmanial agents. Eur J Med Chem 2004;3:547–53. (b) Mayence A, Pietka A, Collins MS, et al. Novel bisbenzimidazoles with antileishmanial effectiveness. Bioorg Med Chem Lett 2008;18:2658–61.
- (a) Torres-Gómez H, Hernández-Núñez E, León-Rivera I, et al. Design, synthesis and in vitro antiprotozoal activity of benzimidazole-pentamidine hybrids. Bioorg Med Chem Lett 2008;1:3147–51. (b) Mendez-Cuesta CA, Herrera-Rueda MA, Hidalgo-Figueroa S, et al. Synthesis, screening and in silico simulations of anti-parasitic propamidine/benzimidazole derivatives. Med Chem 2017;13:137–48.
- (a) Sparatore F, Boido V, Fanelli F. Dialkylaminoalkylbenzimi-dazoles of pharmacological interest. Farmaco Sci 1968;23:344–59. (b) Paglietti G, Sparatore F. Dialkylaminoalkyl-benzimidazoles of pharmacological interest. 3. Farmaco Sci 1972;27:333–42. (c) Boido A, Vazzana I, Sparatore F, et al. Preparation and pharmacological activity of some 1-lupinylbenzimidazoles and 1-lupinylbenzotriazoles. Farmaco 1991;46:775–88.
- (a) Paglietti G, Pirisi MA, Loriga M, et al. Preparation and pharmacologic activity of 2-(4'R')benzyl-5R-benzimidazole. Analgesic activity and effect on conditioned avoidance response. Farmaco Sci 1988;43:203–14. (b) Paglietti G, Pirisi MA, Loriga M, et al. Preparation and pharmacologic activity of 2-(4'R')benzyl-5R-benzimidazole and 2-(4'-pyridinyl)-5R-benzimidazoles. Analgesic activity and effect on conditioned avoidance response. Farmaco Sci 1988;43:215–26.
- (a) Paglietti G, Sparatore F. Preparation of beta-benzimidazolyl- and indazolylbutyric acids as potential choleretic agents. Farmaco Sci 1972;27:471–9. (b) Grella G, Paglietti G, Sparatore F, et al. Synthesis and choleretic activity of 3-(2-aryl-5R-benzimidazol-1-yl)butanoic acids. Farmaco Sci 1987;42:475–90. (c) Grella G, Paglietti G, Sparatore F, et al. Synthesis and choleretic activity of 3-[2-(3-R', 4-R'', 5-R'''-benzyl)-5-R-benzimidazol-1-yl]-butanoic acids. Farmaco Sci 1992;47:21–35. (d) Loriga M, Paglietti G, Piras S, et al. Synthesis and evaluation of gastroprotective and antiulcer activity of some 2-substituted-1H-imidazo[4,5-b] pyridines and -1H-benzimidazoles. Farmaco 1992;47:287–303.
- (a) Tonelli M, Paglietti G, Boido V, et al. Antiviral activity of benzimidazole derivatives. I. Antiviral activity of 1-substituted-2-[(benzotriazol-1/2-yl)methyl]benzimidazoles. Chem Biodivers 2008;5:2386–401. (b) Tonelli M, Simone M, Tasso B, et al. Antiviral activity of benzimidazole derivatives. II. Antiviral activity of 2-phenylbenzimidazole derivatives. Bioorg Med Chem 2010;1;2937–53. (c) Tonelli M, Novelli F, Tasso B, et al. Antiviral activity of benzimidazole derivatives. III. Novel anti-CVB-5, anti-RSV and anti-Sb-1 agents. Bioorg Med Chem 2014;22:4893–909.
- (a) Novelli F, Tasso B, Sparatore F. Synthesis and biological investigations of 2-(tetrahydropyran-2'-yl) and 2-(tetrahydrofuran-2'-yl)benzimidazoles. Farmaco 1997;52:499–507. (b) Tonelli M, Tasso B, Mina L, et al. Primary anti-proliferative activity evaluation of 1-(quinolizidin-1'-yl)methyl- and 1-(ω-tert-amino)alkyl-substituted 2-phenyl-, 2-benzyl- and 2-[(benzotriazol-1/2-yl)methyl]benzimidazoles on human cancer cell lines. Mol. Divers 2013;17:409–19.
- Pool WO, Harwood HJ, Ralston AW. 2-Alkylbenzimidazoles as derivatives for the identification of aliphatic acids. J Am Chem Soc 1937;59:178–9.
- Shi Z, Ta J-T. Synthesis of the β‐keto acids from benzimidazolium iodides and ethyl malonate. Chin J Chem 2000;18:940–1.
- Babu KR, Zhu N, Bao H. Iron-catalyzed C-H alkylation of heterocyclic C-H bonds. Org Lett 2017;19:46–9.
- Guo Y, Lu Z, Yao L, et al. A novel synthetic method for the preparation of aliphatic aldehydes from the corresponding carboxylic acids. Chin J Chem 2011;29:489–92.
- She J, Jiang Z, Wang Y. One-pot synthesis of functionalized benzimidazoles and 1H-pyrimidines via cascade reactions of o-aminoanilines or naphthalene-1,8-diamine with alkynes and p-tolylsulfonyl azide. Synlett 2009;12:2023–7.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Baiocco P, Ilari A, Ceci P, et al. Inhibitory effect of silver nanoparticles on trypanothione reductase activity and leishmania infantum proliferation. ACS Med Chem Lett 2010;2:203–33.
- D'Alessandro S, Gelati M, Basilico N, et al. Differential effects on angiogenesis of two antimalarial compounds, dihydroartemisinin and artemisone: implications for embryotoxicity. Toxicology 2007;241:66–74.
- Tandon VK, Kumar M. BF3·Et2O promoted one-pot expeditious and convenient synthesis of 2-substituted benzimidazoles and 3,1,5-benzoxadiazepines. Tetrahedr Lett 2004;45:4185–7.
- Howarth J, Hanlon K. N-ferrocenylmethyl, N'-methyl-2-substituted benzimidazolium iodide salts with in vitro activity against the Leishmania infantum parasite strain L1. Bioorg Med Chem Lett 2003;13:2017–20.
- Hunger A, Kebrle J, Rossi A, et al. Benzimidazol‐derivate und verwandte Heterocyclen. II. Synthese von 1‐aminoalkyl‐2‐benzyl‐benzimidazolen. Helv Chim Acta 1960;43:800–9.
- Boido V, Sparatore F. Simple molecular analogs of anti-inflammatory 1-lupinyl-2-(p-methoxy)benzyl-5-trifluoromethylbenzimidazole. Farmaco Sci 1974;29:517–25.
- Dorlo TP, Balasegaram M, Beijnen JH, et al. Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J Antimicrob Chemother 2012;67:2576–97.
- Ancelin ML, Vial HJ. Quaternary ammonium compounds efficiently inhibit Plasmodium falciparum growth in vitro by impairment of choline transport. Antimicrob Agents Chemother 1986;29:814–20.
- Bibis SS, Dahlstrom K, Zhu T, et al. Characterization of Leishmania major phosphatidylethanolamine methyltransferases LmjPEM1 and LmjPEM2 and their inhibition by choline analogs. Mol Biochem Parassitol 2014;196:90–9.
- Whittington FM, Enser M, Pratt J, et al. Effect of sodium 2-n-pentadecyl-benzimidazole-5-carboxylate (M & B 35347B), an inhibitor of acetyl-CoA carboxylase, on lipogenesis and fat deposition in obese hyperglycaemic (ob/ob) and lean mice. Int J Obes 1987;11:619–29.
- Chawla B, Madhubala R. Drug targets in Leishmania. J Parasit Dis 2010;34:1–13.
- (a) Fernandes Rodrigues JC, Concepcion JL, Rodrigues C, et al. In vitro activities of ER-119884 and E5700, two potent squalene synthase inhibitors, against Leishmania amazonensis: antiproliferative, biochemical, and ultrastructural effects. Antimicrob Agents Chemother 2008;5:4098–114. (b) de Macedo-Silva ST, Visbal G, Urbina JA, et al. Potent in vitro antiproliferative synergism of combinations of ergosterol biosynthesis inhibitors against Leishmania amazonensis. Antimicrob Agents Chemother 2015;59:6402–18.
- Das P, Alam MN, Paik D, et al. Protease inhibitors in potential drug development for Leishmaniasis. Indian J Biochem Biophys 2013;50:363–76.
- (a) Rodney G, Black ML, Bird OD, The common mode of action of three new classes of inhibitors of cholesterol biosynthesis. Biochem Pharmacol 1965;1:445–56. (b) Black ML, Rodney G, Capps DB. Simultaneous inhibition of alternative pathways of cholesterol biosynthesis by two related hypocholesteremic agents. Biochem Pharmacol 1968;17:1803–14.
- Powell DA, Ramtohul Y, Lebrun ME, et al. 2-Aryl benzimidazoles: human SCD1-specific stearoyl coenzyme-A desaturase inhibitors. Bioorg Med Chem Lett 2010;20:6366–9.
- (a) Maldonado RA, Kuniyoshi RK, Linss JG, et al. Trypanosoma cruzi oleate desaturase: molecular characterization and comparative analysis in other trypanosomatids. J Parassitol 2006;92:1064–74. (b) Ramakrishnan S, Serricchio M, Striepen B, et al. Lipid synthesis in protozoan parasites: a comparison between kinetoplastids and apicomplexans. Prog Lipid Res 2013;52:488–512.