
Abstract
A series of 6-substituted ureido- and thioureido-benzoxaboroles were investigated as inhibitors of carbonic anhydrases from Trypanosoma cruzi (TcCA), and Leishmania donovani chagasi (LdcCA). Both enzymes were inhibited by benzoxaboroles in the micromolar range. Preferential inhibitory potency against the β-CA LdcCA versus the α-CA TcCA was observed with submicromolar inhibitory activities. Some derivatives displayed excellent inhibitory and selectivity profile over the ubiquitous and physiological relevant human off-target hCA II. This study provides a convincing opportunity to study benzoxaborole scaffold for the design of antiprotozoan potential drugs targeting the pathogen’s carbonic anhydrases.
Introduction
Chagas disease (American trypanosomiasis) and leishmaniasis belong to the list of neglected tropical diseases developed by the World Health Organization (WHO). Both of these diseases are caused by parasites belonging to the kinetoplastidae family and belong to the vector-borne diseases which are responsible for more than 17% of infectious diseases, affecting 20 million people and killing more than 50,000 every yearCitation1. Trypasonoma is transmitted by a variety of bedbugs, appeared in Latin America before spreading to other continents. In 30% of cases, it manifests itself in cardiac disorders, and, in 10% of cases, in digestive or neurological disorders. Leishmania, transmitted by the bite of an infected phlebotoma, causes skin or visceral ailments that are very debilitating or even fatal if left untreated. Current treatment used today has many limitations in terms of cost and toxicity, as well as the emergence of resistance phenomena throughout the world. Finding new therapeutic targets to develop new drugs is therefore urgent for these parasitoses, which WHO now classifies as priority infections (category 1: reemerging or uncontrolled infections)Citation2,Citation3.
The availability of the complete genome sequence of both protozoans has given the possibility of large-scale analysis, which lead to consequent identification of novel drug targets. Protozoan carbonic anhydrases (CAs, EC 4.2.1.1) were thus recently identified as novel promising targets for chemotherapeutic interventionsCitation4–6. Because of the universal reaction they catalyze, i.e. reversible hydration of CO2 to bicarbonate with a proton release, the prokaryotic metalloproteins are considered as important components in the growth and virulence of pathogenic microorganisms. In 2013, carbonic anhydrases from the two unicellular protozoans Trypanosoma cruzi (TcCA) and Leishmania donovani chagasi (LdcCA) were cloned and characterizedCitation7,Citation8, paving the way for new opportunities in the design of novel inhibitors exploitable as antiprotozoal agents and acting by a totally new mechanism of action, lacking of cross-resistance to existing drugs.
Important classes of carbonic anhydrase inhibitors (CAIs) have been investigated in detail for their inhibitory profile against these two protozoans carbonic anhydrases.
The α-CA TcCA, which is characterized by very high catalytic activity for the CO2 hydration reaction (kcat of 1.21 × 106 s−1 and kcat/KM of 1.49 × 108 M−1 s−1), was shown to be inhibited in the nanomolar range by many types of aromatic/heterocyclic sulfonamidesCitation7,Citation9,Citation10, sulfamatesCitation7, thiolsCitation7 and hydroxamatesCitation11; the two last families of inhibitors demonstrated anti-trypanosomal activity, inhibiting in vivo the three phases of the pathogen’s life cycleCitation5,Citation7,Citation11. Inorganic anions and other small molecules were also reported to inhibit TcCA, with a simple dithiocarbamate showing low micromolar activity in vitroCitation5,Citation12.
The β-CA LdcCA which also possess an effective catalytic activity for the CO2 hydration reaction (kcat of 9.35 × 105 s−1 and kcat/KM of 5.9 × 107 M−1 s−1), has been reported to be efficiently inhibited by sulfonamides and heterocyclic thiols with nanomolar inhibition constantsCitation8,Citation13. Preliminary in vivo assays demonstrated that selected inhibitors in thiol series possessed anti-leishmania activity, being able to reduce parasites growth and causing their deathCitation8.
Considering the druggability of protozoans CAs, the design of new inhibitors with potent anti-trypasonomal or anti-leishmania activities is worth of investigation.
Previously, we reported a new generation of CAIs based on benzoxaborole scaffold. This new family of inhibitors were shown to act via a new binding mode, and to be effective against human α-CA as well as β-CA from pathogenic fungiCitation14,Citation15.
Encouraged by these results but also by the interesting profile of the trypanocidal orally active benzoxaborole compound SCYX-7158Citation16,Citation17 which enter Phase IIb/III trials in 2016 for the treatment of African trypanosomiasis, we report in this paper, the inhibitory activities of a series of 6-substituted urea/thiourea benzoxaboroles against CAs from the two pathogenic protozoans TcCA and LdcCA in order to detect possible candidates for anti-protozoans studies.
Material and methods
Chemistry
Compounds 1–23 were previously reported by this groupCitation14.
CA inhibition assay
An applied photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activityCitation18. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer for testing the α-CAs and with 20 mM TRIS buffer (pH 8.3) for testing the β-class enzyme. Na2SO4 of 20 mM were also added to the assay system for maintaining constant the ionic strength. The initial rates of the CA-catalyzed CO2 hydration reaction were followed for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation19–23 and represent the mean from at least three different determinations. All CA isoforms used in these experiments were recombinant ones obtained in-house as reported earlierCitation7,Citation8.
Results and discussion
Inhibition data of benzoxaborole 1–23 against TcCA and LdcCA were measured by a stopped flow CO2 hydrase assay and are shown in . Acetazolamide, a clinically used sulfonamide inhibitor, was used as standard. SCYX-7158 being not commercially available, Tavaborole, a commercially benzoxaborole used as topical antifungal medication was also used as standard in our inhibition assays. Inhibitory activities were displayed in comparison with the ones against the two physiologically relevant and off-target human isoforms hCA I and hCA II ().
Table 1. Inhibition data of α-CA isoforms hCA I, hCA II, TcCA and β-CA isoform LdcCA, with benzoxaboroles 1–23, tavaborole and the standard sulfonamide inhibitor acetazolamide by a stopped flow CO2 hydrase assayCitation15. 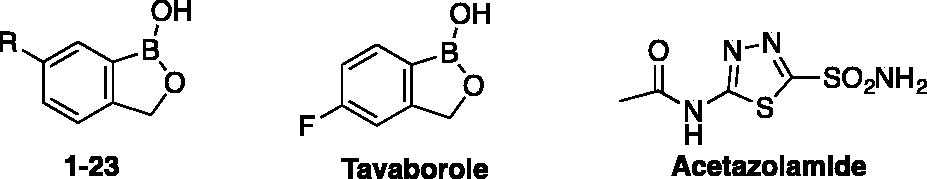
Regarding the data in , the following structure activity relationship for compounds 1–23 may be noted:
Ureido and thioureido benzoxaborole 1–23 exhibited micromolar to low micromolar inhibitory activities against protozoans TcCA and LdcCA, showing a pronounced selectivity against the β-CA LdcCA with inhibition constant ranging from 3 to 0.65 μM, and selectivity ratio (KI(TcCA)/KI(LdcCA)) ranging from 9 to 70, the more selective being compound 13. Against the TcCA isoform, KIs were ranging from 12.6 to 75.1 μM, the best inhibitor detected being the para-methoxy derivative 17.
It is worth noting that simple benzoxaborole 1, 6-nitro-2 and 6-amino benzoxaboroles 3 exhibited low micromolar activities against LdcCA, as the same level of magnitude as against hCA I, with inhibition constants ranging from 2.37 to 4.01 μM. No activity was noticed for these three compounds against TcCA (KI>100 μM). Tavaborole, a benzoxaborole used as standard in this study, showed inhibition potency similar to compound 2, and 3, against LdcCA, and a slight selectivity against human isoforms hCA II with a 5.5 magnitude selectivity ratio (KI(LdcCA)/KI(hCA II)).
From a general point of view, change of the ureido by a thioureido group did not affect significantly inhibitory potency. Inhibitory activity against LdcCA was comparable with the ones observed for the off-targets hCA I and hCA II. A slight change of selectivity may be noticed for some compounds when we compare LdcCA versus hCA II. For example, if we compare inhibition data for the phenylureido benzoxaborole 6 and phenylthioureido benzoxaborole 23, a selectivity of 2.5 magnitude for LdcCA was observed with the thioureido derivative 23 and no selectivity with the ureido compound 6. Substitution in the para position of the phenyl ring (e.g. 4-F, 20 and 4-CF3, 22) also showed a 2.2 magnitude of selectivity against LdcCA over CA II in thioureido series. An inversion of selectivity can be noticed for these two compounds in ureido series where compounds 9 (4-F derivative) and 10 (4-CF3) were, respectively, more selective against hCA II over LdcCA with selectivity ratio of 1.6 and 5.6 (KI(LdcCA)/KI(hCA II)).
The most performing inhibitor was found to be the para-nitrophenyl thioureido derivative 18 with a high-selectivity ratio against the pathogenic isoform LdcCA over hCA II (around 110). This compound could represent a slightly better solution compared with the standard clinically used sulfonamide acetazolamide, as acetazolamide showed selectivity for hCA II over LdcCA and TcCA with selectivity ratio, respectively, of 6 and 9.
Conclusion
We report for the first time the activity of benzoxaborole derivatives against protozoans CAs. 6-Substituted ureido and thioureido benzoxaborole derivatives 4–23 investigated here showed a preferential inhibitory activity against the β-CA from Leishmania donovani chagasi (LdcCA) versus the α-CA from TcCA. Some derivatives such as 18 displayed excellent inhibitory and selectivity profile for LdcCA over the human off-target hCA II.
The present study demonstrates that benzoxaborole chemotype offers interesting opportunities for the inhibition of CA from pathogenic protozoans and for the development of anti-trypasonomal or anti-leishmania compounds with a new mechanism of action, warranting further development.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
Related Research Data
References
- WHO. World Health Organization. Available from: http://www.who.int/chagas/en/ and http://www.who.int/leishmaniasis/en/
- WHO. World Health Organization. Sustaining the drive to overcome the global impact of neglected tropical diseases. Second WHO report on neglected tropical diseases; 2013. Available from: http://www.who.int/neglected_diseases/9789241564540/en/
- Mackey TK, Liang BA, Cuomo R, et al. Emerging and reemerging neglected tropical diseases: a review of key characteristics, risk factors, and the policy and innovation environment. Clin Microbiol Rev 2014;27:949–79.
- Vermelho AB, Capaci GR, Rodrigues IA, et al. Carbonic anhydrases from Trypanosoma and Leishmania as anti-protozoan drug targets. Bioorg Med Chem 2017;25:1543–55.
- Supuran CT. Inhibition of carbonic anhydrase from Trypanosoma cruzi for the management of Chagas disease: an underexplored therapeutic opportunity. Future Med Chem 2016;8:311–24.
- Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704.
- (a) Pan P, Vermelho AB, Capaci Rodrigues G, et al. Cloning, characterization, and sulfonamide and thiol inhibition studies of an α-carbonic anhydrase from Trypanosoma cruzi, the causative agent of Chagas disease. J Med Chem 2013;56:1761–71. (b) de Menezes Dda R, Calvet CM, Rodrigues GC, et al. Hydroxamic acid derivatives: a promising scaffold for rational compound optimization in Chagas disease. J Enzyme Inhib Med Chem 2016;31:964–73.
- Syrjänen L, Vermelho AB, Rodrigues Ide A, et al. Cloning, characterization, and inhibition studies of a β-carbonic anhydrase from Leishmania donovani chagasi, the protozoan parasite responsible for leishmaniasis. J Med Chem 2013;56:7372–81.
- Güzel-Akdemir Ö, Akdemir A, Pan P, et al. A class of sulfonamides with strong inhibitory action against the α-carbonic anhydrase from Trypanosoma cruzi. J Med Chem 2013;56:5773–81.
- Alafeefy AM, Ceruso M, Al-Jaber NA, et al. A new class of quinazoline-sulfonamides acting as efficient inhibitors against the α-carbonic anhydrase from Trypanosoma cruzi. J Enzyme Inhib Med Chem 2015;30:581–5.
- Rodrigues GC, Feijó DF, Bozza MT, et al. Design, synthesis, and evaluation of hydroxamic acid derivatives as promising agents for the management of Chagas disease. J Med Chem 2014;57:298–308.
- Pan P, Vermelho AB, Scozzafava A, et al. Anion inhibition studies of the α-carbonic anhydrase from the protozoan pathogen Trypanosoma cruzi, the causative agent of Chagas disease. Bioorg Med Chem 2013;21:4472–6.
- Ceruso M, Carta F, Osman SM, et al. Inhibition studies of bacterial, fungal and protozoan β-class carbonic anhydrases with Schiff bases incorporating sulfonamide moieties. Bioorg Med Chem 2015;23:4181–7.
- Alterio V, Cadoni R, Esposito D, et al. Benzoxaborole as a new chemotype for carbonic anhydrase inhibition. Chem Commun (Camb) 2016;52:11983–6.
- Nocentini A, Cadoni R, del Prete S, et al. Benzoxaboroles as efficient inhibitors of the β-carbonic anhydrases from pathogenic fungi: activity and modelling study. ACS Med Chem Lett 2017;8:1194–8.
- Jacobs RT, Plattner JJ, Nare B, et al. Benzoxaboroles: a new class of potential drugs for human African trypanosomiasis. Future Med Chem 2011;3:1259–78.
- Singh Grewal A, Pandita D, Bhardwaj S, et al. Recent updates on development of drug molecules for human African Trypanosomiasis. Curr Top Med Chem 2016; 16:2245–65.
- Khalifah RG. The carbon dioxide hydration activity of Carbonic Anhydrase I. Stop-flow kinetic studies on the native human Isoenzymes B and C. J Biol Chem 1971; 246:2561–73.
- (a) Aspatwar A, Hammarén M, Koskinen S, et al. β-CA-specific inhibitor dithiocarbamate Fc14-584B: a novel antimycobacterial agent with potential to treat drug-resistant tuberculosis. J Enzyme Inhib Med Chem 2017;32:832–40. (b) Supuran CT. Bortezomib inhibits bacterial and fungal β-carbonic anhydrases. Bioorg Med Chem 2016; 24:4406–9.
- (a) De Vita D, Angeli A, Pandolfi F, et al. Inhibition of the α-carbonic anhydrase from Vibrio cholerae with amides and sulfonamides incorporating imidazole moieties. J Enzyme Inhib Med Chem 2017;32:798–804. (b) Supuran CT, Capasso C. New light on bacterial carbonic anhydrases phylogeny based on the analysis of signal peptide sequences. J Enzyme Inhib Med Chem 2016; 31:1254–60.
- (a) Perfetto R, Del Prete S, Vullo D, et al. Cloning, expression and purification of the α-carbonic anhydrase from the mantle of the Mediterranean mussel, Mytilus galloprovincialis. J Enzyme Inhib Med Chem 2017;32:1029–35. (b) Abdoli M, Angeli A, Bozdag M, et al. Synthesis and carbonic anhydrase I, II, VII, and IX inhibition studies with a series of benzo[d]thiazole-5- and 6-sulfonamides. J Enzyme Inhib Med Chem 2017;32:1071–8.
- (a) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11. (b) Nocentini A, Vullo D, Del Prete S, et al. Inhibition of the β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa with monothiocarbamates. J Enzyme Inhib Med Chem 2017;32:1064–70.
- (a) Kumar R, Sharma V, Bua S, et al. Synthesis and biological evaluation of benzenesulphonamide-bearing 1,4,5-trisubstituted-1,2,3-triazoles possessing human carbonic anhydrase I, II, IV, and IX inhibitory activity. J Enzyme Inhib Med Chem 2017;32:1187–94. (b) Stanica L, Gheorghiu M, Stan M, et al. Quantitative assessment of specific carbonic anhydrase inhibitors effect on hypoxic cells using electrical impedance assays. J Enzyme Inhib Med Chem 2017;32:1079–90.