
Abstract
Zinc binding groups (ZBGs) play a crucial role in targeting histone deacetylase inhibitors (HDACIs) to the active site of histone deacetylases (HDACs), thus determining the potency of HDACIs. Due to the high affinity to the zinc ion, hydroxamic acid is the most commonly used ZBG in the structure of HDACs. An alternative ZBG is benzamide group, which features excellent inhibitory selectivity for class I HDACs. Various ZBGs have been designed and tested to improve the activity and selectivity of HDACIs, and to overcome the pharmacokinetic limitations of current HDACIs. Herein, different kinds of ZBGs are reviewed and their features have been discussed for further design of HDACIs.
Introduction
Histone deacetylases are a family of enzymes that are responsible for removing acetyl groups from the ɛ-N-acetyl group of histone lysine residuesCitation1–3. A total of 18 different isoforms of HDACs (which are classified into four classes) have been discovered. Class I (HDAC1, 2, 3, and 8), II (HDAC4–7, 9, and 10), and IV (HDAC11). HDACs are zinc-dependent enzymes which require the zinc ion for the catalytic reaction. On the other hand, class III HDACs (Sirt1–7) are a group of nicotinamide adenine dinucleotide (NAD+)-dependent enzymes. Enzymes in class I and II are known as classical HDACs, which attracted enormous attentions in the HDAC function exploration and inhibitor development.
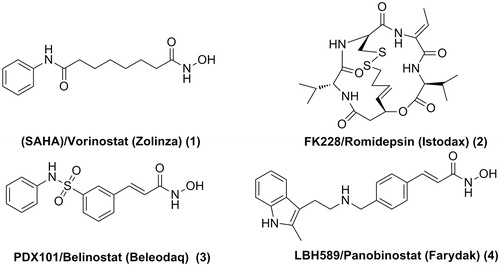
Overexpression and aberrant recruitment of HDACs play crucial roles in a variety of diseases such as neurodegenerative diseasesCitation4,Citation5, infectionCitation6,Citation7, human immunodeficiency virus (HIV)Citation8–10, cardiac diseasesCitation11,Citation12, and especially tumourCitation13. Inhibitors of HDACs have received considerable attention, in particular due to their potential as antitumor agents. Four HDAC inhibitors (HDACIs) have been approved by US Food and Drug Administration (FDA) for the treatment of cancer (). Suberoylanilide hydroxamic acid (SAHA)/Vorinostat (Zolinza) was approved in 2006 for the treatment of refractory and relapsed Cutaneous T-cell lymphoma (CTCL)Citation14. The cyclic tetrapeptide HDACI, FK228/Romidepsin (Istodax)Citation15, was also approved for treatment of CTCL. PDX101/Belinostat (Beleodaq)Citation16 and LBH589/Panobinostat (Farydak)Citation17 were approved for the treatment of peripheral T-cell lymphoma (PTCL) and multiple myeloma, respectively ().
Figure 1. Structures of HDACIs approved by the US FDA.
Structures of HDACIs are generally characterised by a zinc binding group (ZBG), a cap, and a linker that combines the above two parts together (). The cap and linker bind to residues in the active site of HDACs, thus contribute to the ligand–receptor interactions and affect the selectivity of HDACIs. Meanwhile, binding of ZBGs to the zinc ion and surrounding residues play the decisive role in the inhibitory activity of HDACIsCitation18. Although different ZBGs have been evaluated and reported, modifications in the cap and linker still received more attention in the development of potent and selective HDACIsCitation19. Structural modification in the ZBGs of current HDACIs might be a more efficient way of getting HDACIs to the market. In consideration of the importance of ZBGs as functional moieties in the structure of HDACIs, different kinds of ZBGs as well as their advantages and disadvantages are systematically reviewed in the present work.
Figure 2. Pharmacophore of HDACIs.

Classic ZBGs
The commonly used ZBGs, such as hydroxamic acid and benzamide, along with other ZBGs from HDACIs that had been approved by FDA or currently being investigated in clinical trials (such as carboxylic acid and thiol), are classified as classic ZBGs. These ZBGs have been widely accepted in the development of HDACIs, and their characteristics have been extensively studiedCitation20. The classic ZBGs are characterised by high activity, selectivity, but also off-target effects, potential toxicities and instability in vivoCitation21.
Hydroxamic acid
Hydroxamic acid is the most commonly used ZBG for its strong binding affinity with the zinc ion. By analysis of the crystal structure of the inhibitor–receptor complex, it was elucidated that hydroxamic acid binds to the zinc ion in a chelating manner (), which is considered as a guarantee of inhibitor activity. The naturally derived compound Trichostatin A (TSA), which showed very high HDAC inhibitory and antiproliferative activities, was equipped with hydroxamic acid as ZBGCitation22. The first HDAC inhibitor approved by the FDA, SAHA, also utilised the hydroxamic acid as the ZBG. Moreover, most HDACIs currently in clinical trials shared structural similarities with TSA and SAHA, using the hydroxamic acid as ZBGsCitation23.
Figure 3. The binding pattern of TSA in the active site of HDAC8 (PDB entry: 1T64).
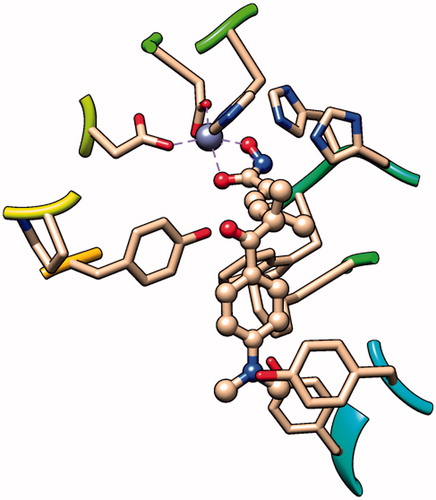
The advantages of hydroxamic acid group as ZBG include superior zinc binding ability, fair in vitro stability, good solubility, and easy synthesis. Therefore, in most cases, this group was the preferred ZBG in the design of new HDACIs. Various groups have been designed to mimic the hydroxamic acid group, but few exhibited higher potencyCitation24. The disadvantages of hydroxamic acid group as ZBG in HDACIs are also obvious. The selectivity of hydroxamic acid group is often questioned in the drug design, because of its high binding affinity to the zinc and other ionsCitation25. Undesirable side effects can also result from the binding of hydroxamic acid group to other zinc-dependent enzymes such as aminopeptidases, matrix metalloproteinases, and carbonic anhydrase. The common approaches to improve the selectivity of HDACIs with hydroxamic acid group are structural modifications in the linker and cap regions. Poor pharmacokinetics (rapid degradation and clearance in vivo) were also associated with HDACIs with hydroxamic acid as ZBGCitation26.
Benzamide
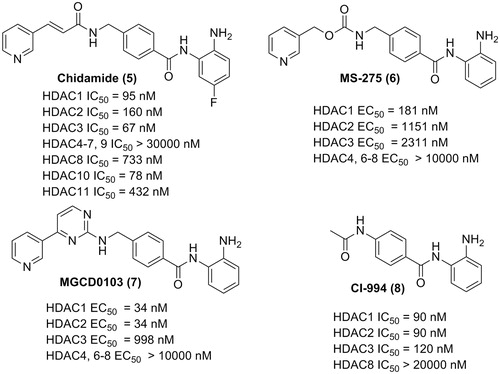
Benzamide derivatives are a big class of HDACIs with superior class I selectivity compared with hydroxamic acid HDACIs. The crystal structure analysis revealed chelation of the zinc ion by the amino group in the benzamide. Additionally, the distance between the carbonyl oxygen and zinc is more than 2.5 Å, indicating weak interactions. Chidamide (Epidaza)Citation27 is a benzamide HDACI approved by Chinese Food and Drug Administration (CFDA) for the treatment of relapsed or refractory PTCL (). MS-275Citation28–31 (HDAC1 selective) and MGCD0103Citation32–36 (HDAC1, 2 selective) are two benzamide HDACIs designed for the treatment of both hematologic malignancies and solid cancers, which are currently in clinical trial as mono therapies or in combination with other drugs. Good performances in clinical trials were also observed for another benzamide derivative, CI994Citation37–40.
Figure 4. Structures of the benzamide HDACIs.
The most significant feature of benzamide as ZBG in HDACIs is class I selectivity or individual HDAC isoform selectivity. Side effects of HDACIs are supposed to be reduced with the improvement of selectivity. The highly selective HDACIs could also serve as probe molecules in the diagnosis and pathogenetic research of diseases involving only a specific isoform of HDACs. The major disadvantage of benzamide derivatives is relatively compromised therapeutic benefits in clinical trials, which might explain the fact that, none of the benzamide HDACIs has been approved by the US FDA yet. Although high selectivity implies safety in clinical application, the exposed amino group in the benzamide structure could potentially induce in vivo toxicityCitation41. Furthermore, tumour cells are more likely to develop drug resistance against highly selective inhibitors, which might compromise the therapeutic effects of benzamide HDACIs in long-term therapiesCitation42.
Carboxylic acid
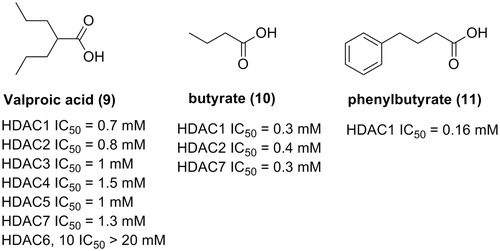
Carboxylic acid derivatives are a small group of HDACIs. Due to their weak zinc ion binding abilities, carboxylic acid HDACIs have not attracted as much attention as HDACIs with hydroxamic acid or benzamide ZBGs. The short chain fatty acids in clinical trials, such as valproic acidCitation43,Citation44, butyric acidCitation45, and phenylbutyrateCitation46, only exhibited IC50 in the magnitude of mM, as revealed by the in vitro enzymatic inhibitory assay (). Nonetheless, the marketing potential of compounds in this group is still under evaluation. Current activity data derived from the clinical trials indicated the potential of application of these fatty acids in tumour treatment.
Figure 5. Carboxylic HDACIs in clinical trials.
Thiols
Thiol group is commonly used for metal binding in the drug design, with the most famous example of Captopril. The first natural HDACI approved by US FDA, FK228, was regarded as a pro-drug, which can be metabolised to its active form via glutathione conjugation. The thiol group exposed in this process was supposed to chelate the zinc ion. Many thiol derivatives have been reported by structural modification of SAHACitation47 and potent natural productsCitation48 or by de novo designCitation49–53. However, none of them has been gained access to the clinical research so far.
Since sulfydryl group is a strong ZBG which makes few contributions to the selectivity of HDACIs, the selectivity can be adjusted by structural modifications in the linkers and caps of HDACIs. Introduction of thiol group to HDACIs also increases the risk of side effects caused by binding to other metal-dependent targets in vivo.
Novel ZBGs
Since classic ZBGs possess both obvious advantages and disadvantages, efforts have been made in discovery of more ideal ZBGs for HDACIs. Classes of HDACIs with novel ZBGs featured with high selectivity, potency, and stability have been designed and synthesisedCitation19. Although none of the HDACIs with novel ZBG has gained access to clinical trial currently, such investigations guaranteed the rise of new generations of HDACIs with improved pharmacological and pharmacokinetic profiles.
An imidazole thione containing molecule (12), which showed HDAC8 selectivity over HDAC1 and HDAC3, was discovered by an enzyme based screening approach ()Citation54. The imidazole thione group supposedly could bind to the zinc ion, thus could serve as a candidate ZBG for the further design of selective HDACIs. Molecules with pyrimido[1,2-c][1,3]benzothiazin-6-imine scaffold were also discovered to selectively target HDAC8Citation55.
Figure 6. HDACIs with novel ZBGs.
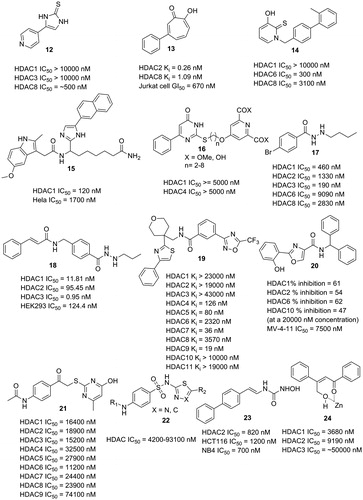
A series of tropolone derivatives were reported by Wright and co-workers as HDAC2 selective inhibitorsCitation56. Molecule 13 displayed high levels of selectivity for HDAC2 (Ki = 0.06 nM) comparing with HDAC1 (not active), HDAC4 (Ki = 10860 nM), HDAC5 (not active), HDAC6 (not active), and HDAC8 (Ki = 1.47 nM). It can also potently inhibit the growth of several tumour cell lines in the antiproliferative assay. In the molecular docking analysis, simulation results revealed that the tropolone group is capable of zinc ion binding. These investigations into tropolone-based HDACIs could give rise to a new chemotype of HDACIs with high potency and selectivity.
Oyelere and co-workers identified 3-hydroxypyridin-2-thione (3-HPT) as a novel ZBG, and the 3-HPT derivatives were HDAC6 selective inhibitors with no inhibition of HDAC1Citation57. In this series, molecule 14 exhibited HDAC6 selectivity with inhibitory IC50 value of 300 nM compared with HDAC8 (IC50 value of 3100 nM). Docking studies revealed that the 3-HPT group functions as ZBG. It is valuable for the 3-HPT derivatives in the development of specifically selective HDACIs for the treatment of tumour and other diseases without interference with HDAC1.
Several hydroxamic acid substitutes have been designed and synthesised as ZBGs by Attenni and co-workers, and the inhibitory activities were evaluatedCitation58. Primary amide containing molecules exhibited good performance in the enzymatic inhibition and antitumor assays. Molecule 15 with primary carboxamide moiety functioned as ZBG showed selective inhibitory pattern for HDAC1, and demonstrated antitumor efficacy in a xenograft model comparable to vorinostat. These findings revealed that primary carboxamide group could be used as an alternative ZBG for further HDACI design.
Chelidamic derivatives have been reported as novel HDACIs by Mai and co-workersCitation59. Molecules with chelidamic scaffold represented as the zinc binding group (16) exhibited inhibitory effects on both HDAC1 and HDAC4. Multiple analyses, including cell cycle analysis, apoptosis induction, and granulocytic differentiation analysis, have revealed that the representative molecules have the potential to serve as leading compounds in the cancer treatment. Chelidamic group as ZBG is considered as a promising candidate in the development of selective HDACIs by further structural modification.
Liao and co-workers discovered a class of HDACIs with benzoylhydrazide scaffold as the ZBGsCitation60. Molecule 17 exhibited class I selectivity, especially effective against HDAC3 (IC50 value of 0.06 μM). Subsequent in vitro assays with molecule 17 revealed potent anti-proliferative activities along with less overt cytotoxicity comparing with both SAHA and MS-275. Other in vitro evaluations emphasised the potential of benzoylhydrazide as new chemotype of HDACIs as well. Recently, Chou and co-workers also reported a series of hydrazide derivatives as potent class I HDACIsCitation61. The SAR (structure–activity relationship) analysis revealed that a 3-carbon-length β-nitrogen alkyl substituent chain provides ideal activity. One of the most potent molecule, 18, exhibited HDAC3 inhibitory activity of 0.95 nM (IC50 value), and EC50 values of 36.37, 76.64, and 151.7 nM against MV4–11, Molm14, and RS4–11 cell lines, respectively.
HDACIs with hydrazide motif are of significant importance as a new generation. Without the dependence on the zinc binding interactions, these molecules are supposed to exert less off-target effects. Moreover, the reported hydrazide derivatives showed class I selectivity (especially HDAC3), indicating the potential of successful disease treatment without serious adverse reactions. However, long-term use safety is yet to be confirmed for those HDACIs with hydrazide group.
A distinct class of HDACIs using trifluoromethyloxadiazolyl (TFMO) moiety as ZBG was reported by Nolan and co-workersCitation62. Binding patterns of these molecules were elucidated to be a non-chelating manner by crystallographic approaches. Zinc binding of TFMO derivatives is mediated by a fluorine atom in the trifluoromethyl group and aided by an oxygen atom in the oxadiazole heterocycle. In the enzymatic inhibition assay, these TFMO containing HDACIs exhibited high selectivity for HDAC class IIa. Molecule 19 displayed Ki values of 0.126, 0.080, 0.036, and 0.019 μM against HDAC4, 5, 7, and 9, respectively. In contrast, the Ki values of molecule 19 against HDAC6, HDAC8, and the rest HDACs (HDAC1, 2, 3, 10, 11) are 2.32, 3.57, and >10 μM, respectively. These class IIa selective HDACIs were further utilised to reveal the gene regulation mechanisms of class IIa HDACs. The discovery of TFMO-based HDACIs supports the design of selective HDACIs being of importance in treatment of a specific disease, and exploration of underlying mechanisms as probe molecules.
Woster and co-workers reported the 2-(oxazol-2-yl)phenol moiety as a novel ZBG that can be used for the design of potent HDACIsCitation63. A series of 2-(oxazol-2-yl)phenol derivatives were synthesised and tested in the activity assay. The derived molecules exhibited potent HDAC1, HDAC6 and HDAC10 inhibitory activities. Among these compounds, molecule 20 (IC50 7.5 μM against MV-4–11 leukemia cell line) could efficiently induce the acetylation of histone 3 lysine 9 (H3K9) and p21Waf1/CIP1 compared with SAHA. Molecular modelling analysis revealed that 2-(oxazol-2-yl)phenol group shows a similar zinc-binding pattern as the benzamide group in the ligand of a crystal structure (PDB entry: 4LY1). Although no remarkable selectivity was observed, the 2-(oxazol-2-yl)phenol derivatives are considered promising candidates as ZBG in the development of novel and potent HDAC inhibitory drugs.
Hydroxypyrimidines (21) without contribution to the selectivity of HDACs were discovered to be a new ZBGCitation64. The hydroxyl group and the pyrimidine group in the ZBG are both critical for activity, as revealed by SAR studies. Park and co-workers have identified N-[1,3,4]thiadiazol-2-yl and N-thiazol-2-yl sulfonamide groups as promising ZBG (21) with probably better physicochemical properties than hydroxamic acids, as demonstrated by the clinical studiesCitation65. The oxygen atoms of the sulfonamide group and the nitrogen atom of the thiadiazol or thiazol rings were predicted to bind to the zinc ion as revealed by the molecular modelling studies. Dallavallea and co-worker reported phenyl-4-yl-acrylohydroxamic acid derivatives as potent HDACIs with cinnamic-based hydrazones and amino or hydroxyureas as ZBGs (23)Citation66. In the activity studies, the derived molecules were discovered to be less effective than the parent hydroxamic acid derivatives. Wu and co-worker designed HDAC2 selective inhibitor β-hydroxymethyl chalcone (24), which exhibited about 20-fold isoform selectivity of HDAC2 (IC50 0.17 μM) over HDAC1 (IC50 2.74 μM)Citation67. A reaction-mechanism-based pattern was proposed in the binding of the derived inhibitor to zinc ion by intramolecular cyclisation which was catalysed by the zinc atom.
Selectivity is considered to play important role in the safety and potency of HDACIs. The novel ZBGs not only functioned as zinc chelators in the structures of HDACIs, but also can influence the selectivity of the enclosed molecules. The imidazole thione and pyrimido[1,2-c][1,3]benzothiazin-6-imine derivatives exhibited inhibitory selectivity of HDAC8, which is implicated in cancer, schistosomiasis, and Cornelia de Lange syndromeCitation68. The tropolone derivatives can selectively inhibit the bioactivity of both HDAC2 and HDAC8. The primary amide derivatives showed inhibitory selectivity of HDAC1, and the 3-HPT derivatives exhibited selectivity of HDAC6 over HDAC1. The chelidamic and hydrazide containing HDACIs featured selectivity of HDAC1, 4, and HDAC3, respectively. Two chemotypes of HDACIs even exhibited group-specific inhibitory selectivity of HDACs, such as the TFMO (with selectivity of class IIa HDACs) and 2-(oxazol-2-yl)phenol (with selectivity of HDAC1, 6, and 10) derivatives. The mentioned ZBGs can be applied to the design of HDACIs for the treatment of diseases or mechanistic studies by targeting a specific HDAC isoform or a specific group of HDACs.
Conclusion and discussion
Inhibition of HDACs has achieved rapid development in recent years, and HDACIs have exhibited enormous therapeutic potential in the treatment of cancer and other diseases. More than 15 HDACIs have been investigated in clinical trials, and four of them have gained approval from the US FDA. As targeted antitumor drugs, the efficacy and safety of HDACIs in tumour treatment had been confirmedCitation69. Therefore, development of HDACIs has attracted extreme attentions in the field of drug discovery. However, current development efforts focused on structural reorganisation and modification in the cap and linker motifs, while the ZBGs did not receive much attention, the classical ZBGs such as hydroxamic acid and benzamide groups were usually directly adopted into the new compounds.
Hydroxamic acid group is widely used as ZBGs due to its high zinc chelating ability, and benzamide is chosen for its class I HDACs selectivity. The application of carboxylic acid and thiol groups were limited by the lack of potency and only found in a small group of HDACIs. The potency and safety of HDACIs with the above ZBGs have been evaluated by various studies including different stages of clinical trials. Discovery of HDACIs with classic ZBGs, and especially the hydroxamic acid and benzamide derivatives, plays significant roles in the drug development by the inhibition of HDACs. Substituting the hydroxamic acid and benzamide groups with other groups usually resulted in reduction of activities comparing to the current highly active HDACIs. Moreover, the HDACIs designed with novel ZBGs from the beginning also have difficulties to show improved activities than those with the classical hydroxamic acid group.
In spite of the difficulties mentioned above, introduction of novel ZBGs is still necessary for the development of HDACIs of new chemotypes so that the pharmacokinetic and safety issues of current agents could be improved. Significant achievements have been gained in the development of HDACIs with novel ZBGs. Many have exhibited superior selectivity, such as the tropolone, 3-HPT, and TFMO derivatives. Some molecules also displayed surprisingly high potency in the activity assay, such as the hydrazide HDACIs discovered by Chou and co-worker (exhibited enzyme inhibitory activity with IC50 in the pM range and antiproliferative activity with IC50 in the nM range). Although none of the HDACIs with novel ZBGs has been approved by FDA or even gained access to the clinical trials, the future of these HDACIs as new generations is promising. The encouraging outcomes of these investigations also guaranteed further explorations on new ZBGs in HDACI development. Wide application of novel ZBGs in the HDACIs design will contribute to the emergence of new HDACIs.
Perspectives
Zinc binding groups play significant role in the potency of HDACIs. The wide application of current ZBGs is restricted by the poor pharmacokinetics or selectivity. Thus, it is necessary to develop new types of ZBGs for the design of HDACIs with improved druggability. In order to be selected in the design of HDACIs, the new types of ZBGs should exhibit advantages in potency, selectivity, pharmacokinetics, or safety compared with the classic ZBGs. However, it is still a challenge for the new designed ZBGs to show better performance than the classic ZBGs in the design of HDACIs. Therefore, a vast amount of further research work is needed in the discovery of novel ZBGs with exploitable value. Structural derivatisation of current ZBGs, such as hydroxamic acid and benzamide groups, is believed to be an effective way of rapid discovery of ZBGs with development potential. Novel ZBGs can be efficiently derived by computing methods, reference to or modification of natural structures, screening of molecular data base, isostere substitution, and de novo design. Study on the metabolic patterns of current ZBGs also provides valuable information for the design of new ZBGs.
Acknowledgements
Some of the materials in this work were supported by Qingdao Applied Basic Research Program (Youth Special, No. 16-5-1-60-jch).
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Bernstein BE, Tong JK, Schreiber SL. Genomewide studies of histone deacetylase function in yeast. Proc Natl Acad Sci USA 2000;97:13708–13.
- De Ruijter AJM, Van Gennip AH, Caron HN, et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003;370:737–49.
- Foglietti C, Filocamo G, Cundari E, et al. Dissecting the biological functions of Drosophila histone deacetylases by RNA interference and transcriptional profiling. J Biol Chem 2006;281:17968–76.
- Soragni E, Xu C, Cooper A, et al. Evaluation of histone deacetylase inhibitors as therapeutics for neurodegenerative diseases. Method Mol Biol 2011;793:495–508.
- Mai A, Rotili D, Valente S, Kazantsev AG. Histone deacetylase inhibitors and neurodegenerative disorders: holding the promise. Curr Pharm Design 2009;15:3940–57.
- Roger T, Lugrin J, Le Roy D, et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood 2011;117:1205–17.
- Valenzuela-Fernandez A, Alvarez S, Gordon-Alonso M, et al. Histone deacetylase 6 regulates human immunodeficiency virus type 1 infection. Mol Biol Cell 2005;16:5445–54.
- Saiyed ZM, Gandhi N, Agudelo M, et al. HIV-1 Tat upregulates expression of histone deacetylase-2 (HDAC2) in human neurons: implication for HIV-associated neurocognitive disorder (HAND). Neurochem Int 2011;58:656–64.
- Margolis DM. Histone deacetylase inhibitors and HIV latency. Curr Opin HIV AIDS 2011;6:25–9.
- Ylisastigui L, Archin NM, Lehrman G, et al. Coaxing HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. Aids 2004;18:1101–8.
- Gallo P, Latronico MV, Gallo P, et al. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardiovasc Res 2008;80:416–24.
- Kook H, Lepore JJ, Gitler AD, et al. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J Clin Invest 2003;112:863–71.
- Marks PA, Rifkind RA, Richon VM, et al. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 2001;1:194–202.
- Richon VM, Emiliani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci USA 1998;95:3003–7.
- Ueda H, Nakajima H, Hori Y, et al. Fr901228, a novel antitumor bicyclic depsipeptide produced by chromobacterium-violaceum No-968.1. Taxonomy, fermentation, isolation, physicochemical and biological properties, and antitumor-activity. J Antibiot 1994;47:301–10.
- Yang L, Xue XW, Zhang YH. Simple and efficient synthesis of belinostat. Syn Commun 2010;40:2520–4.
- Neri P, Bahlis NJ, Lonial S. Panobinostat for the treatment of multiple myeloma. Expert Opin Investig Drugs 2012;21:733–47.
- Manal M, Chandrasekar MJN, Priya JG, Nanjan MJ. Inhibitors of histone deacetylase as antitumor agents: a critical review. Bioorg Chem 2016;67:18–42.
- Roche J, Bertrand P. Inside HDACs with more selective HDAC inhibitors. Eur J Med Chem 2016;121:451–83.
- Zhang L, Han YT, Jiang QX, et al. Trend of histone deacetylase inhibitors in cancer therapy: isoform selectivity or multitargeted strategy. Med Res Rev 2015;35:63–84.
- Clawson GA. Histone deacetylase inhibitors as cancer therapeutics. Ann Transl Med 2016;4:287
- Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem 1990;265:17174–9.
- Bian J, Zhang LH, Han YT, et al. Histone deacetylase inhibitors: potent anti-leukemic agents. Curr Med Chem 2015;22:2065–74.
- Giannini G, Cabri W, Fattorusso C, Rodriquez M. Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med Chem 2012;4:1439–60.
- Benedetti R, Conte M, Altucci L. Targeting histone deacetylases in diseases: where are we? Antioxid Redox Signal 2015;23:99–126.
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov 2008;7:854–68.
- Dong M, Ning Z, Newman MJ, et al. Phase I study of chidamide (CS055/HBI-8000), a novel histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. J Clin Oncol 2009;27:3529a.
- Gojo I, Jiemjit A, Trepel JB, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood 2007;109:2781–90.
- Hauschild A, Trefzer U, Garbe C, et al. A phase II multicenter study on the histone deacetylase (HDAC) inhibitor MS-275, comparing two dosage schedules in metastatic melanoma. J Clin Oncol 2006;24:463s.
- Gojo I, Gore SD, Jiemjit A, et al. Phase I study of histone deacetylase inhibitor (HDI) MS-275 in adults with refractory or realpsed hematologic malignancies. Blood 2003;102:388a.
- Connolly RM, Jankowitz RA, Zahnow CA, et al. A phase 2 study investigating the safety, efficacy and surrogate biomarkers of response of 5-azacitidine (5-AZA) and entinostat (MS-275) in patients with advanced breast cancer. Cancer Res 2011;71:Abstract nr OT3-01-06.
- Bonfils C, Kalita A, Dubay M, et al. Evaluation of the pharmacodynamic effects of MGCD0103 from preclinical models to human using a novel HDAC enzyme assay. Clin Cancer Res 2008;14:3441–9.
- Martell RE, Younes A, Assouline SE, et al. Phase II study of MGCD0103 in patients with relapsed follicular lymphoma (FL): study reinitiation and update of clinical efficacy and safety. J Clin Oncol 2010;28:8086a.
- Blum KA, Advani A, Fernandez L, et al. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br J Haematol 2009;147:507–14.
- Garcia-Manero G, Assouline S, Cortes J, et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood 2008;112:981–9.
- DiPersio J, Stadtmauer EA, Nademanee AP, et al. Investigators, Phase I/II study of MGCD0103, an oral isotype-selective histone deacetylase (HDAC) inhibitor, in combination with 5-azacitidine in higher-risk myelodysplastic syndrome (MDS) and acute myelogenous leukemia (AML). Blood 2007;110:137a.
- Undevia SD, Kindler HL, Janisch L, et al. A phase I study of the oral combination of CI-994, a putative histone deacetylase inhibitor, and capecitabine. Ann Oncol 2004;15:1705–11.
- Pauer LR, Olivares J, Cunningham C, et al. Phase I study of oral CI-994 in combination with carboplatin and paclitaxel in the treatment of patients with advanced solid tumors. Cancer Invest 2004;22:886–96.
- Nemunaitis JJ, Orr D, Eager R, et al. Phase I study of oral CI-994 in combination with gemcitabine in treatment of patients with advanced cancer. Cancer J 2003;9:58–66.
- Prakash S, Foster BJ, Meyer M, et al. Chronic oral administration of CI-994: a phase 1 study. Invest New Drugs 2001;19:1–11.
- Verna L, Whysner J, Williams GM. 2-acetylaminofluorene mechanistic data and risk assessment: DNA reactivity, enhanced cell proliferation and tumor initiation. Pharmacol Ther 1996;71:83–105.
- Wagner JM, Hackanson B, Lubbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics 2010;1:117–36.
- Krauze AV, Myrehaug SD, Chang MG, et al. A phase 2 study of concurrent radiation therapy, temozolomide, and the histone deacetylase inhibitor valproic acid for patients with glioblastoma. Int J Radiat Oncol 2015;92:986–92.
- Kuendgen A, Strupp C, Hildebrandt B, et al. Phase 2 trial of the histone deacetylase inhibitor valproic acid as a monotherapy or in combination with all-trans retinoic acid in 24 patients with acute myeloid leukemia. Blood 2004;104:501a.
- Aviram A, Zimrah Y, Shaklai M, et al. Comparison between the effect of butyric-acid and its prodrug pivaloyloxymethylbutyrate on histones hyperacetylation in an Hl-60 leukemic-cell line. Int J Cancer 1994;56:906–9.
- Gore SD, Weng LJ, Yu K, Fu S. Phenylbutyrate in myeloid malignancies: relationship between inhibition of histone deacetylase, cytostasis and differentiation. Clin Cancer Res 1999;5:3817s.
- Suzuki T, Kouketsu A, Matsuura A, et al. Thiol-based SAHA analogues as potent histone deacetylase inhibitors. Bioorg Med Chem Lett 2004;14:3313–17.
- Nishino N, Jose B, Okamura S, et al. Cyclic tetrapeptides bearing a sulfhydryl group potently inhibit histone deacetylases. Org Lett 2003;5:5079–82.
- Wen JC, Niu Q, Liu J, et al. Novel thiol-based histone deacetylase inhibitors bearing 3-phenyl-1H-pyrazole-5-carboxamide scaffold as surface recognition motif: design, synthesis and SAR study. Bioorg Med Chem Lett 2016;26:375–9.
- Suzuki T, Kouketsu A, Itoh Y, et al. Highly potent and selective histone deacetylase 6 inhibitors designed based on a small-molecular substrate. J Med Chem 2006;49:4809–12.
- Giannini G, Vesci L, Battistuzzi G, et al. ST7612AA1, a thioacetate-omega(gamma-lactam carboxamide) derivative selected from a novel feneration of oral HDAC inhibitors. J Med Chem 2014;57:8358–77.
- Vesci L, Bernasconi E, Milazzo FM, et al. Preclinical antitumor activity of ST7612AA1: a new oral thiol-based histone deacetylase (HDAC) inhibitor. Oncotarget 2015;6:5735–48.
- Battistuzzi G, Giannini G. Synthesis of ST7612AA1, a novel oral HDAC inhibitor, via radical thioacetic acid addition. Curr Bioact Compd 2016;12:282–8.
- Hu ED, Dul E, Sung CM, et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther 2003;307:720–8.
- Kleinschek A, Meyners C, Digiorgio E, et al. Non-hydroxamate histone deacetylase 8 inhibitors. ChemMedChem 2016;11:2598–606.
- Ononye SN, VanHeyst MD, Oblak EZ, et al. Tropolones as lead-like natural products: the development of potent and selective histone deacetylase inhibitors. Acs Med Chem Lett 2013;4:757–61.
- Patil V, Sodji QH, Kornacki JR, et al. 3-Hydroxypyridin-2-thione as novel zinc binding group for selective histone deacetylase inhibition. J Med Chem 2013;56:3492–506.
- Attenni B, Ontoria JM, Cruz JC, et al. Histone deacetylase inhibitors with a primary amide zinc binding group display antitumor activity in xenograft model. Bioorg Med Chem Lett 2009;19:3081–4.
- Valente S, Conte M, Tardugno M, et al. Developing novel non-hydroxamate histone deacetylase inhibitors: the chelidamic warhead. MedChemComm 2012;3:298–304.
- Wang YF, Stowe RL, Pinello CE, et al. Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem Biol 2015;22:273–84.
- McClure J, Zhang C, Inks E, et al. Development of allosteric hydrazide-containing class I histone deacetylase inhibitors for use in acute myeloid leukemia. J Med Chem 2016;59:9942–59.
- Lobera M, Madauss KP, Pohlhaus DT, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol 2013;9:319–25.
- Li YX, Woster PM. Discovery of a new class of histone deacetylase inhibitors with a novel zinc binding group. MedChemComm 2015;6:613–18.
- Kemp MM, Wang Q, Fuller JH, et al. A novel HDAC inhibitor with a hydroxy-pyrimidine scaffold. Bioorg Med Chem Lett 2011;21:4164–9.
- Park H, Kim S, Kim YE, Lima SJ. A structure-based virtual screening approach toward the discovery of histone deacetylase inhibitors: identification of promising zinc-chelating groups. ChemMedChem 2010;5:591–7.
- Musso L, Cincinelli R, Zuco V, et al. Investigation on the ZBG-functionality of phenyl-4-yl-acrylohydroxamic acid derivatives as histone deacetylase inhibitors. Bioorg Med Chem Lett 2015;25:4457–60.
- Zhou JW, Li M, Chen NH, et al. Computational design of a time-dependent histone deacetylase 2 selective inhibitor. Acs Chem Biol 2015;10:687–92.
- Chakrabarti A, Oehme I, Witt O, et al. HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol Sci 2015;36:481–92.
- Mann BS, Johnson JR, Cohen MH, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007;12:1247–52.