
Abstract
Synthetic or natural carbazole derivatives constitute an interesting class of heterocycles, which showed several pharmaceutical properties and occupied a promising place as antitumour tools in preclinical studies. They target several cellular key-points, e.g. DNA and Topoisomerases I and II. The most studied representative, i.e. Ellipticine, was introduced in the treatment of metastatic breast cancer. However, because of the onset of dramatic side effects, its use was almost dismissed. Many efforts were made in order to design and synthesise new carbazole derivatives with good activity and reduced side effects. The major goal of the present study was to synthesise a series of new N-thioalkylcarbazole derivatives with anti-proliferative effects. Two compounds, 5a and 5c, possess an interesting anti-proliferative activity against breast and uterine cancer cell lines without affecting non-tumoural cell lines viability. The most active compound (5c) induces cancer cells death triggering the intrinsic apoptotic pathway by inhibition of Topoisomerase II.
Graphical Abstract
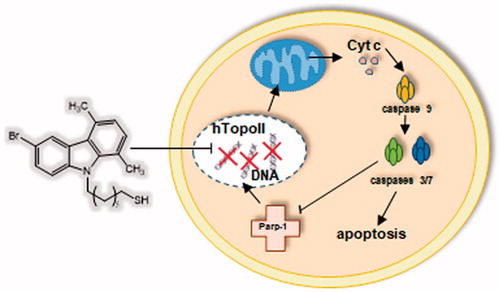
Introduction
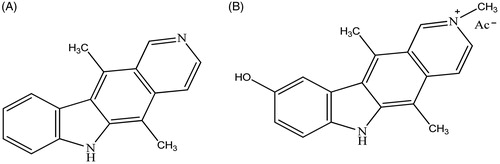
Recently, one of the principal targets of the pharmaceutical research is represented by the design of new antitumoural drugs with higher selectivity on neoplastic cells, few side effects and able to overcome resistance onsetCitation1. In this scenario, carbazoles were widely studied since they possess not only antitumour activity but also antimicrobic, antiepileptic, antihistaminic, antioxidant, anti-inflammatory, analgesic and neuroprotective propertiesCitation2–4. In recent studies, N-substituted carbazoles were reported as neuroprotective agents with strong anti-oxidant activity and some of them were also used as antifungal, photoconductive, antioxidant, antibacterial, anti-malaria, anti-tuberculosis, anti-HIV agents and in the treatment of obesityCitation5–7. Regarding their antitumour activity, several key-points, namely telomerases, topoisomerase I and II, tubulin were identifiedCitation8–11. The most studied representative of this class of compounds, i.e. Ellipticine () was introduced in the treatment of metastatic breast cancerCitation1,Citation12–15, then dismissed because of poor solubility in water and dramatic side effectsCitation16,Citation17. An improved solubility was achieved through the formation of salts as, for instance, in the case of 9-hydroxyellipticine (Celiptium, ), which was widely used, alone or in combination with other chemotherapeutics, possesses a higher DNA affinity than Ellipticine and lacks of toxicity at therapeutic dosesCitation18–20.
Figure 1. Structures of Ellipticine (A) and Celiptium (B).
In other previous studies, the carbazole scaffold was variously functionalised obtaining 6-aryl, 3-acyl (or 3-aroyl) and 9-alkyl-1,4-dimethyl-9H-carbazolesCitation21–26, with improved pharmacokinetic properties. Furthermore, benzofuroquinazolinones derivatives were synthesised as carbazole bioisostersCitation27 and they showed interesting antitumour properties at sub-micromolar concentrationsCitation28. In this paper, we report the synthesis of a new series of N-thioalkylcarbazoles differently substituted at the 1, 4, 6 and 9 positions of the carbazole core. In these molecules, the carbazole nitrogen bears alkyl chains with seven, eight and nine methylene groups, linked to a thiol terminal group.
Two of these molecules have demonstrated an interesting anti-proliferative activity against two breast cancer cell lines, MCF-7 and MDA-MB-231, and, to a greater extent, against cervical HeLa and endometrial ISHIKAWA cells. Moreover, the assayed compounds did not show effects on the proliferation of non-tumoural MCF-10A and 3T3-L1 cells. The antitumour activity resides in the ability to trigger the apoptotic intrinsic pathway, also known as mitochondrial pathway, and to inhibit the human Topoisomerase II (hTopo II) activity, as shown by direct enzyme assays and docking simulations. hTopoII is an essential enzymes playing a crucial role during transcription and replication and is over-expressed in tumour cells, thus it is a valid target for the development of new molecules for the treatment of a wide spectrum of tumoursCitation29,Citation30. Our outcomes set the compound 5c as a valid lead compound to be useful for cancer treatment.
Materials and methods
Synthesis
Commercial reagents were purchased from Sigma-Aldrich (St. Louis, MO) and used without additional purification. Silica gel 60 (300 − 400 mesh, Merck, Kenilworth, NJ) was used for flash chromatography. Preparative TLC was performed on 20 × 20 cm glass plates coated with a 2 mm layer of silica gel PF254 Merk. 1HNMR and 13CNMR spectra were recorded on a Bruker Avance 400 spectrometer (Bruker Corporation, Billerica, MA). Chemical shifts are expressed in parts per million downfield from tetramethylsilane as an internal standard. Multiplicities are represented by s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Coupling constants (J) are reported in Hertz (Hz). Mass spectra were obtained using an ESI mass spectrometer: LC-ADVANTAGE MAX (ESI). High-resolution mass spectra (HRMS) were recorded on a high-resolution mass spectrometer equipped by electrospray (ESI) and nanospray sources, and a quadrupole-time of flight hybrid analyser, coupled with capillary UPLC system (Q-TOF Premier/nanoAquity, Waters, Milford, MA) in positive mode, and either protonated molecular ions [M + H]+ were used for empirical formula confirmation, unless otherwise stated. The 9-(bromoalkyl)-9H-carbazoles (2, 3, 4 a–c) intermediates were prepared as described in the literatureCitation28 by the corresponding 1a–c carbazolesCitation31–34.
9-(7-Bromoheptyl)-9H-carbazole (2a). Colourless oil (145 mg, 71%). 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 7.7 Hz, 2H), 7.54–7.51 (m, 2H), 7.45 (d, J = 8.1 Hz, 2H), 7.31–7.28 (m, 2H), 4.35–4.33 (m, 2H), 3.42–3.40 (m, 2H), 1.95–1.87 (m, 2H), 1.85–1.82 (m, 2H), 1.47–1.36 (m, 6H) ppm. HRMS (ESI-Q-TOF) m/z: Calcd. for C19H22BrN 343.0936, found 344.0939 [M + H+]+.
9-(8-Bromooctyl)-9H-carbazole (3a). Colourless oil (129 mg, 61%). 1H NMR (400 MHz, CDCl3) δ: 7.51–7.37 (m, 2H), 7.36–7.22 (m, 2H), 7.17–7.12 (m, 2H), 7.11–7.06 (m, 2H), 4.10–4.06 (m, 4H), 3.37–3.33 (m, 2H), 1.86–1.82 (m, 2H), 1.68–1.64 (m, 2H), 1.40–1.29 (m, 6H) ppm. HRMS (ESI-Q-TOF) m/z: Calcd. for C20H24BrN 357.1092, found 359.1095 [M + 2]+.
9-(9-Bromononyl)-9H-carbazole (4a). Colourless oil (144 mg, 65%). 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 7.7 Hz, 2H), 7.54–7.50 (m, 2H), 7.45 (d, J = 8.1 Hz, 2H), 7.30–7.27 (m, 2H), 4.35–4.32 (m, 2H), 3.44–3.42 (m, 2H), 1.94–1.87 (m, 2H), 1.85–1.83 (m, 2H), 1.44–1.30 (m, 10H) ppm. HRMS (ESI-Q-TOF) m/z: Calcd. for C21H26BrN 371.1249, found 373.1245 [M + 2]+.
9-(7-Bromoheptyl)-1,4,6-trimethyl-9H-carbazole (2b). Colourless oil (75 mg, 81%). 1H NMR 400 MHz, CDCl3) δ 8.00 (s, 1H), 7.35–7.28 (m, 2H), 7.09 (d, J = 7.3 Hz, 1H), 6.90 (d, J = 7.3 Hz, 1H), 4.53–4.50 (m, 2H), 3.45–3.40 (m, 2H), 2.88 (s, 3H), 2.81 (s, 3H), 2.59 (s, 3H), 1.92–1.81 (m, 4H), 1.51–1.35 (m, 6H) ppm. HRMS (ESI-Q-TOF) m/z: Calcd. for C22H28BrN 385.1405, found 387.1402 [M + 2]+.
9-(8-Bromooctyl)-1,4,6-trimethyl-9H-carbazole (3b). Colourless oil (70 mg, 73%). 1H NMR (400 MHz, CDCl3) δ 7.96 (s, 1H), 7.28–7.24 (m, 2H), 7.03 (d, J = 7.3 Hz, 1H), 6.85 (d, J = 7.3 Hz, 1H), 4.44–4.41 (m, 2H), 3.36–3.34 (m, 2H), 2.82 (s, 3H), 2.74 (s, 3H), 2.53 (s, 3H), 1.81–1.72 (m, 4H), 1.40–1.25 (m, 8H) ppm. HRMS (ESI-Q-TOF) m/z: Calcd. for C23H30BrN 399.1562, found 401.1565 [M + 2]+.
9-(9-Bromononyl)-1,4,6-trimethyl-9H-carbazole (4b). Colourless oil (64 mg, 64%). 1H NMR (400 MHz, CDCl3) δ 8.01 (s, 1H), 7.34–7.28 (m, 2H), 7.09 (d, J = 7.3 Hz, 1H), 6.90 (d, J = 7.2 Hz, 1H), 4.52–4.49 (m, 2H), 3.44–3.41 (m, 2H), 2.87 (s, 3H), 2.80 (s, 3H), 2.58 (s, 3H), 1.89–1.78 (m, 4H), 1.58–1.28 (m, 10H) ppm. HRMS (ESI-Q-TOF) m/z: Calcd. for C24H32BrN 413.1718; found 415.1721 [M + 2]+.
6-Bromo-9-(7-bromoheptyl)-1,4-dimethyl-9H-carbazole (2c). Colourless oil (102 mg, 41%). 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 1H), 7.40–7.37 (m, 1H), 7.13 (d, J = 8.9 Hz, 1H), 6.96 (d, J = 7.3 Hz, 1H), 6.77 (d, J = 7.3 Hz, 1H), 4.35–4.31 (m, 2H), 3.25–3.22 (m, 2H), 2.67 (s, 3H), 2.63 (s, 3H), 1.70–1.61 (m, 4H), 1.30–1.18 (m, 6H) ppm. HRMS (ESI-Q-TOF) m/z: Calcd. for C21H25Br2N 449.0354; found 451.0355 [M + 2]+.
6-Bromo-9-(8-bromooctyl)-1,4-dimethyl-9H-carbazole (3c). Colourless oil (95 mg, 37%). 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 1H), 7.39–7.36 (m, 1H), 7.13–7.11 (m, 1H), 6.96 (d, J = 7.3 Hz, 1H), 6.76 (d, J = 7.3 Hz, 1H), 4.32–4.28 (m, 2H), 3.29–3.23 (m, 4H), 2.66 (s, 3H), 2.62 (s, 3H), 1.75–1.63 (m, 4H), 1.29–1.17 (m, 6H). HRMS (ESI-Q-TOF) m/z: Calcd. for C22H27Br2N 463.0510; found 465.0513 [M + 2]+.
6-Bromo-9-(9-bromononyl)-1,4-dimethyl-9H-carbazole (4c). Colourless oil (105 mg, 61%). 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 1H), 7.38 (d, J = 8.6 Hz, 1H), 7.13 (d, J = 8.8 Hz, 1H), 6.96 (d, J = 7.3 Hz, 1H), 6.77 (d, J = 7.2 Hz, 1H), 4.33–4.30 (m, 2H), 3.25 (t, J = 6.8 Hz, 2H), 2.67 (s, 3H), 2.63 (s, 3H), 1.73–1.60 (m, 4H), 1.26–1.12 (m, 10H). HRMS (ESI-Q-TOF) m/z: Calcd. for C23H29Br2N 477,0667; found 479.1599 [M + 2]+.
General procedure for the synthesis of N-thioalkylcarbazole derivatives (5, 6, 7a–c). To a mixture of the 9-(bromoalkyl)-9H-carbazoles 2, 3, 4a–c (0.21 mmol) in i-PrOH (13 ml) thiourea (0.84 mmol) was added and the solution was refluxed for 12 h under nitrogen atmosphereCitation35. At the residue obtained, after removal of the solvent, was added 6 N NaOH (27 ml), the resulting mixture was refluxed for 5 h, then it was neutralised by dropwise addition of 3 N HCl. Finally, by extraction with CH2Cl2 (3 × 100 ml), we obtained the crude compounds, purified by preparative thin layer chromatography (PTLC) (n-hexane/ethyl acetate 97:3) to give pure compounds.
7-(9H-Carbazol-9-yl)heptane-1-thiol (5a). Yellow oil (17 mg, 25%). 1H NMR (400 MHz, CDCl3) δ: 8.14 (d, J = 7.6 Hz, 2H, Ar), 7.51–7.48 (m, 2H, Ar), 7.43 (d, J = 8.1 Hz, 2H, Ar), 7.27 (m, 2H, Ar), 4.33–4.31 (m, 2H, NCH2), 2.66–2.63 (m, 2H, CH2S), 1.92–1.86 (m, 2H, NCH2CH2), 1.66–1.64 (m, 2H, HSCH2CH2), 1.41–1.37 (m, 7H, 3CH2, SH) ppm. 13C NMR (100 MHz, CDCl3) δ: 140.73, 126.45, 124.15, 121.68, 120.48, 109.63, 47.43, 33.25, 28.96, 28.61, 27.95, 27.55, 25.38 ppm. MS(ESI): C19H23NS (298.46) [M + H+]+. HRMS (ESI-Q-TOF) m/z: Calcd. for C19H23NS 297.1551, found 298.1549 [M + H+]+.
8-(9H-Carbazol-9-yl)octane-1-thiol (6a). Yellow oil (14 mg, 23%).1H NMR (300 MHz, CDCl3) δ: 8.10 (d, J = 7.8 Hz, 2H, Ar), 7.49–7.32 (m, 4H, Ar), 7.24–7.19 (m, 2H, Ar), 4.33–4.24 (m, 2H, NCH2), 2.64–2.59 (m, 2H, CH2S), 1.87–1.82 (m, 2H NCH2CH2), 1.63–1.55 (m, 2H, HSCH2CH2), 1.47–1.25 (m, 9H, 4CH2, SH) ppm. 13C NMR (100 MHz, CDCl3) δ: 140.73, 126.45, 124.15, 121.68, 120.48, 109.63, 47.43, 33.25, 28.96, 28.61, 27.95, 27.55, 25.38 ppm. MS (ESI): C20H25NS (312.48) [M + H+]+. HRMS (ESI-Q-TOF) m/z: Calcd. for C20H25NS 311.1708, found 312.4844 [M + H+]+.
9-(9H-Carbazol-9-yl)nonane-1-thiol (7a). Yellow oil (13 mg, 40%). 1H NMR (300 MHz, CDCl3) δ: 7.95 (d, J = 7.7 Hz, 2H, Ar), 7.33–7.24 (m, 4H, Ar), 7.11–7.06 (m, 2H Ar), 4.16–4.12 (t, 2H, NCH2), 2.51–2.47 (m, 2H, CH2S), 1.73–1.67 (m, 2H, CH2), 1.53–1.44 (m, 2H, CH2), 1.20–0.98 (m, 11H, 5CH2, SH) ppm. 13C NMR (100 MHz, CDCl3) δ 140.73, 126.45, 124.15, 121.68, 120.48, 109.63, 47.43, 33.25, 28.96, 28.61, 27.95, 27.55, 25.38 ppm. MS (ESI): C21H27NS (326.51) [M + H+]+. HRMS (ESI-Q-TOF) m/z: Calcd. for C21H27NS 325.1864, found 326.1844 [M + H+]+.
7-(1,4,6-Trimethyl-9H-carbazol-9-yl)-heptane-1-thiol (5b). Yellow oil (23 mg, 52%). 1H NMR (400 MHz, CDCl3) δ: 8.05 (s, 1H, Ar), 7.36–7.28 (m, 2H, Ar), 7.12 (d, J = 7.2 Hz, 1H, Ar), 6.94 (d, J = 7.2 Hz, 1H, Ar), 4.57–4.45 (m, 2H, NCH2), 2.83 (s, 3H, CH3), 2.77 (s, 3H, CH3), 2.74–2.71 (m, 2H, CH2S), 2.60 (s, 3H, CH3), 1.83–1.77 (m, 2H, CH2), 1.76–1.74 (m, 2H, CH2), 150–1.27 (m, 7H, 3CH2, SH) ppm. 13C NMR (100 MHz, CDCl3) δ: 139.01, 136.81, 131.45, 130.73, 127.92, 127.11, 125.67, 123.58, 123.13, 122.63, 120.23, 111.57, 48.35, 33.25, 28.96, 28.61, 27.95, 27.55, 25.38, 21.21, 20.58, 18.02 ppm. MS (ESI): C22H29NS (340.54) [M + H+]+. HRMS (ESI-Q-TOF) m/z: [M + H]+ Calcd. for C22H29NS 339.2021, found 340.2017 [M + H+]+.
8-(1,4,6-Trimethyl-9H-carbazol-9-yl)-octane-1-thiol (6b). Yellow oil (14 mg, 31%). 1H NMR (400 MHz, CDCl3) δ: 8.07 (s, 1H, Ar), 7.36–7.34 (m, 2H, Ar), 7.14 (d, J = 7.2 Hz, 1H, Ar), 6.95 (d, J = 7.2 Hz, 1H, Ar), 4.54–4.50 (m, 2H, NCH2), 2.93 (s, 3H, CH3), 2.84 (s, 3H, CH3), 2.75–2.72 (m, 2H CH2S), 2.64 (s, 3H, CH3), 1.85–1.82 (m, 2H, CH2), 1.75–1.71 (m, 2H, CH2), 1.65–1.33 (m, 9H, 4 CH2, SH) ppm. 13C NMR (100 MHz, CDCl3) δ: 139.01, 136.81, 131.45, 130.73, 127.92, 127.11, 125.67, 123.58, 123.13, 122.63, 120.23, 111.57, 48.35, 33.25, 28.96, 28.61, 27.95, 27.55, 25.38, 21.21, 20.58, 18.02 ppm. MS (ESI): C23H31NS (354.56) [M + H+]+. HRMS (ESI-Q-TOF) m/z: [M + H]+ Calcd. for C23H31NS 353.2177, found 354.2178 [M + H+]+.
9-(1,4,6-Trimethyl-9H-carbazol-9-yl)-nonane-1-thiol (7b). Yellow oil (18 mg, 51%). 1H NMR (400 MHz, CDCl3) δ: 8.03 (s, 1H, Ar), 7.36–7.32 (m, 2H, Ar), 7.11 (d, J = 7.2 Hz, 1H, Ar), 6.92 (d, J = 7.2 Hz, 1H, Ar), 4.53–4.49 (m, 2H, NCH2), 2.90 (s, 3H, CH3), 2.82 (s, 3H, CH3), 2.73–2.70 (m, 2H, CH2S), 2.60 (s, 3H, CH3), 1.83–1.77 (m, 2H, CH2), 1.72–1.62 (m, 2H, CH2), 1.43–1.31 (m, 11H, 5CH2, SH) ppm. 13C NMR (100 MHz, CDCl3) δ: 139.01, 136.81, 131.45, 130.73, 127.92, 127.11, 125.67, 123.58, 123.13, 122.63, 120.23, 111.57, 48.35, 33.25, 28.96, 28.61, 27.95, 27.55, 25.38, 21.21, 20.58, 18.02 ppm. MS (ESI): C24H33NS (368.59) [M + H+]+. HRMS (ESI-Q-TOF) m/z: Calcd. for C24H33NS 367.2334, found 368.2336 [M + H+]+.
7-(6-Bromo-1, 4-dimethyl-9H-carbazol-9-yl)-heptane-1-thiol (5c). Yellow oil (22 mg, 48%). 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H, Ar), 7.54–7.51 (m, 1H, Ar), 7.29–7.26 (m, 1H, Ar), 7.12–7.10 (m, 1H, Ar), 6.91 (d, J = 7.3 Hz, 1H, Ar), 4.49–4.46 (m, 2H, NCH2), 2.81 (s, 3H, CH3), 2.77 (s, 3H, CH3), 2.53–2.47 (m, 2H, CH2S), 1.81–1.77 (m, 2H, CH2), 1.60–1.55 (m, 2H, CH2), 1.41–1.25 (m, 7H, 3CH2, SH) ppm. 13C NMR (100 MHz, CDCl3) δ: 142.37, 139.01, 130.73, 128.14, 127.83, 127.11, 124.71, 123.13, 122.63, 120.23, 118.45, 112.31, 48.35, 33.25, 28.96, 28.61, 27.95, 27.55, 25.38, 20.58, 18.02 ppm. MS (ESI): C21H26BrNS (405.41) [M + 2]+. HRMS (ESI-Q-TOF) m/z: Calcd. for C21H26BrNS 403.0969, found 405.4066 [M + 2]+.
8-(6-Bromo-1, 4-dimethyl-9H-carbazol-9-yl)-octane-1-thiol (6c). Yellow oil (14 mg, 31%). 1H NMR (400 MHz, CDCl3) δ 8.26 (s, 1H, Ar), 7.53–7.51 (m, 1H, Ar), 7.28–7.26 (m, 1H, Ar), 7.11–7.09 (m, 1H, Ar), 6.91–6.89 (m, 1H, Ar), 4.48–4.44 (m, 2H, NCH2), 2.81 (s, 3H, CH3), 2.77 (s, 3H, CH3), 2.68–2.63 (m, 2H, CH2S), 1.80–1.76 (m, 2H, CH2), 1.66–1.62 (m, 2H, CH2), 1.36–1.30 (m, 9H, 4CH2, SH) ppm. 13C NMR (100 MHz, CDCl3) δ: 142.37, 139.01, 130.73, 128.14, 127.83, 127.11, 124.71, 123.13, 122.63, 120.23, 118.45, 112.31, 48.35, 33.25, 28.96, 28.61, 27.95, 27.55, 25.38, 20.58, 18.02 ppm. MS (ESI): C22H28BrNS (419.43) [M + 2]+. HRMS (ESI-Q-TOF) m/z: Calcd. for C22H28BrNS 417.1126, found 418.1129 [M + H+]+.
9-(6-Bromo-1,4-dimethyl-9H-carbazol-9-yl)-nonane-1-thiol (7c). Yellow oil (12 mg, 29%). 1H NMR (400 MHz, CDCl3) δ 8.26 (s, 1H, Ar), 7.53–7.51 (m, 1H, Ar), 7.28–7.26 (m, 1H, Ar), 7.11–7.09 (m, 1H, Ar), 6.91–6.89 (m, 1H, Ar), 4.46–4.44 (m, 2H, NCH2), 2.81 (s, 3H, CH3), 2.77 (s, 3H, CH3), 2.68–2.64 (m, 2H, CH2S), 1.81–1.74 (m, 2H, CH2), 1.69–1.65 (m, 2H, CH2), 1.37–1.26 (m, 11H, 5CH2, SH) ppm. 13C NMR (100 MHz, CDCl3) δ 142.37, 139.01, 130.73, 128.14, 127.83, 127.11, 124.71, 123.13, 122.63, 120.23, 118.45, 112.31, 48.35, 33.25, 28.96, 28.61, 27.95, 27.55, 25.38, 20.58, 18.02 ppm. MS (ESI): C23H30BrNS (433.46) [M + 2]+. HRMS (ESI-Q-TOF) m/z: Calcd. for C23H30BrNS 431.1282, found 432.1129 [M + H+]+.
Biological assay
Cell cultures
The six cell lines used in this work were purchased from American Type Culture Collection (ATCC, Manassas, VA). MCF-7 human breast cancer cells, oestrogen receptor (ER) positives, were maintained in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 Ham (DMEM/F12), supplemented with 5% foetal bovine serum (FBS) and 100 U/ml penicillin/streptomycin, as previously describedCitation36. MDA-MB-231, human breast cancer cells, oestrogen receptor (ER) negative, were maintained in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 Ham (DMEM/F12), supplemented with 5% Newborn Calf Serum (NCS) and 100 U/ml penicillin/streptomycin. HeLa human epithelial cervix carcinoma cells, oestrogen receptor (ER) negative, and ISHIKAWA human endometrial adenocarcinoma cell, oestrogen receptor positive, were maintained in minimum essential medium (MEM), supplemented with 10% FBS, 100 U/ml penicillin/streptomycin and 1% non-essential amino acid. 3T3-L1 cells line of murine fibroblasts of embryonic type, were maintained in DMEM, supplemented with 10% NCS and 100 U/ml penicillin/streptomycin. MCF-10A human mammary epithelial cells were cultured in DMEM/F12 medium, supplemented with 5% horse serum (HS) (Eurobio, Les Ullis, Cedex, France), 100 U/ml penicillin/streptomycin, 0.5 mg/ml hydrocortisone, 20 ng/ml hEGF (human epidermal growth factor), 10 μg/ml insulin and 0.1 mg/ml cholera enterotoxin (Sigma-Aldrich, Milano, Italy). Cells were maintained at 37 °C in a humidified atmosphere of 95% air and 5% CO2 and periodically screened for contaminationCitation37.
Cell viability
MDA-MB-231, MCF-7, HeLa and ISHIKAWA cells were grown in complete medium and, before being treated, they were serum deprived for 24 h, to allow cell cycle synchronisation. Then cells were grown in phenol red-free medium supplemented with 1% DCC (destran-coated charcoal treated) FBS, then treated with increasing concentrations (0.1, 1, 10, 20, 40, 100 µM) of each compound for 72 h. Compounds were dissolved in dimethylsulphoxide (DMSO) (Sigma, St. Louis, MO) and diluted in each cell medium. Untreated cells, used as a control, were added with DMSO alone (final concentration 0.1%). Cell viability was assessed using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide reagent (MTT), according to the protocol from manufacturer (Sigma-Aldrich, Milan, Italy), as previously describedCitation37,Citation38. For each sample, the mean absorbance, measured at 570 nm, was expressed as a percentage of the control and plotted versus drug concentration to determine for each cell line the IC50 values (namely, drugs concentrations able to reduce cell viability of 50% with respect to the control), using GraphPad Prism 5 Software (GraphPad Inc., San Diego, CA)Citation39. Each experiment was carried out in quadruplicate. Standard deviations (SD) were shown.
Cell proteins extraction and immunoblot analysis
Cells were serum deprived for 24 h, treated for 24, 48 and 72 h with the compound to be tested. At the end, cells were rinsed twice with ice cold PBS and immediately lysed in lysis buffer (50 mM Tris-HCl, pH 7, 5–8; 150 mM NaCl, 1% Igepal CA-630; 0.1% SDS; 0.5% sodium deoxycholate) containing 1% sodium orthovanadate and 1% phenylmethylsulphonyl fluoride (PMSF). Cell lysates were cleared by centrifugation at 14,000 g for 10 min at 4 °C. Protein concentrations were determined using the Bradford protein assayCitation40 (Bio-Rad Laboratories, Milan, Italy) according to the directions from the manufacturer. Equal amounts of cell extracts (about 20 µg) were resolved under denaturing conditions by electrophoresis in 10% polyacrylamide gels containing SDS (SDS-PAGE) and transferred to nitrocellulose membranes by electroblotting (GE Healthcare, Piscataway, NJ). Membranes were first stained with Ponceau S, washed with 1× TBST (Tween-20 0,1%, Tris/HCl 10 mM e NaCl 100 mM), incubated with TBST containing 5% milk for 1 h (blocking solution) and, then, incubated overnight at 4 °C with the primary antibody raised against Parp-1(7150) or GADPH (25778), purchased from Santa Cruz-Biotechnology Inc. (Santa Cruz, CA) and used at 1:500 and 1:2000 dilutions, respectively. After incubation with the appropriate secondary antibody (VWR International PBI, MI, Italy) for 1 h at room temperature, the proteins of interest were detected on the membranes by using enhanced chemiluminescence (Amersham ECL Prime Western Blotting Detection Reagent, GE Healthcare, Piscataway, NJ).
TUNEL assay
Apoptosis was detected by the TUNEL assay, according to the guidelines of the manufacturer (CF™488A TUNEL Assay Apoptosis Detection Kit, Biotium, Hayward, CA)Citation8. Briefly, cells were grown on glass coverslips and, after treatment, they were washed trice with PBS, then methanol-fixed at −20 °C for 15 min. Fixed cells were washed trice with 0.01% (V/V) Triton X-100 in PBS and incubated with 100 µL of TUNEL equilibration buffer for 5 min. After its removal, 50 µL of TUNEL reaction mixture containing 1 µL of terminal deoxynucleotidyl transferase (TdT) were added to each sample and incubated in a dark and humidified chamber for 2 h at 37 °C. Samples were washed trice with ice-cold phosphate-buffered saline (PBS) containing 0.1% Triton X-100 and 5 mg/mL bovine serum albumin (BSA). 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) (0.2 µg/mL) counterstain was performed (10 min, 37 °C, dark and humidified conditions). After three additional washes with cold PBS, one drop of mounting solution was added, then they were observed and imaged under a fluorescence microscope (Leica DM 6000, Leica, Frankfurt am Main, Germany) (20× magnification) with excitation/emission wavelength maxima of 490 nm/515 nm (CFTM488A) or 350 nm/460 nm (DAPI). Representative fields were shown. The experiments were repeated three timesCitation41.
Immunofluorescence
Cells were grown on glass coverslips in full media, then serum-deprived for 24 h and exposed to compound to test, at the indicated time. Then, they were PBS-washed, fixed with cold methanol (15 min/−20 °C) and washed three times (10 min/room temperature) with cold PBS containing 0.01% TritonX-100. After incubation (30 min/room temperature) with blocking solution (PBS, 2% BSA), they were incubated with primary antibody diluted in blocking solution (4 °C/overnight). The mouse anti-cytochrome c (556433) was purchased from BD Biosciences (Franklin Lakes, NJ) and used at 1:100 dilution. Coverslips were then washed three times with PBS, then fixed cells were incubated with the secondary antibody Alexa Fluor® 568 conjugate goat-anti-mouse (1:500, Thermo Fisher Scientific, Waltham, MA). Nuclei were stained using DAPI (Sigma-Aldrich, Milan, Italy) for 10 min at a concentration of 0.2 µg/mL then washed three times with PBS. Fluorescence was detected using a fluorescence microscope (Leica DM 6000 Leica, Frankfurt am Main, Germany). LASX software was used to acquire and process all imagesCitation42.
Caspase assay
Caspases-3/7, -8 and -9 activities were measured with the Caspase-Glo Assay, according to the guidelines of manufacturer (Caspase-Glo® 3/7, 8 and 9 Assay Systems, Promega Corporation, Madison, WI) as described in Iacopetta et al.Citation43.
Statistical analysis
Data were analysed for statistical significance (p < .001) using one-way ANOVA followed by Dunnett’s test performed by GraphPad Prism 5 (GraphPad Software, La Jolla, CA). Standard deviations (SD) are shown.
Human topoisomerase I relaxation assay: hTopoI relaxation assays were performed as reported in Iacopetta et al.Citation8.
Human topoisomerase II decatenation assay: hTopoII decatenation assays were performed as reported in Iacopetta et al.Citation8.
Computer modelling and docking simulations: The crystal structure of hTopoII in complex with a short DNA fragment and the ATP-analogue AMPPNP [PDB Code 4GFH] has been used as a template to build the complete three-dimensional models of hTopoII, as previously describedCitation8. The atomic coordinates of the three-dimensional model obtained were then used as a target for all the following molecular docking simulations. The ligand structures have been built and energy minimised using the program MarvinSketch [ChemAxon ltd, Budapest, Hungry]. In order to investigate the possible binding modes of 5c to the protein, calculations were carried out with “Achilles” Blind Docking Server, available at: http://bio-hpc.eu/software/blind-docking-server/. Using a “blind docking” approach, the docking of the small molecule to the targets is done without a priori knowledge of the location of the binding site by the systemCitation44. Figures were drawn using the program ChimeraCitation45.
Results and discussion
Chemistry
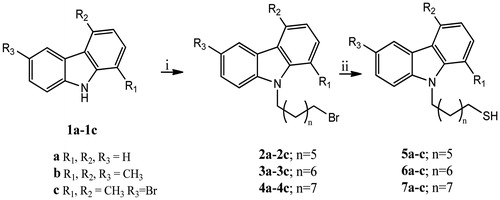
The N-thioalkylcarbazole derivatives (5, 6, 7a-c) were synthesised by the reaction of appropriate 9-(bromoalkyl)-9H-carbazoles (2, 3, 4a-c) with thiourea in i-PrOH (at reflux for 12 h), under a nitrogen atmosphere (Scheme 1). The obtained residue, after removal of the solvent, was first treated with 6 N NaOH (reflux for 5 h), and then with 3 N HCl (at room temperature, hereafter rt). The obtained crude was purified by PTLC (n-hexane/ethyl acetate 97:3) to give the pure compounds. The intermediates 9-(bromoalkyl)-9H-carbazoles (2, 3, 4a–c) were prepared following general synthetic methodsCitation28. In particular, carbazole derivatives (1a–c) and dry DMF were stirred at rt until became clear. Then, NaH 60% oil dispersion and, successively, the appropriate dibromoalkane were added at 0 °C, and the mixture was stirred for 5 h at rt. Finally, water was added and the resulting mixture was extracted with EtOAc. The obtained residue was purified by silica gel column chromatography (Et2O/hexane, 2/3 as eluent) to give the pure compounds.
Scheme 1. Synthesis of N-9H-alkyltiocarbazoles 5–7a–c. Reagents and conditions: (i) 1, N-dibromoalkane, NaH 60%, DMF, rt; (ii) Thiourea, i-PrOH, reflux, 12 h; NaOH 6 N, reflux, 5 h; HCl 3 N, rt, 10 min.
Antiproliferative activity
All synthesised carbazole derivatives (5a–c, 6a–c, 7a–c) were examined to test their antiproliferative activity against two human breast cancer cell lines, namely the MCF-7 cells expressing the oestrogen receptor α (ERα), and the MDA-MB-231 triple negative cells (ER-, PR- and HER-2/Neu not amplified), and two cell lines of uterine cancer, which are the Hela cells, from uterine cervix epithelium (ER−), and endometrial ISHIKAWA cells (ER+). Cells were subjected to 72 h of continuous exposure to the compounds to test and, at the end, their viability was measured by MTT assay. Ellipticine, which represents the main natural molecule containing the carbazole moiety, was used as reference molecule for these studies. The results, summarised in , clearly indicate that only two compounds, 5a and 5c, possess an antitumour activity. In both breast cancer cells, a moderate antitumour activity was noticed; indeed the IC50 values for the compounds 5a and 5c in MCF-7 cells were found to be, respectively, 50.4 ± 0.96 µM and 54.9 ± 1.00 µM. In MDA-MB-231 cells, the IC50 values resulted lower than those determined for the MCF-7 cells and were 39.9 ± 0.52 µM and 27.2 ± 0.75 µM, respectively. However, a higher antitumour activity was found against HeLa and ISHIKAWA cells, where compound 5a showed IC50 values of 12.8 ± 0.62 µM and 19.2 ± 1.13 µM, respectively. Compound 5c showed IC50 values of 11.3 ± 0.63 µM (HeLa) and 15.7 ± 0.95 µM (ISHIKAWA). These data set the compound 5c the most active and selective for uterine tumour cells. Moreover, both compounds did not affect the proliferation of MCF-10A cells, human non-malignant breast epithelial cells, and 3T3-L1 murine fibroblast-like cells, at least at doses lower than 100 µM. Finally, Ellipticine demonstrates a higher antitumour activity in all cell lines used in these assays with respect to compounds 5a and 5c, but together with dramatic cytotoxic effects on normal cells (). Thus, a relevant antitumour activity was obtained for compound 5c, which hold two methyl substituents at the position 1 and 4, a bromine at position 6 and a 7 terms chain on the carbazole ring nitrogen. The replacement of bromine at position 6 with a methyl group (5b)Citation12,Citation22,Citation24,Citation25,Citation28 and the elongation of the chain produced a loss of the antitumour activity (6c and 7c). With respect to compound 5c, a lesser antitumour activity was noticed for compound 5a. The latter does not possess substituents on the carbazolic scaffold and holds a 7 terms chain on the nitrogen at position 9.
Table 1. Antiproliferative activity: IC50 values of carbazole derivatives (5a–c, 6a–c, 7a–c) and Ellipticine, expressed in micromolar (µM).
Human topoisomerases I and II inhibition assays
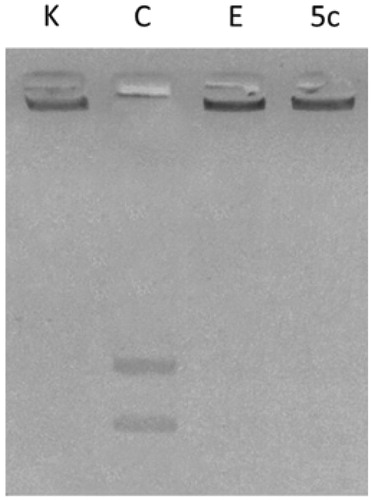
Several studies reported that carbazole derivatives and Ellipticine are inhibitors of human Topoisomerases I and II. In particular, Ellipticine exhibits multi-modal mechanism of action with DNA intercalation and hTopoII inhibitionCitation46–48. For these reasons, the most active compound 5c, structurally correlated to Ellipticine, was evaluated for its ability to inhibit DNA metabolising enzymes, i.e. hTopoI and II direct in vitro assays. The latter evidenced no inhibition of the hTopoI (data not shown) upon 5c exposure, at least under the adopted experimental conditions. On the contrary, in hTopo II decatenation assays, the same compound was able to inhibit hTopoII activity at 10 µM concentration (, lane 5c), preventing the hTopoII from cutting the catenated kinetoplast DNA (KDNA), as instead happens in the experiment control (, lane C) where the enzyme releases the nicked open circular minicircles and fully closed circular rings (decatenation products) visible at the bottom of gel as two distinct DNA bands. As well in these experiments, Ellipticine was used as reference molecule (, lane E) at the concentration of 50 µM to obtain a complete inhibitionCitation27.
Figure 2. hTopoII inhibition assay. kDNA was incubated with human Topoisomerase II in the absence (lane C, vehicle DMSO) or presence of compound 5c (lane 5c) or Ellipticine (lane E) at 10 and 50 µM, respectively. Lane K, kinetoplast DNA (kDNA).
Docking simulations
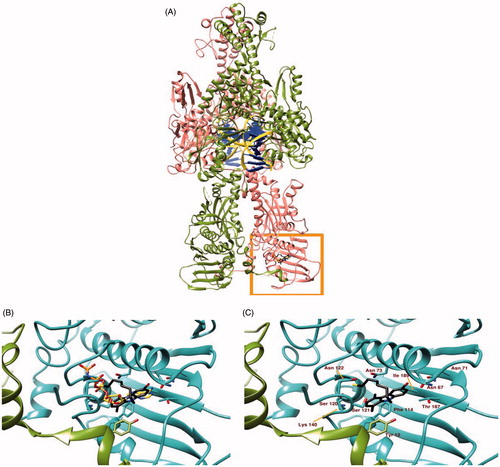
The binding site of 5c to hTopoII was identified by our docking simulation as coincident to the ATP-binding site (, panel A), with the docked moiety almost perfectly superposed to the one of ANP-PNP (a non-hydrolysable ATP analogue), determined experimentally through X-ray diffraction (, panel B). The binding site includes different amino acids from both chains of hTopoII, which contribute to binding stability mostly through polar and hydrophobic interactions (, panel C). The best pose for molecule in the most populated cluster has a binding energy of −7.10 kcal/mol, as calculated by the server “Achilles”. A visual inspection of the crystallographic structure of hTopoII in complex with AMPPNP indicates that (1) the adenine moiety is hydrogen bonded to residues Asn 67, Asn 92 and Thr 187; (2) the ribose ring is stabilised by hydrogen bonds with Ser 120 and Ser 121; (3) the phosphates tail interacts with the peptidic nitrogen of Gly 138 and with the side chains of Asn 63 Ser 120, Asn 122, Lys 140, Gln 348 and Lys 350 through hydrogen bonds. Our simulation poses 5c with the carbazole moiety almost superposed to the adenine of the crystallographic AMPPNP, with the bromine interacting with Asn 92 and Thr 187 through halogen bonds and the SH group forming a weak hydrogen bond to Asn 122. Residues Asp 66, Ile 97, Ile 113, Phe 114, and Ala 139 further contribute to stabilise 5c binding mode to hTopoII by hydrophobic interactions.
Figure 3. Panel (A) Ribbon representation of a dimeric hTopoII (chains A and B coloured in pink and olive green respectively) in complex with DNA (yellow ribbon and blue lego). The area within the orange rectangle highlights ATP binding site. Panel (B) A close up of the ATP binding site showing the different binding modes of AMP-PNP, a non-hydrolisable ATP analogue, (orange) and 5c (black sticks) as suggested by docking simulations. Panel (C) shows the protein residues involved in 5c binding.
Compound 5c induces parp-1 cleavage
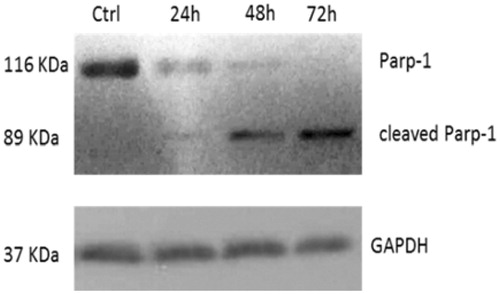
The observed inhibition of hTopoII pushed us to evaluate whether cells were undergoing cell death by apoptosis. The Topoisomerase II, indeed, plays an important role in DNA replication, transcription, recombination, thus its inhibition provokes genotoxic and mutagenic effects, leading to cell death. In response to DNA damage, cells may utilise the poly-(ADP) ribose polymerase (Parp-1), a nuclear enzyme that catalyses the transfer of the ADP-ribose portion of NAD+ into a specific subgroup of nuclear substratesCitation49. Parp-1 is an important regulator of the DNA base excision repair (BER) pathway and it is involved in the maintenance of genomic integrity and survival following genotoxic insults. Parp-1 is specifically proteolysed by executioner caspases (caspases 3 and 7) to an 89 kDa catalytic fragment during the final steps of apoptosisCitation50,Citation51. Indeed, Parp-1 cleavage promotes apoptosis by preventing DNA repair-induced survival and across the block of energy depletion induced by necrosisCitation52. With this in mind, we treated HeLa cells with compound 5c at a concentration of 10 µM and at three different times (24, 48 and 72 h). At the end of the treatment, the cells were lysed and the total cellular protein content was extracted and used for western blot analyses. The obtained results, reported in , showed the presence of the cleaved form of Parp-1 (about 89 KDa) in cells treated with compound 5c, visible at 24 h and increased at 48 and 72 h, together with a decrease of the native protein, due to the cleavage. At 72 h, the only appreciable form is the cleaved one, whereas, on the contrary, in vehicle-treated cells only the whole form is present and not the cleaved one. In , the shown control is referred only to 72 h, but same results were obtained at 24 and 48 h (data not shown).
Figure 4. Parp-1 cleavage: time course of PARP-1 cleavage in uterine cancer Hela cells treated with 5c, used at a concentration equal to its IC50 value. The control shown is referred to 72 h of vehicle exposure, but same results was obtained for 24 and 48 h. GAPDH was used as loading normalisation.
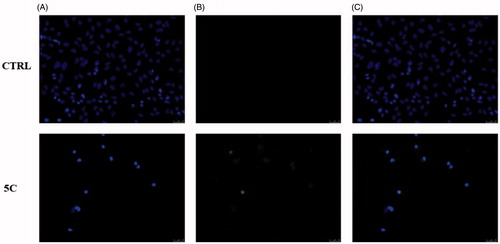
In order to assess whether the observed cytotoxic effects could depend on the induction of apoptosis under treatment, we exposed Hela cells to 5c compound, at the concentration equal to its IC50 value for 24 h. As in , a green nuclear fluorescence (5c, panel B) indicates DNA damage and that cells have undergone the apoptotic process due to the exposure to the 5c compound, whereas no fluorescence resulted in the vehicle-treated cells used as experiment control (, CTRL, panel B). These data confirm that the selected compound induced DNA damage and led to cell death triggering the apoptotic mechanism.
Figure 5. TUNEL assay of HeLa cells. Cells were treated for 24 h with compound 5c (at the concentration equal to its IC50) or with vehicle (CTRL). Then they were fixed, subjected to TdT reaction, washed, dyed with DAPI and imaged under a fluorescence microscope (20× magnification, excitation/emission wavelength 490 nm/515 nm for CF™488 A, panels B, and 350 nm/460 nm for DAPI, panels A). Panels C show the merge.
Compound 5c induces apoptosis by the intrinsic pathway
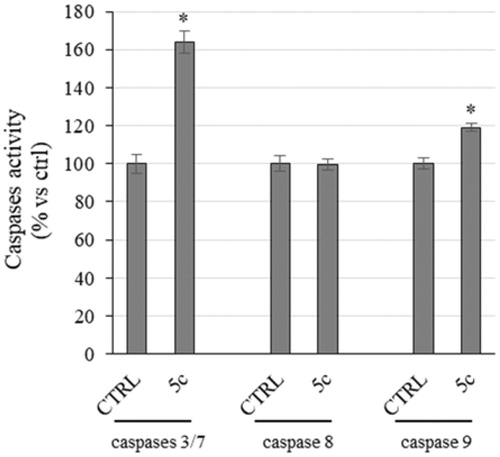
Even though the mechanisms of apoptosis are complexes and involve an energy-dependent cascade of molecular events, it is noteworthy that there are at least two main pathways,Citation53 not necessarily independent from each other, i.e. the extrinsic or death receptor pathway and the intrinsic or mitochondrial oneCitation54. In order to establish which pathway was involved, we evaluate the activity of initiators caspases, namely caspases 8 and 9, that are involved, respectively, in the extrinsic and intrinsic pathway, and effectors or executioner caspases 3 and 7, by means of a luminescent assay. Caspases are usually expressed in an inactive form and after activation they may initiate a proteolytic cascade,Citation55 in which one caspase may activate other caspases or cleave cell proteins, included Parp-1, amplifying the apoptotic cell deathCitation53. Effectively, we noticed a little, but significant, rise of caspase-9 activity in HeLa cells treated with compound 5c for 24 h, whereas no increase of caspase-8 activity was evidenced (). Moreover, we observed a clear increase of caspases 3 and 7 activity (), which are cleaved by the initiator caspase-9 and that, in turn, may activate other pro-apoptotic proteins and cleave Parp-1, as we already observed (). Summing up, the exposure of HeLa cells to compound 5c induces the activation of caspase-9 that activates caspases 3/7, responsible of the observed Parp-1 cleavage that provokes the inability of DNA reparation processes and leads to cell death by apoptosis.
Figure 6. Caspases activity. Activation of caspases 3/7 and 9 following to the treatment of HeLa cells with the compound 5c at a concentration equal to its IC50 value for 24 h. Columns mean, bars SD, *p < .001.
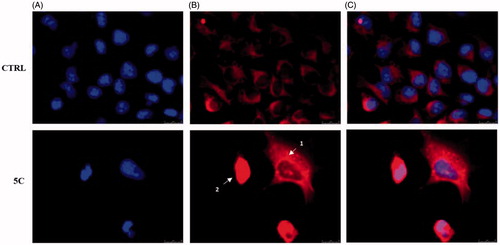
Another important event of the intrinsic apoptotic pathway is represented by the release of Cytochrome c from mitochondria to the cytosolCitation56. Thus, we wanted to evaluate the localisation of Cytochrome c, by means of immunofluorescence assays in HeLa cells treated with the compound 5c for 24 h at the concentration equal to its IC50, and fixed (see experimental section for details). These assays confirmed that compound 5c determines the release of Cytochrome c from mitochondria to the cytosol (). Indeed, the fluorescence (red colour in ) is diffused and delocalised throughout the cell cytoplasm, whereas in vehicle-treated cells (CTRL), the fluorescence is strictly localised in well-defined areas of the cell, given that Cytochrome c is normally localised in the mitochondrial compartment. In , it should be evidenced how it is possible, as already reportedCitation57, to identify the different stages of mitochondria disruption during apoptosis. In fact, a punctiform fluorescence in the cytosol (white arrow 1) indicates mitochondria fragmentation and partial release of Cytochrome c into the cytosol, which occurs during the early stages of apoptosis. Successively, the release of Cytochrome c is complete (white arrow 2, ) and the fluorescence (red) is greatly increased and spread throughout the cytosol. On the contrary, in cells treated with the vehicle only, the fluorescence (red) is defined and organised in an ordered and filamentous way, indicating that mitochondria are intact.
Figure 7. Cytochrome c release during apoptosis in Hela cells. In vehicle-treated cells Cytochrome c is localised within the mitochondria (panel B, CTRL). During the early stages of apoptosis, induced by compound 5c treatment at the concentration equal to its IC50 for 24 h, mitochondria fragmentation occurs (as shown by the red dots indicated by arrow 1 in panel B, 5c) and Cytochrome c is partially released. In the last apoptotic cell stage, a full release of Cytochrome c is observed (diffused red fluorescence, indicated by the white arrow 2). Panel A, DAPI (excitation/emission wavelength 350 nm/460 nm), panel B, Alexa Fluor® 568 (excitation/emission wavelength 578 nm/603 nm), panel C, overlay. Images were acquired at 63× magnification and are representative of three separated experiments.
Conclusions
Cancer is one of the leading causes of death in economically developed countries, probably due to population aging and lifestyle choices, such as smoking, lack of physical activity and unbalanced dietsCitation58. Among the human cancers, breast and uterine cancers represent the death cause of almost the 26% and 7% of women, respectivelyCitation59. Even though the earlier diagnosis, as result of the publicity surrounding the disease, and its prevention increased the survival, the availability of new and improved chemotherapies facing the resistance occurrence and the toxic side effects is still a major challenge. Under this point of view, the preclinical research may offer a panel of new potential anticancer molecules that may be useful as valid alternative to the classic therapies or in co-treatment. Herein, we have studied a series of new N-thioalkylcarbazole derivatives, structurally correlated to the most known Ellipticine, which exert interesting antiproliferative effects on two in vitro models of the most common cancer types in women. Our outcomes indicates that two derivatives, 5a and 5c, possess a good antitumour activity against two models of breast cancer cells and a higher selectivity toward the cervical and endometrial cancer cells. Even though Ellipticine, the ancestor of carbazoles, possess a higher, but non-selective, activity compared with these derivatives, it should be evidenced how these properties come together with very high cytotoxic effects on non-tumoural cells used in these studies. On the contrary, the two lead molecules, did not affect the viability of non-tumoural cells, at least at the doses lower than 100 µM. The underlying molecular mechanism by which the most active compound exerts its antiproliferative activity is represented by the inhibition of hTopoII and the activation of the mitochondrial or intrinsic apoptotic pathway. Indeed, the treatment of HeLa cells with compound 5c induces caspase-9 activation, which targets several proteins, amongst them caspases 3 and 7, known as executioner caspases. The latter are able to continue the cascade of proteolytic cleavages, as we observed for Parp-1, one of the most important protein involved in DNA damage repair. Since Parp-1 is cleaved, cells are unable to repair their DNA, which increased damage, together with hTopoII inhibition, is the major stimuli that direct cells to die by apoptosis. Effectively, Hela cells treated with 5c are clearly undergoing apoptosis, as demonstrated by TUNEL assay and, particularly, through the mitochondrial mechanism. Indeed, immunofluorescences studies evidenced as Cytochrome c is released from its natural site, namely mitochondria, to the cytosol of treated Hela cells. Considered the biological features of compound 5c, we are confident that this lead compound may be a useful and interesting tool in the treatment of uterine cancer.
Acknowledgements
C. R. would like to acknowledge networking contribution by the COST Action CM1407 “Challenging organic syntheses inspired by nature – from natural products chemistry to drug discovery”.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
Related Research Data
References
- Caruso A, Iacopetta D, Puoci F, et al. Carbazole derivatives: a promising scenario for breast cancer treatment. Mini Rev Med Chem 2016;16:630–43.
- Caruso A, Chimento A, El-Kashef H, et al. Antiproliferative activity of some 1,4-dimethylcarbazoles on cells that express estrogen receptors: Part I. J Enzyme Inhib Med Chem 2012;27:609–13.
- Gluszynska A. Biological potential of carbazole derivatives. Eur J Med Chem 2015;94:405–26.
- Carocci A, Catalano A, Sinicropi MS. Melatonergic drugs in development. Clin Pharmacol 2014;6:127–37.
- Saturnino C, Iacopetta D, Sinicropi MS, et al. N-Alkyl carbazole derivatives as new tools for Alzheimer's disease: preliminary studies. Molecules 2014;19:9307–17.
- Zhu D, Chen M, Li M, et al. Discovery of novel N-substituted carbazoles as neuroprotective agents with potent anti-oxidative activity. Eur J Med Chem 2013;68:81–8.
- Bashir M, Bano A, Ijaz AS, Chaudhary BA. Recent developments and biological activities of N-substituted carbazole derivatives: a review. Molecules 2015;20:13496–517.
- Iacopetta D, Rosano C, Puoci F, et al. Multifaceted properties of 1,4-dimethylcarbazoles: Focus on trimethoxybenzamide and trimethoxyphenylurea derivatives as novel human topoisomerase II inhibitors. Eur J Pharmaceut Sci: Off J Eur Feder Pharmaceut Sci 2017;96:263–72.
- Vann KR, Ergun Y, Zencir S, et al. Inhibition of human DNA topoisomerase IIalpha by two novel ellipticine derivatives. Bioorg Med Chem Lett 2016;26:1809–12.
- Maji B, Kumar K, Muniyappa K, Bhattacharya S. New dimeric carbazole-benzimidazole mixed ligands for the stabilization of human telomeric G-quadruplex DNA and as telomerase inhibitors. A remarkable influence of the spacer. Org Biomol Chem 2015;13:8335–48.
- Andrews WJ, Panova T, Normand C, et al. Old drug, new target: ellipticines selectively inhibit RNA polymerase I transcription. J Biol Chem 2013;288:4567–82.
- Sinicropi MS, Lappano R, Caruso A, et al. Bromo-1,4-dimethyl-9H-carbazol-3-yl-methylene)-hydrazine (carbhydraz) acts as a GPER agonist in breast cancer cells. Curr Top Med Chem 2015;15:1035–42.
- Parisi OI, Morelli C, Puoci F, et al. Magnetic molecularly imprinted polymers (MMIPs) for carbazole derivative release in targeted cancer therapy. J Mater Chem B 2014;2:6619–25.
- Stiborova M, Frei E. Ellipticines as DNA-targeted chemotherapeutics. Curr Med Chem 2014;21:575–91.
- Isah T. Rethinking Ginkgo biloba L.: medicinal uses and conservation. Pharmacogn Rev 2015;9:140–8.
- Deane FM, O'Sullivan EC, Maguire AR, et al. Synthesis and evaluation of novel ellipticines as potential anti-cancer agents. Organ Biomol Chem 2013;11:1334–44.
- Miller CM, O'Sullivan EC, Devine KJ, McCarthy FO. Synthesis and biological evaluation of novel isoellipticine derivatives and salts. Organ Biomol Chem 2012;10:7912–21.
- Deane FM, O'Sullivan EC, Maguire AR, et al. Synthesis and evaluation of novel ellipticines as potential anti-cancer agents. Org Biomol Chem 2013;11:1334–44.
- Harding MM, Grummitt AR. 9-hydroxyellipticine and derivatives as chemotherapy agents. Mini Rev Med Chem 2003;3:67–76.
- Saturnino C, Buonerba M, Boatto G, et al. Synthesis and preliminary biological evaluation of a new pyridocarbazole derivative covalently linked to a thymidine nucleoside as a potential targeted antitumoral agent. I. Chem Pharm Bull (Tokyo) 2003;51:971–4.
- Caruso A, Voisin-Chiret AS, Lancelot JC, et al. Efficient and simple synthesis of 6-aryl-1,4-dimethyl-9H-carbazoles. Molecules 2008;13:1312–20.
- Caruso A, Sinicropi MS, Lancelot JC, et al. Synthesis and evaluation of cytotoxic activities of new guanidines derived from carbazoles. Bioorg Med Chem Lett 2014;24:467–72.
- Sinicropi MS, Caruso A, Conforti F, et al. Synthesis, inhibition of NO production and antiproliferative activities of some indole derivatives. J Enzyme Inhib Med Chem 2009;24:1148–53.
- Cresteil T, Rault S, Aubert G, Lancelot JC, Caruso A, A. L, inventors. 2012.
- Panno A, Sinicropi MS, Caruso A, et al. New trimethoxybenzamides and trimethoxyphenylureas derived from dimethylcarbazole as cytotoxic agents. J Heterocycl Chem 2014;51:E294–302.
- Caruso A, Lancelot JC, El-Kashef H, et al. A rapid and versatile synthesis of novel pyrimido[5,4-b]carbazoles. Tetrahedron 2009;65:10400–5.
- Rizza P, Pellegrino M, Caruso A, et al. 3-(Dipropylamino)-5-hydroxybenzofuro[2,3-f]quinazolin-1(2H)-one (DPA-HBFQ-1) plays an inhibitory role on breast cancer cell growth and progression. Eur J Med Chem 2016;107:275–87.
- Saturnino C, Caruso A, Longo P, et al. Crystallographic study and biological evaluation of 1,4-dimethyl-N-alkylcarbazoles. Curr Top Med Chem 2015;15:973–9.
- Galsky MD, Hahn NM, Wong B, et al. Phase 2 trial of the topoisomerase II inhibitor, amrubicin, as second-line therapy in patients with metastatic urothelial carcinoma. Cancer Chemother Pharmacol 2015;76:1259–65.
- Yao BL, Mai YW, Chen SB, et al. Design, synthesis and biological evaluation of novel 7-alkylamino substituted benzo[a]phenazin derivatives as dual topoisomerase I/II inhibitors. Eur J Med Chem 2015;92:540–53.
- Andre V, Boissart C, Lechevrel M, et al. Mutagenicity of nitro- and amino-substituted carbazoles in Salmonella typhimurium. I. Monosubstituted derivatives of 9H-carbazole. Mutat Res 1993;299:63–73.
- Saturnino C, Palladino C, Napoli M, et al. Synthesis and biological evaluation of new N-alkylcarbazole derivatives as STAT3 inhibitors: preliminary study. Eur J Med Chem 2013;60:112–9.
- Sopkova-de Oliveira Santos J, Caruso A, Lohier JF, et al. 9-Ethyl-1,4-dimethyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9H-carbazole and 6-bromo-9-ethyl-1,4-dimethyl-9H-carbazole. Acta Crystallogr C 2008;64:o453–5.
- Lohier JF, Caruso A, Sopkova-de Oliveira Santos J, et al. tert-Butyl 6-bromo-1,4-dimethyl-9H-carbazole-9-carboxyl-ate. Acta Crystallogr Sect E Struct Rep Online 2010;66:o1971–2.
- Paesano N, Marzocco S, Vicidomini C, et al. Synthesis and biological evaluation of 3-benzyl-1-methyl- and 1-methyl-3-phenyl-isothioureas as potential inhibitors of iNOS. Bioorg Med Chem Lett 2005;15:539–43.
- Carocci A, Catalano A, Bruno C, et al. N-(Phenoxyalkyl)amides as MT(1) and MT(2) ligands: antioxidant properties and inhibition of Ca(2+)/CaM-dependent kinase II. Bioorg Med Chem 2013;21:847–51.
- Sirignano E, Saturnino C, Botta A, et al. Synthesis, characterization and cytotoxic activity on breast cancer cells of new half-titanocene derivatives. Bioorg Med Chem Lett 2013;23:3458–62.
- Sala M, Chimento A, Saturnino C, et al. Synthesis and cytotoxic activity evaluation of 2,3-thiazolidin-4-one derivatives on human breast cancer cell lines. Bioorg Med Chem Lett 2013;23:4990–5.
- Chimento A, Saturnino C, Iacopetta D, et al. Inhibition of human topoisomerase I and II and anti-proliferative effects on MCF-7 cells by new titanocene complexes. Bioorg Med Chem 2015;23:7302–12.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54.
- Iacopetta D, Grande F, Caruso A, et al. New insights for the use of quercetin analogs in cancer treatment. Future Med Chem 2017;9:2011–28.
- Iacopetta D, Lappano R, Cappello AR, et al. SLC37A1 gene expression is up-regulated by epidermal growth factor in breast cancer cells. Breast Cancer Res Treat 2010;122:755–64.
- Iacopetta D, Mariconda A, Saturnino C, et al. Novel Au and Ag carbene complexes exert anti-tumor effects triggering the ROS-dependent intrinsic apoptotic pathway. ChemMedChem 2017;12:2054–65.
- Sanchez-Linares I, Perez-Sanchez H, Cecilia JM, Garcia JM. High-throughput parallel blind virtual screening using BINDSURF. BMC Bioinformatics 2012;13(Suppl 14):S13.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12.
- Monnot M, Mauffret O, Simon V, et al. DNA-drug recognition and effects on topoisomerase II-mediated cytotoxicity. A three-mode binding model for ellipticine derivatives. J Biol Chem 1991;266:1820–9.
- Froelich-Ammon SJ, Patchan MW, Osheroff N, Thompson RB. Topoisomerase II binds to ellipticine in the absence or presence of DNA. Characterization of enzyme–drug interactions by fluorescence spectroscopy. J Biol Chem 1995;270:14998–5004.
- Kizek R, Adam V, Hrabeta J, et al. Anthracyclines and ellipticines as DNA-damaging anticancer drugs: recent advances. Pharmacol. Ther 2012;133:26–39.
- de Murcia JM, Niedergang C, Trucco C, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci USA 1997;94:7303–7.
- D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 1999;342: 249–68.
- Kaufmann SH, Desnoyers S, Ottaviano Y, et al. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res 1993;53:3976–85.
- Soldani C, Scovassi AI. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis 2002;7:321–8.
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007;35:495–516.
- Ouyang L, Shi Z, Zhao S, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif 2012;45:487–98.
- Cullen SP, Martin SJ. Caspase activation pathways: some recent progress. Cell Death Differ 2009;16:935–8.
- Kadam CY, Abhang SA. Apoptosis markers in breast cancer therapy. Adv Clin Chem 2016;74:143–93.
- Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev 2008;22:1577–90.
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.