
Abstract
GH20 human β-N-acetylhexosaminidases (hsHex) and GH84 human O-GlcNAcase (hOGA) are involved in numerous pathological processes and emerged as promising targets for drug discovery. Based on the catalytic mechanism and structure of the catalytic domains of these β-N-acetylhexosaminidases, a series of novel naphthalimide moiety-bearing thioglycosides with different flexible linkers were designed, and their inhibitory potency against hsHexB and hOGA was evaluated. The strongest potency was found for compound 15j (Ki = 0.91 µM against hsHexB; Ki > 100 µM against hOGA) and compound 15b (Ki = 3.76 µM against hOGA; Ki = 30.42 µM against hsHexB), which also exhibited significant selectivity between these two enzymes. Besides, inhibitors 15j and 15b exhibited an inverse binding patterns in docking studies. The determined structure–activity relationship as well as the established binding models provide the direction for further structure optimizations and the development of specific β-N-acetylhexosaminidase inhibitors.
1. Introduction
β-N-acetylhexosaminidases (EC 3.2.1.52) are widely distributed exo-glycosidases that catalyze the release of a β-linked N-acetyl-d-hexosamine unit from non-reducing ends of glycoconjugates. These enzymes are classified into three glycosyl hydrolase family (GH3, GH20 and GH84) based on amino acid sequence similarities in the CAZy classification system (http://www.cazy.org)Citation1,Citation2. And they are involved in various physiological functions, such as energy metabolismCitation3, cell communicationCitation4, cell proliferationCitation5 and inflammationCitation6. Among these, the GH20 human β-N-acetylhexosaminidases (hsHex) and GH84 human O-GlcNAcase (hOGA) are the most promising targets for drug development.
GH20 human β-N-acetylhexosaminidases (hsHex) have gained many attentions owing to their pivotal actions in osteoarthritis and lysosomal storage disorders. Research has shown that hexosaminidase is a dominant enzyme released into the extracellular compartment by chondrocytes in patients with osteoarthritis. Moreover, inhibition of the enzyme can prevent cartilage matrix degradation, providing a new avenue for treatments of osteoarthritisCitation6,Citation7. Likewise, lysosomal hexosaminidase can degrade GM2 gangliosides in neuronal cells. Dysfunction of this enzyme not only causes severe neurodegenerative lipid storage disorders, but also leads to Tay-Sachs or Sandhoff diseaseCitation8. Inhibitors of lysosomal hsHex can therefore be used as pharmacological chaperones to increase activity of mutant lysosomal enzyme while maintain its metabolic function at a normal levelCitation9,Citation10.
GH84 human O-GlcNAcase (hOGA) catalyzes the removal of O-GlcNAc from serine or threonine residues in glycoproteins, and has been found to link to Alzheimer’s disease (AD)Citation11. Evidences have shown that AD is closely associated with tau hyperphosphorylation in patient’s brain, while such site remains protected by O-GlcNAc in a healthy neuronCitation12. Thus, human O-GlcNAcase inhibitors that block tau hyperphosphorylation can help in the treatments of ADCitation13,Citation14.
A number of small molecule inhibitors against β-N-acetylhexosaminidases have been reported. These include PUGNAcCitation15 (1), NagstatinCitation16 (2), NAG-thiazolineCitation17 (3), pyrimethamineCitation18 (4), iminocyclitolsCitation19,Citation20, and naphthalimidesCitation21–24. Among these, PUGNAc, Nagstatin, and NAG-thiazoline are the three classic potent inhibitors, which however show non-selectivity between GH20 and GH84 β-N-acetylhexosaminidasesCitation4,Citation25. Pyrimethamine has been approved by FDA to be a safe and a suitable pharmacological chaperone of human lysosomal hsHex ACitation18. Although the well-studied inhibitors, iminocyclitols, always become handicapped in complex synthetic methodsCitation19,Citation20. It is worth mentioning that Ho has reported a series of iminocyclitol derivatives, in which alkylamine chains were attached to the iminocyclitol ringCitation26. The most potent compound 4 (5), containing two methoxyphenyl groups, has a Ki value of 0.69 nM against hsHexB and exhibits 250,000-fold higher selectivity toward GH84 hOGACitation26. Further docking analysis has revealed that a hydrophobic cleft located near −1 subsite (active pocket) and alkylamine chains that extend out into the cleft can acquire additional interactionsCitation26. Naphthalimides are β-N-acetylhexosaminidases inhibitors discovered during a high-throughput screening. One of these inhibitors, M-31850 (6) exhibits IC50 values of 6.0 µM and 3.1 µM against hsHexA and hsHexB, respectivelyCitation21. Following this discovery, a large number of non-carbohydrate-based naphthalimide derivatives are synthesized and evaluated as β-N-acetylhexosaminidases inhibitors by Qian and YangCitation22–24. An example of these is compound 7a (7), which exhibits high inhibitory potency with a Ki value of 0.63 µM against hsHex, 3.3 folds lower than that of M-31850Citation22. In addition, compound Q1 (8) shows inhibitory potency against GH20 insect β-N-acetylhexosaminidase OfHex1 with Ki values of 4.28 µM. The complex crystal structure of OfHex1-Q1 (PDB ID: 3WMB) reveals that the methylthiadiazole group of Q1 binds the −1 subsite of OfHex1, whereas the napthalimide group is sandwiched by the amino acid residues of the +1 subsite (outside of −1 subsite)Citation23.
These observations prompted us to choose N-acetyl-d-glucamine as the structural fragment to provide strong affinity toward −1 subsite of β-N-acetylhexosaminidases and convert the glycosidic bond to thioglycosidic bond to prevent hydrolysis by the enzymes. Then, the napthalimide groups with different linkers were further introduced into the structure to acquire additional interactions outside of the −1 binding subsite. Influences of length and nitrogen atom of the linkers between N-acetyl-d-glucamine and napthalimide group could also be compared. Thus, in this work, we synthesized a series of naphthalimide-bearing thioglycoside derivatives. Their inhibitory potency was evaluated in vitro against two enzymes, GH20 human hsHexB and GH84 hOGA. The kinetic and molecular docking studies were carried out to further explore their interaction mechanisms with hsHexB and hOGA ( and ).
Figure 1. Reported β-N-acetylhexosaminidases inhibitors.
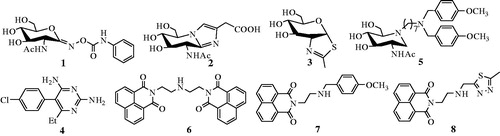
Figure 2. Design of thioglycoside inhibitors.
2. Experimental
2.1. Materials
All chemicals, reagents, and solvents were purchased from commercial sources. The solvents were dried prior to use. Reaction progress was monitored using thin layer chromatography (TLC) on pre-coated silica gel GF254 plates, in which the spots were detected by charring with 30% (v/v) H2SO4 in MeOH or by UV light (254 nm). Melting points were determined using a Ry-1 G melting point instrument. 1H NMR and 13 C NMR spectra were carried out on a Bruker AVANCE600 spectrometer in CDCl3 or DMSO-d6 at 25 °C and referenced to TMS. High-resolution mass spectra (HRMS) were collected by the Beijing Launcher Scientific Co. Ltd.
2.2. Chemical synthesis
2.2.1. Synthesis of compounds 10, 13a–13c, 19a–19e
Thiol 10 was prepared according to procedures described in the literatureCitation27,Citation28. Compounds 13a–13c were synthesized from 1,8-naphthalic anhydride as described previouslyCitation29. Compounds 19a–19e were prepared from 1,8-naphthalimide according to previous methodsCitation30. Data for compounds 10, 13a–13c and 19a–19e can be found in Supporting Information.
2.2.2. Synthesis of compounds 11a–11d and 16
Following thiol 10 (5.5 mmol, 1.0 eq) was dissolved in acetone (20 ml) and H2O (10 ml), solid potassium carbonate (6.6 mmol, 1.2 eq) and α,ω-dibromoalkane (44 mmol, 8.0 eq) were added. The mixture was stirred for 20 h at room temperature and subsequently concentrated in vacuo. The resulting residue was diluted with DCM (50 ml), washed with H2O (100 ml), brine (100 ml), dried over Na2SO4, and concentrated in vacuo. Finally, the residue was subjected to chromatography, from which the compounds 11a–11d and 16 were obtained.
Data for compounds 11a–11d and 16 can be found in Supporting Information.
2.2.3 Synthesis of compound 11e
Thiol 10 (1.0 g, 2.8 mmol) was first dissolved in 1,4-dibromobutane (10 ml, 83.7 mmol) and DBU (0.5 ml, 3.4 mmol) was then added. The reaction mixture was stirred at room temperature for 3 h, until TLC (EtOAc) indicated all starting materials were consumed and formed the final product. Following phase separation, the organic layer was diluted with DCM (60 ml), washed with H2O (100 ml), brine (100 ml), dried over Na2SO4, and concentrated in vacuo. In the final step, the residue was purified by column chromatography to yield 11e as a white solid. (0.65 g, 46.7%)yield; [α]D25–78.8 (c = 0.50, CHCl3); mp152–154 °C; 1H NMR (300 MHz, CDCl3) δ 5.73 (d, J = 9.4 Hz, 1H, NH), 5.17 (t, J = 9.7 Hz, 1H, H-3), 5.08 (t, J = 9.6 Hz, 1H, H-4), 4.59 (d, J = 10.3 Hz, 1H, H-1), 4.28–4.04 (m, 3H, H-6a, H-6 b, H-2), 3.70 (ddd, J = 9.5, 4.8, 2.3 Hz, 1H, H-5), 3.43 (t, J = 6.6 Hz, 2H, CH2Br), 2.85–2.61 (m, 2H, SCH2), 2.08, 2.03, 2.02 (3 s, 9H, 3 OAc), 1.98 (s, 3H, NAc), 1.98–1.91 (m, 2H, CH2), 1.83–1.71 (m, 2H, CH2); HRMS (ESI) calcd for C18H29BrNO8S (M + H+) 498.0797, found 498.0791.
2.2.4. Synthesis of compounds 14a–14l
A mixture of compounds 11a–11d (2 mmol, 1 eq), compounds 13a–13c (3 mmol, 1.5 eq) and potassium carbonate (2.4 mmol, 1.2 eq) in acetonitrile (50 ml) was refluxed for 4 h, until TLC (EtOAc/MeOH = 6/1) indicated that the reaction was complete. After the undissolved solid was removed by filtration, the filtrate was concentrated in vacuo. The resulting solid residue was further purified by silica gel column chromatography using EtOAc/CH3OH (12:1), in which compounds 14a–14l were obtained.
Data for compounds 14a–14l can be found in Supporting Information.
2.2.5. Synthesis of compounds 15a–15l
After compounds 14a–14l solution (1 mmol) was mixed with MeOH (15 ml), saturated solution of NH3 in MeOH (6 ml) was added. The reaction mixture was stirred for 40 h at room temperature, and until TLC (EtOAc:MeOH:H2O = 8:1:1) indicated that the reaction was completed. The mixture was then concentrated in vacuo, purified by flash column chromatography (EtOAc:MeOH = 6:1) to obtain compounds 15a–15l.
2-[2-[2-[(2-acetamido-β-d-glucopyranosyl)thio]ethylamino]ethyl]-1H-benzo[de] isoquinoline-1,3(2H)-dione (15a)
White solid; (0.46 g, 92.0%) yield; [α]D25–10.7 (c = 0.10, DMF); mp 223–225 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.53–8.40 (m, 4H, ArH), 7.91–7.81 (m, 2H, ArH), 7.71 (d, J = 9.3 Hz, 1H, NHAc), 5.01 (d, J = 4.7 Hz, 1H, OH), 4.96 (d, J = 5.3 Hz, 1H, OH), 4.49 (br s, 1H, OH), 4.36 (d, J = 10.3 Hz, 1H, H-1), 4.17–4.08 (m, 2H, H-3, H-4), 3.66 (dd, J = 11.4, 3.4 Hz, 1H, H-6 b), 3.57–3.38 (m, 2H, H-2, H-6a), 3.31–3.20 (m, 1H, H-5), 3.15–3.05 (m, 2H, CH2NC=O), 2.85–2.72 (m, 4H, 2 CH2), 2.72–2.54 (m, 2H, SCH2), 1.79 (s, 3H, NAc); 13 C NMR (75 MHz, DMSO-d6) δ 169.05, 163.64, 134.36, 131.41, 130.82, 127.53, 127.32, 122.26, 84.37, 81.26, 75.71, 70.63, 61.35, 54.73, 48.90, 46.39, 39.57, 29.83, 23.17; HRMS (ESI) calcd for C24H30N3O7S (M + H+) 504.1804, found 504.1805.
Data for compounds 15b–15l can be found in Supporting Information.
2.2.6. Synthesis of compound 17
Compound 16 (0.78 g, 1 mmol) was first suspended in MeOH (15 ml), and saturated solution of NH3 in MeOH (10 ml) was then added. The reaction mixture was stirred for 50 h at room temperature, until TLC (EtOAc:MeOH:H2O = 8:3:1) indicated that the reaction was complete. The mixture was further concentrated in vacuo and recrystallized from MeOH/ether, which resulted in compound 17 (0.43 g, 81.3%) as a white solid. [α]D25–33.4(c = 0.90, DMF); mp 236–238 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.70 (d, J = 9.2 Hz, 2H, 2 NHAc), 4.99 (d, J = 4.1 Hz, 2H, 2 OH), 4.95 (d, J = 5.3 Hz, 2H, 2 OH), 4.51 (t, J = 5.6 Hz, 2H, 2 OH), 4.32 (d, J = 10.3 Hz, 2H, 2 H-1), 3.67 (dd, J = 11.4, 5.5 Hz, 2H, 2 H-6 b), 3.56–3.38 (m, 4H, 2 H-2, 2 H-6a), 3.31–3.20 (m, 2H, 2 H-5), 3.13–3.01 (m, 4H, 2 H-3, 2 H-4), 2.69–2.53 (m, 4H, 2 CH2), 1.79 (s, 6H, 2 NAc), 1.62–1.49 (m, 4H, 2 CH2). 13 C NMR (75 MHz, DMSO-d6) δ 169.12, 84.13, 81.28, 75.64, 70.67, 61.35, 54.71, 28.74, 28.36, 23.19; HRMS (ESI) calcd for C20H37N2O10S2 (M + H+) 529.1890, found 529.1895.
2.2.7. Synthesis of compounds 20a–20e
After thiol 10 (2.8 mmol, 1.0 eq) and N-(ω-bromoalkyl)-1,8-naphthalimides 19a–19e (2.8 mmol, 1.0 eq) were dissolved in acetone (20 ml), potassium carbonate (3.4 mmol, 1.2 eq) and H2O (10 ml) were added. The mixture was stirred for 20 h at room temperature, until TLC (EtOAc) showed that the reaction was completed. The mixture was then concentrated in vacuo; and the resulting residue was diluted with DCM (40 ml), washed with H2O (80 ml), brine (80 ml), dried over Na2SO4, and concentrated in vacuo. Chromatography was carried out to purify the resulting compounds 20a–20e.
Data for compounds 20a–20e can be found in Supporting Information.
2.2.8. Synthesis of compounds 21a–21e
Compounds 21a–21e were synthesized by deacetylation of compounds 20a–20e (1 mmol) using the procedure described for the synthesis of compound 17.
2-[2-[(2-acetamido-β-d-glucopyranosyl)thio]ethyl]-1H-benzo[de]isoquinoline-1,3(2H)-dione (21a)
White solid; (0.42 g, 90.9%) yield; [α]D25 –13.5(c = 0.2, DMF); mp170–172 °C;1H NMR (300 MHz, DMSO-d6) δ 8.57–8.43 (m, 4H, ArH), 7.94–7.84 (m, 2H, ArH), 7.74 (d, J = 9.4 Hz, 1H, NHAc), 5.05 (d, J = 4.8 Hz, 1H, OH), 5.01 (d, J = 5.3 Hz, 1H, OH), 4.50 (d, J = 10.3 Hz, 1H, H-1), 4.45 (t, J = 6.0 Hz, 1H, OH), 4.34–4.21 (m, 2H, H-3, H-4), 3.70 (dd, J = 11.3, 6.0 Hz, 1H, H-6 b), 3.56 (dd, J = 19.5, 9.6 Hz, 1H, H-2), 3.47 (dd, J = 11.4, 5.6 Hz, 1H, H-6a), 3.33–3.26 (m, 1H, H-5), 3.22–3.10 (m, 2H, CH2N), 2.99 –2.74 (m, 2H, SCH2), 1.74 (s, 3H, NAc); 13 C NMR (75 MHz, DMSO-d6) δ 169.07, 163.39, 134.51, 131.43, 130.91, 127.53, 127.33, 122.14, 84.86, 81.40, 75.62, 70.54, 61.35, 54.64, 39.98, 27.59, 23.11; HRMS (ESI) calcd for C22H25N2O7S (M + H+) 461.1382, found 461.1377.
Data for compounds 21b–21e can be found in Supporting Information.
2.3. Enzyme preparation
Gene encoding hsHexB, which was engineered to contain a His6-tagged fusion protein, was cloned into pPIC9 expression vector (Invitrogen, Carlsbad, CA, USA), and subsequently transformed into Pichia pastoris GS115 (Invitrogen, Carlsbad, CA, USA) by electroporation. The cells were first cultured in BMGY broth (1% yeast extract, 1% glycerol, 2% peptone, 0.2% biotin, 1.34% yeast nitrogen, 0.1 M potassium phosphate, pH 6.0) at 30 °C. Methanol was added into the culture daily at a final concentration of 1% (v/v). When OD600 value reached 2.0 (after ∼120 h), the cells were harvested by centrifugation at 6000g for 10 min. While the pellet was resuspended in BMMY broth (1% yeast extract, 1% methanol, 2% peptone, 0.2% biotin, 1.34% yeast nitrogen, 0.1 M potassium phosphate, pH 6.0), the supernatant was subjected to ammonium sulfate precipitation (75% saturation) at 4 °C, in which the precipitate was further resuspended in distilled water and then desalted in buffer A (20 mM sodium phosphate, 0.5 M sodium chloride, pH 7.4). Subsequently, the resuspension solution was centrifuged at 17,000g for 30 min at 4 °C and passed through a 0.2-µm filter prior to loading onto a 5-ml HisTrap FF affinity column (GE Healthcare, Chicago, IL, USA), which was pre-equilibrated with buffer A. To remove nonspecific binding proteins, the column was sequentially washed with buffer A-containing 20 mM imidazole and buffer A-containing 50 mM imidazole. Finally, the target protein was eluted with buffer B (20 mM sodium phosphate, 0.5 M sodium chloride, 150 mM imidazole, pH 7.4). Purity of the protein was further analyzed by SDS–PAGE. hOGA was overexpressed in Escherichia coli BL21(DE3) and purified as described previouslyCitation31.
2.4. Inhibitory potency assay
Potencies of hsHexB and hOGA were assayed by end-point experiment, in which 4-methylumbelliferyl N-acetyl-β-d-glucosaminide (4-MU-GlcNAc; Sigma, St. Louis, MO, USA) was used as a substrate. Various concentrations of inhibitors and substrates (50 µM, 25 µM, and 12.5 µM) were mixed with Britton-Robinson buffer (hsHexB, pH 4.0; hOGA, pH 6.0), dimethylsulfoxide at a final concentration of 2%, enzyme, and 40 µM 4-MU-GlcNAc in a reaction of 100-µl final volume, and incubated at 30 °C for 30 min. Then, the enzymatic reaction was terminated by the addition of 100 µl of 0.5 M sodium carbonate solution. The fluorescence of the released 4-methylumbelliferone was quantified (excitation at 366 nm, emission at 445 nm) on a Varioskan Flash microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). To determine the inhibition constant (Ki), the reciprocal plots of 1/velocity versus inhibitor were constructed. Data analysis was performed using Graph Pad Prism software (Graph Pad Software Inc., San Diego, CA, USA).
2.5. Molecular docking studies
The Sybyl Software (Version7.3; Tripos Associates, St. Louis, IL, USA, 2006) was used in molecular docking studies. The complex crystal structures of hsHexB-pyrimethamine (PDB ID: 3LMY) and hOGA-iminocyclitol-type inhibitor (PDB ID: 5M7U) were taken from Protein Data Bank (http://www.rcsb.org/pdb) and used as the starting model. Prior to docking calculations, the structures were first optimized using MMFF94 force field to gain corresponding low energy conformations. All water molecules were then removed and missing hydrogen atoms were added. After that, the ligand docking mode was performed to generate appropriate putative ligand pose, so called “protomol”, based on the Hammerhead scoring function with the molecular similarity algorithm in the active domain of the receptorCitation32–34. Finally, molecular dockings between the ligands and the optimized crystal structure of hOGA or HsHexB were performed using the Surflex–Dock algorithm in the Sybyl 7.3 software.
3. Results and discussion
3.1. Chemical synthesis
3.1.1. Synthesis of compounds 15a–15l
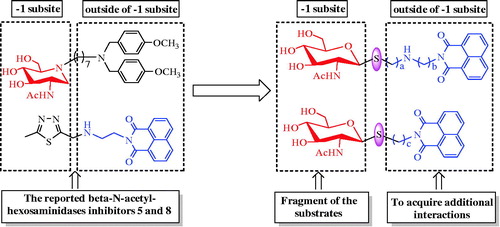
As shown in Scheme 1, the key intermediate thiol 10 was obtained from N-acetyl-d-glucosamine as the starting material. In the procedure, acetyl chloride was first used for acetylation and chlorination. Thiourea was then used in substitution prior to removing carbamimidoyl by Na2S2O5 in DCM and H2O. These three steps can conveniently be carried out without chromatographic purification and has made large-scale preparation of compound 10 possible. Then, compound 10 was reacted with α,ω-dibromoalkane in the presence of potassium carbonate in acetone and H2O to obtain mono-bromide precursors 11a–11d. Meanwhile, 1,8-naphthalic anhydride 12 was refluxed with α,ω-diaminoalkane in ethanol to yield 13a–13c. Subsequently, the preparations of acetyl-protected compounds 14a–14l were completed by the reactions of bromides 11a–11d with excess naphthalimide derivatives 13a–13c under the condition of potassium carbonate and acetonitrile with 65–72% yield. Finally, deacetylation of hydroxyl groups by methanol-ammonia catalysis resulted in the target compounds 15a–15l.
Scheme 1. Synthesis of thioglycosides 15a–15l. (i) AcCl; (ii) thiourea, acetone; (iii) Na2S2O5, DCM, H2O; (iv) α, ω-dibromoalkane, K2CO3, acetone, H2O; (v) α, ω- diaminoalkane, EtOH (vi) K2CO3, CH3CN; (vii) NH3, MeOH.
3.1.2. Synthesis of compounds 11e and 17
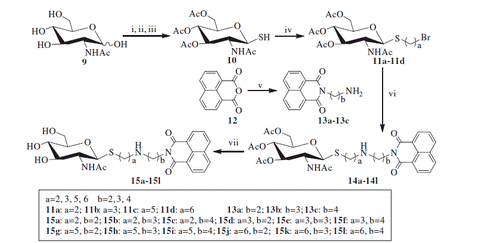
Unlike the synthesis of mono-bromide precursors 11a–11d, treatment of thiol 10 with 1,4-dibromobutane under the condition of potassium carbonate in acetone and H2O was not able to form the desired mono-bromide 11e. Instead, 1,4-bisthiobutane derivatives 16 was obtained (Scheme 2). The resulting compound 16 was further deprotected by employing methanol-ammonia to gain 1,4-bis[(2-acetamido-2-deoxy-β-d-glucopyranosyl)thio] butane 17.
Scheme 2. Synthesis of compounds 11e and 17. (i) 1,4- dibromobutane, K2CO3, acetone, H2O; (ii) 1,4- dibromobutane, DBU; (iii) K2CO3, CH3CN; (iv) NH3, MeOH.
In order to achieve the desired compound 11e, 1,4-dibromobutane was selected to use as the reaction solvent and DBU was used as the acid-accepter. Under this condition, thiol 10 was successfully converted to 11e at room temperature for 3 h with 47% yield. However, the reaction of 11e with 13a (in the preparation of 14a–14l) did not yield the desired compound 14m. Presumably, this may be because compound 11e was not stable, and self-decomposition may take place under strong alkaline conditions (i.e. in the presence of potassium carbonate).
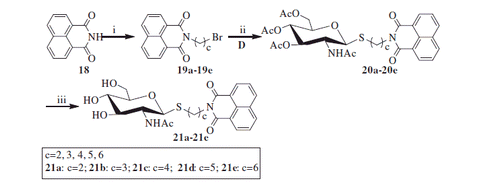
3.1.3. Synthesis of compounds 21a–21e
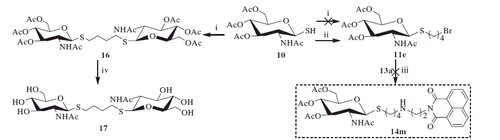
As shown in Scheme 3, reaction of the starting material 1,8-naphthalimide 18 with α, ω-dibromoalkane was carried out to yield bromoalkyl naphthalimides 19a–19e. Its further reaction with thiol 10 in acetone and H2O resulted in acetyl-protected precursors 20a–20e with 80–86% yield. Finally, the resulting naphthalimide derivatives 21a–21e were deprotected by methanol-ammonia catalysis with 88–92% yield.
Scheme 3. Synthesis of thioglycosides 21a–21e. (i) α, ω-dibromoalkane, K2CO3, CH3CN; (ii) K2CO3, acetone, H2O; (iii) NH3, MeOH.
3.2. Bioevaluation of inhibitory potency
3.2.1. Inhibition studies
The target compounds 15a–15l, 17, and 21a–21e, each at 20 µM, were evaluated for their inhibition potency against hsHexB and hOGA. As shown in , compounds 21a–21e (nitrogen atom is absent in the linker) exhibited lower inhibitory potency compared with that of compounds 15a–15l. Compound 17 without naphthalimide moiety hardly showed activity, suggesting that naphthalimide moiety may be involved in the improved binding affinity.
Figure 3. Inhibition of hsHexB in comparison to hOGA.
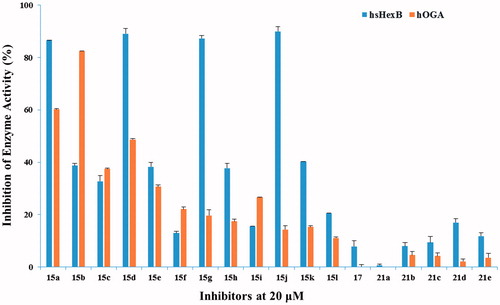
Analysis of compounds 15a–15l against hsHexB revealed a significant correlation between the efficiency of the inhibitors and the position of nitrogen atom in the linker. Specifically, compounds 15a, 15d, 15g, and 15j containing 2-aminoethyl-naphthalimide moiety exhibited the highest potencies. Increasing number of carbon atom (from 2-aminoethyl to 4-aminobutyl) brought the potency to steadily dwindle compared with compounds 15a, 15b, 15c. Besides, the glycosyl moieties with different length of linkers (Scheme 1, with the value b) played partial inhibitory role against hsHexB. Further Ki determination showed that compound 15j (Ki = 0.91 µM against hsHexB; Ki > 100 µM against hOGA) exhibited excellent selectivity and good inhibition potency against hsHexB.
Analysis of compounds 15a–15l against hOGA showed that the inhibitory potency of these compounds was associated with the length of the linker between glycosyl and naphthalimide group. The linker containing five to seven atoms in compounds 15a–15e exhibited higher inhibitory potency than those with longer chains in compounds 15f–15l. This suggests that the existed hydrophobic domain is in the proximity of −1 binding-site pocket so that it can bind to naphthalimide groupCitation35. Position of nitrogen atom in the linker appears to have an effect on the efficiency of the inhibitors. For instance, although compounds 15b and 15d had the same length of linker, compound 15b showed the highest potency against hOGA compared with those of compounds 15a–15l. This suggests that nitrogen atom that is in a suitable position may lead to such molecular conformation that improves the binding affinity. Moreover, compound 15b had Ki values of 3.76 µM against hOGA and 30.42 µM against hsHexB, indicating that it could be used as a new leading compound against hOGA in further research.
3.2.2. Kinetic studies
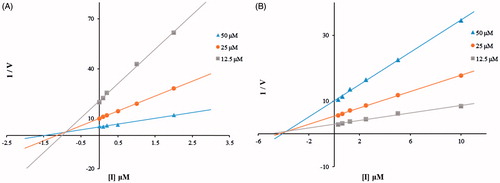
To explore inhibitory mechanisms toward hsHexB and hOGA by the thioglycoside derivatives, the most potent inhibitors (compound 15j against hsHexB; and 15b against hOGA) were selected for kinetic studies by Dixon plots. As shown in , the Dixon plots of compound 15j against hsHexB and compound 15b against hOGA revealed that these active thioglycosides were competitive inhibitors. Thus, compounds 15j and 15b were able to bind to the active pockets of hsHexB and hOGA, respectively.
Figure 4. (A) Dixon plots for inhibition of hsHexB by compound 15j. (B) Dixon plots for inhibition of hOGA by compound 15b.
3.3. Molecular docking results
To further explore possible binding modes of compound 15j to hsHexB and 15b to hOGA, the molecular docking studies were carried out using Sybyl 7.3 software.
The molecular docking for compound 15j-hsHexB complex in showed that naphthalimide group of compound 15j bound to the −1 subsite (active pocket) of hsHexB via aromatic π–π stacking interactions with His294, Trp489, and Trp405. Moreover, oxygen of naphthalimide ring formed hydrogen bonds with the catalytic Arg211. These interactions are coherent with those found in the complex structure of hsHexB-PyrimethamineCitation36. In addition, NH in alkylamine linker of 15j bound to Glu491 via hydrogen bonds at a distance of 2.0 Å. As supported by the enzymatic activity assays, this H-bonding interaction is an important factor contributing to inhibitory potency. When the linker lengths were increased (i.e. from ethylamino in 15j to propylamino in 15k or butylamino in 15l), the inhibitory potency was considerably decreased. Furthermore, the glycosyl moiety of 15j, which extended out from the active pocket could help improve the affinity by forming a hydrogen bond with Ala447 at a distance of 2.0 Å. And the additional H-bonding interaction could explain its increasing inhibitory potency when compared with the structure of its parent compound, 2-aminoethyl-naphthalimide (Ki = 2.09 µM against hsHex)Citation22.
Figure 5. (A) Molecular docking of 15j with hsHexB (PDB ID: 3LMY). The enzyme is presented as cartoon representation, the catalytic residues and compound 15j are shown as sticks, and hydrogen bonds are highlighted as red dashed lines. Atom colors of 15j: light pink-carbon atoms, red-oxygen atoms, blue-nitrogen atoms, dark yellow-sulfur atoms. (B) Molecular docking of 15b with hOGA (PDB ID: 5M7U). The enzyme is presented as cartoon representation, the catalytic residues and compound 15b are shown as sticks, hydrogen bonds are highlighted as red dashed lines. Atom colors of 15b: yellow-carbon atoms, red-oxygen atoms, blue-nitrogen atoms, dark yellow-sulfur atoms. The molecular models were created using software PyMOL.
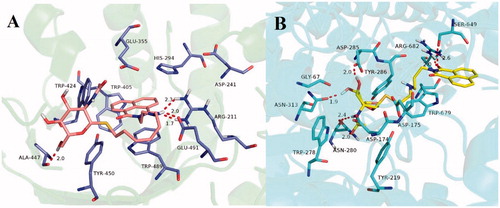
Unlike the interactions between compound 15j and hsHexB, as shown in , the glycosyl moiety from 15b was found to be tightly bound to the −1 subsite from hOGA via H-bonding interactions with catalytic Asn313, Asp285, and Asn280. This finding is in incoherent with those found in the complex structure of hOGA–VV347, confirming the accuracy of the docking resultCitation35. In addition, the naphthalimide group of compound 15b interacted with residues of domains that were outside the −1 binging-site pocket, notably through hydrogen bondings with Ser649 and Arg682, as well as π–π stacking interaction with Trp679.
4. Conclusions
In conclusion, we have presented the design and synthesis of thioglycoside derivatives-containing naphthalimide moiety, and their inhibitory potency against GH20 hsHexB and GH84 hOGA. The efficiency evaluation by enzymatic assay showed that compounds 15j (Ki = 0.91 µM against hsHexB; Ki > 100 µM against hOGA) and 15 b (Ki = 3.76 µM against hOGA; Ki = 30.42 µM against hsHexB) exhibited the highest inhibitory potency against hsHexB and hOGA, respectively. The structure–activity relationship as well as the molecular docking studies provided some insight into the rational design for the two human β-N-acetylhexosaminidases. For hsHexB, the 2-aminoethyl- naphthalimide moiety, tightly bound to the −1 binging-site pocket, was found to be the critical factor in maintaining the inhibitory potency. For hOGA, while the glycosyl moiety was bound to the −1 subsite, naphthalimide group with five- to seven-atom linker contain a nitrogen atom that was in such position that led to additional H–bonding and π–π stacking interactions outside of the −1 binding subsite, and these interactions contributed to the selectivity and inhibitory potency. These thioglycoside derivatives may prove to be valuable leading compounds for further developments of new selective inhibitors against hsHexB or hOGA.
IENZ_1419217_Supplementary_Materials.pdf
Download PDF (1.9 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Henrissat B, Daviest G. Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol 1997;7:637–44.
- Kong HC, Chen W, Lu HZ, et al. Synthesis of NAG-thiazoline-derived inhibitors for β-N-acetyl-D-hexosaminidases. Carbohyd Res 2015;413:135–44.
- Hattie M, Cekic N, Aleksandra, et al. Modifying the phenyl group of PUGNAc: reactivity tuning to deliver selective inhibitors for N-acetyl-D-glucosaminidases. Org Biomol Chem 2016;14:3193–7.
- Liu T, Yan J, Yang Q. Comparative biochemistry of GH3, GH20 and GH84 β-N-acetyl-D-hexosaminidases and recent progress in selective inhibitor discover. Curr Drug Targets 2012;13:512–25.
- Zhou FX, Huo JW, Liu Y, et al. Elevated glucose levels impair the WNT/b-catenin pathway via the activation of the hexosamine biosynthesis pathway in endometrial cancer. J Steroid Biochem 2016;159:19–25.
- Shikhman AR, Brinson DC, Lotz M, et al. Profile of glycosaminoglycan-degrading glycosidases and glycoside sulfatases secreted by human articular chondrocytes in homeostasis and inflammation. Arthritis Rheum 2000;43:1307–14.
- Liu JJ, Shikhman AR, Lotz MK, Wong C-H. Hexosaminidase inhibitors as new drug candidates for the therapy of osteoarthritis. Chem Biol 2001;8:701--11.
- Mahuran DJ. Biochemical consequences of mutations causing the GM2 gangliosidose. Biochim Biophys Acta 1999;1455:105–38.
- Tropak MB, Mahuran D. Lending a helping hand, screening chemical libraries for compounds that enhance β-hexosaminidase A activity in GM2 gangliosidosis cells. Febs J 2007;274:4951–61.
- Tropak MB, Reid SP, Guiral M, et al. Pharmacological enhancement of β-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff patients. J Biol Chem 2004;279:13478–87.
- Arnold CS, Johnson GVW, Cole RN, et al. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J Biol Chem 1996;271:28741–4.
- Lim S, Haque MM, Nam G, et al. Monitoring of intracellular tau aggregation regulated by OGA/OGT inhibitors. Int J Mol Sci 2015;16:20212--24.
- Bergeron-Brlek M, Goodwin-Tindall J, Cekic N, et al. A convenient approach to stereoisomeric iminocyclitols: generation of potent brain-permeable OGA inhibitors. Angew Chem Int Ed 2015;54:15429–33.
- Kim EJ, Perreira M, Craig J, et al. An O-GlcNAcase-specific inhibitor and substrate engineered by the extension of the N-Acetyl moiety. J Am Chem Soc 2006;128:4234–5.
- Horsch M, Hoesch L, Vasella A, et al. N-Acetylglucosaminono-1,5-lactone oxime and the corresponding (phenylcarbamoyl)oxime. Novel and potent inhibitors of beta-N-acetylglucosaminidase. Eur J Biochem 1991;197:815–8.
- Aoyama T, Naganawa H, Suda H, et al. The structure of nagstatin, a new inhibitor of N-acetyl-beta-D-glucosaminidase. J Antibiot 1992;45:1557–8.
- Knapp S, Vocadlo D, Gao ZN, et al. NAG-thiazoline, An N-acetyl-β-hexosaminidase inhibitor that implicates acetamido participation. J Am Chem Soc 1996;18:6804–5.
- Maegawa GH, Tropak M, Buttner J, et al. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J Biol Chem 2007;282:9150–61.
- Ayers BJ, Glawar AFG, Martínez RF, et al. Nine of 16 stereoisomeric polyhydroxylated proline amides are potent β-N-acetylhexosaminidase inhibitor. J Org Chem 2014;79:3398–409.
- Crabtree EV, Martínez F, Nakagawa S, et al. Synthesis of the enantiomers of XYLNAc and LYXNAc: comparison of β-N-acetylhexosaminidase inhibition by the 8 stereoisomers of 2-Nacetylamino-1,2,4-trideoxy-1,4-iminopentitols. Org Biomol Chem 2014;12:3932–43.
- Tropak MB, Blanchard JE, Withers SG, et al. High-throughput screening for human lysosomal β-N-acetylhexosaminidase inhibitors acting as pharmacological chaperones. Chem Biol 2007;14:153–64.
- Guo P, Chen Q, Xu L, et al. Development of unsymmetrical dyads as potent noncarbohydrate-based inhibitors against human β-N-acetyl-D-hexosaminidase. ACS Med Chem Lett 2013;4:527–31.
- Liu T, Zhou Y, Wang J, et al. A crystal structure-guided rational design switching non-carbohydrate inhibitors’ specificity between two β-GlcNAcase homologs. Sci Rep 2014;4:6188–93.
- Chen Q, Guo P, Xu L, et al. Exploring unsymmetrical dyads as efficient inhibitors against the insect b-N-acetyl-D-hexosaminidase OfHex2. Biochimie 2014;97:152–62.
- Liu T, Chen L, Ma Q, et al. Structural insights into chitinolytic enzymes and inhibition mechanisms of selective inhibitors. Curr Drug Targets 2014;20:754–70.
- Ho CW, Popat SD, Liu TW, et al. Development of GlcNAc-Inspired iminocyclitiols as potent and selective N-acetyl-Hexosaminidase Inhibitors. ACS Chem Biol 2010;5:489–97.
- Floyd N, Vijayakrishnan B, Koeppe JR, et al. Thiyl glycosylation of olefinic proteins: S-linked glycoconjugate synthesis. Angew Chem Int Ed 2009;48:7789–802.
- Paul B, Korytnyk W. S-, N-, and O-glycosyl derivatives of 2-acetamido-2-deoxy-D-glucose with hydrophobic aglycons as potential chemotherapeutic agents and N-acetyl-beta- D-glucosaminidase inhibitors. Carbohydr Res 1984;126:27–43.
- Landeyálvarez MA, Ochoaterán A, Pinaluis G, et al. Novel naphthalimide–aminobenzamide dyads as OFF/ON fluorescent supramolecular receptors in metal ion binding. Supramol Chem 2016;28:892–906.
- Ramchander J. Synthesis of 3(N(1,3dioxo 1H benzo[de]isoquinolin- 2(3H)-yl)alkyl)-2- (4-substituted) phenylthiazolidine-4- carboxylic acid. Chem Sci Trans 2016;5:163–70.
- Kong HC, Chen W, Liu T, et al. Synthesis of NAM-thiazoline derivatives as novel O-GlcNAcase inhibitors. Carbohydr. Res 2016;429:54–61.
- Jain AN. Scoring non-covalent protein–ligand interactions: acontinuous differentiable function tuned to compute binding. J Comput Aided Mol Des 1996;10:427–40.
- Babu S, Nagarajan SK, Lee SH, et al. Structural characterization of human CRTh2: a combined homology modeling, molecular docking and 3D-QSAR-based in silico approach. Med Chem Res 2016;25:653–71.
- Li DL, Du SQ, Tan WM, et al. Computational insight into the structure-activity relationship of novel N-substituted phthalimides with gibberellin-like activity. J Mol Model 2015;21:271–80.
- Roth C, Chan S, Offen WA, et al. Structural and functional insight into human O-GlcNAcase. Nat Chem Bio 2017;13:610–14.
- Bateman KS, Cherney MM, Mahuran DJ, et al. Crystal structure of β-hexosaminidase B in complex with pyrimethamine, a potential pharmacological chaperone. J Med Chem 2011; 54:1421–9.