
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Primaquine (PQ) ureidoamides 5a–f were screened for antimicrobial, biofilm eradication and antioxidative activities. Susceptibility of the tested microbial species towards tested compounds showed species- and compound-dependent activity. N-(diphenylmethyl)-2-[({4-[(6-methoxyquinolin-8-yl)amino]pentyl}carbamoyl)amino]-4-methylpentanamide (5a) and 2-(4-chlorophenyl)-N-(diphenylmethyl)-2-[({4-[(6-methoxyquinolin-8-yl)amino]pentyl}carbamoyl)amino]acetamide (5d) showed antibacterial activity against S. aureus strains (MIC = 6.5 µg/ml). Further, compounds 5c and 5d had weak antibacterial activity against Escherichia coli and Pseudomonas aeruginosa. None of the tested compounds showed a wide spectrum of antifungal activity. In contrast, most of the compounds exerted strong activity in a biofilm eradication assay against E. coli, P. aeruginosa and Candida albicans, comparable to or even higher than gentamycin, amphotericin B or parent PQ. The most active compounds were 5a and 5b. Tested compounds were inactive against biofilm formation by C. parapsylosis, Enterococcus faecalis, C. tropicalis and C. krusei. Compounds 5b–f significantly inhibited lipid peroxidation (80–99%), whereas compound 5c presented interesting LOX inhibition.
Introduction
Primaquine (8-[(4-amino-1-methylbutyl)amino]-6-methoxyquinoline, PQ) is a synthetic antimalarial drug that eliminates exoerythrocytic infection. It prevents the development of dormant liver forms of the parasite, which are responsible for relapses in vivax and ovale malaria. PQ is also active against gametocytes of all parasite species causing human malaria, including chloroquine-resistant Plasmodium falciparumCitation1. In addition, PQ and other antimalarial drugs (artemisinin, artesunate, tetracyclines and chloroquine) show significant anticancer activity, reduce IC50 of anticancer drugs, affect their ADME properties leading to improvement of tumour therapy or inhibit development of drug-resistant cancer cells. Numerous reports corroborate anticancer properties of antimalarial drug classes and their usefulness in adjuvant chemotherapiesCitation2–6.
Our scientific efforts are directed towards new anticancer agents derived from PQ. In the last few years we have designed, prepared and biologically evaluated approximately a hundred PQ-derivatives of amide, urea, bis-urea, hydroxyurea, semicarbazide and acylsemicarbazide type. Many of them have shown a strong antiproliferative effect against a number of tumour cell lines and/or significant antioxidative propertiesCitation7–12. In addition, PQ-cinnamamide, PQ-bis-trifluoromethylated cinnamamide and PQ-urea derivatives with hydroxyl, halogen and trifluoromethyl substituents on the benzene ring exerted very strong antimycobacterial activity towards three Mycobacterium species, stronger than PQ and the standard antitubercular drugsCitation13. PQ-aryl urea derivative, in particular that with a trifluoromethyl moiety, exhibited significant antimicrobial activity with MIC values from 1.6 to 12.5 µg/mlCitation11. Antimalarial activity of our compounds against the erythrocytic stage of drug-sensitive Plasmodium falciparum was evaluated as well. After detailed QSAR studies, a series of ureidoamides 5a–f bearing PQ and amino acid moieties were designed, prepared and evaluated against the same parasite speciesCitation14.
In order to get a better insight into the pharmacological potential of novel ureidoamides, their antimicrobial and antioxidative activities were evaluated and are reported herein. In addition, we were especially interested in their biofilm eradication potential. Biofilms are structured communities of microorganisms formed on biotic or abiotic (medical devices and biomaterials) surfaces, which are often responsible for therapy failures in clinicsCitation15. Biofilm formation starts with microbial attachment, its reproduction and micro-colony formation and leads to microorganism enclosure through self-formation of protective matrices (extracellular polymeric structures), which results in increased resistance to antimicrobial agents and host immune factorsCitation16. Thus, biofilm-associated infections are, in general, chronic infections with high morbidity and mortality ratesCitation17,Citation18. Therefore, many efforts have been made to find drugs effective in inhibition of biofilm formation and its eradicationCitation19.
Materials and methods
Chemistry
General information
Melting points were measured on a Stuart Melting Point (SMP3) apparatus (Barloworld Scientific, UK) in open capillaries with uncorrected values. IR spectra were recorded on FTIR Spectrum One spectrophotometer (Perkin-Elmer, UK). NMR (1H and 13C) were recorded at 25 °C on NMR Avance 600 spectrometer (Bruker, Germany) at 300 and 150 MHz for 1H and 13C nuclei, respectively. All compounds were routinely checked by TLC with Merck silica gel 60 F-254 glass plates using the following solvent systems: petrolether/ethyl acetate/methanol 30:10:5, cyclohexane/ethyl acetate 1:1, cyclohexane/dichloromethane/methanol 10:18:2, dichloromethane/methanol 97:3 and 95:5. Spots were visualised by short-wave UV light and iodine vapour. Column chromatography was performed on silica gel 0.063–0.200 mm (Kemika, Croatia) and 0.040–0.063 mm (Merck, Germany), with the same eluents used in TLC. 1H-benzo[d][1,2,3]triazole (BtH), triphosgene, triethylamine (TEA), hydrazine hydrate, L-leucine, D-phenylglycine, DL-p-chlorophenylglycine and N-methyl-1,1-diphenylmethanamine were purchased from Sigma-Aldrich, USA. Primaquine, diphenylmethanamine and (4-chlorophenyl)(phenyl)methanamine were prepared from commercially available salts (Sigma-Aldrich) prior the use. All reactions with primaquine were run light protected.
Synthesis
Ureidoamides 5a–f with primaquine and amino acid moieties were prepared according to a procedure developed by our research groupCitation14. Synthetic pathway leading to compounds 5a–f included preparation of several precursors: 1-benzotriazole carboxylic acid chloride (benzotriazolecarbonyl chloride, BtcCl, 1), Btc-amino acids 2a–c, Btc-amino acid chlorides 3a–c and Btc-amino acid amides 4a–f. The following PQ-ureidoamides were prepared: N-(diphenylmethyl)-2-[({4-[(6-methoxyquinolin-8-yl)amino]pentyl}carbamoyl)amino]-4-methylpentanamide (5a), N-(diphenylmethyl)-2-[({4-[(6-methoxyquinolin-8-yl)amino]pentyl}carbamoyl)amino]-N-methyl-2-phenylacetamide (5b), N-[(4-chlorophenyl)(phenyl)methyl]-2-[({4-[(6-methoxyquinolin-8-yl)amino]pentyl}carbamoyl)amino]-2-phenylacetamide (5c), 2–(4-chlorophenyl)-N-(diphenylmethyl)-2-[({4-[(6-methoxyquinolin-8-yl)amino]pentyl}carbamoyl)amino]acetamide (5d), 2–(4-chlorophenyl)-N-[(4-chlorophenyl)(phenyl)methyl]-2-[({4-[(6-methoxyquinolin-8-yl)amino]pentyl}carbamoyl)amino]acetamide (5e) and 3-[(4-chlorophenyl)({N'-[(4-methoxyphenyl)(phenyl)methylidene]hydrazinecarbonyl})methyl]-1-{4-[(6-methoxyquinolin-8-yl)amino]pentyl}urea (5f). Their analytical and spectral data were fully in agreement with the previously obtained data.
Biological screening
General information
Absorbances were measured on a Lambda 20 double-beam spectrophotometer (Perkin-Elmer, UK). Centrifuge 5810 R (Eppendorf, Germany), microplate reader (iEMS, Finland) and Multiscan plate reader (Thermo) were used. Linoleic acid sodium salt, soybean lipoxygenase (LOX), 1,1-diphenyl-2-picrylhydrazyl (DPPH), nordihydroguaiaretic acid (NDGA), 2,2’-azobis(2-amidinopropane) dihydrochloride (AAPH), 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox), phosphate-buffered saline (PBS), Triton X100 and foetal bovine serum (FBS) were purchased from Sigma-Aldrich.
In vitro antimicrobial susceptibility assay (MIC determination)
Before analysis, compounds 5a–f were dissolved in 96% ethanol in a concentration of 2 mg/ml as stock solutions. Inoculums were prepared with fresh cultures of microbial strains, cultured on tryptic-soy agar or Sabouraud 2% (m/v) dextrose agar for 18 h (48–72 h for fungi) at 30 °C with physiological saline containing approximately 3 × 106 CFUs/ml. Inoculum density (0.5 McFarland units) was adjusted with a bio Mériex (France) densitometer. Minimum inhibitory concentration (MIC) was determined by the twofold micro-dilution method in Mu¨ller–Hinton broth for bacterial strains and RPMI 1640 + 2% m/v dextrose for fungi according to the EUCAST recommendations in a sterile 96-well flat-bottom plastic tissue culture plateCitation20. MIC was determined using a linear regression curve after incubation of compounds and reading the optical density at 570 nm using an iEMS microplate reader. MIC was defined as the lowest concentration of extract that allows no more than 50% growth of microbes in comparison with intact microbial cells (negative control). All tests were performed in triplicate and expressed as the mean.
Minimum biofilm eradication assay
We tested the susceptibility of different bacterial and yeast strains to compounds 5a–f by determining the minimum biofilm eradication concentration (MBEC). The test was performed in a sterile 96-well flat-bottom plastic tissue culture plate (TPP, Switzerland). Each well was filled with 100 µl of bacterial (107 CFUs/ml) or yeast (5 × 106 CFUs/ml) suspension. When the possibility of inhibition of yeast biofilm formation was tested, the wells were pre-treated with FBS (250 µl per well). Negative controls contained broth only. Positive controls were performed using standard antibacterial and antifungal drugs gentamycin and amphotericin B, respectively. The plates were covered and incubated aerobically for 24 h (bacteria) or 48 h (yeast) at 37 °C. Following the incubation period, each well was aspirated and washed three times with 250 µl of PBS and vigorously shaken in order to remove all non-adherent bacteria/yeast. The remaining attached cells were fixed with methanol (15 min) and the plates were left to dry overnight. The formed biofilm was stained with crystal violet (1%, 5 min). Excess stain was rinsed by placing the plate under running tap water and the plates were left to dry for 24 h. Adherent cells were resolubilized with acetic acid (33%, v/v). The optical density of each well was measured at 570 nm using a Multiscan plate reader. The MBEC value represents the lowest compound dilution at which microorganisms fail to grow.
Antioxidative activity
Interaction of the PQ-ureidoamides 5a–f with 1,1-diphenyl-2-picrylhydrazyl (DPPH)
To a solution of DPPH in absolute ethanol, an appropriate volume of the tested compound solution (0.1 mM final concentration) dissolved in DMSO was added. Absorbance was recorded at 517 nm after 20 and 60 min at room temperature (Lambda 20 double beam spectrophotometer, Perkin-Elmer, UK). Experiments were repeated at least in triplicate and the standard deviation of absorbance was less than 10% of the meanCitation10. NDGA was used under the same experimental conditions as a reference compound.
In vitro soybean lipoxygenase (LOX) inhibition study
In vitro LOX inhibition assay was accomplished as described previouslyCitation10. Test compounds (stock solutions 10 mM in DMSO) were incubated at room temperature with sodium linoleate (0.1 mM) and 0.2 ml of LOX solution (0.1 mg in 10 ml saline). Conversion of sodium linoleate to 13-hydroperoxylinoleic acid was measured at 234 nm and compared to the reference inhibitor. Several concentrations were used for IC50 determination. The assays were repeated at least in triplicate and the standard deviation of absorbance was less than 10% of the mean. NDGA was used under the same experimental conditions as a reference compound.
Inhibition of linoleic acid peroxidation (LP)
To initiate lipid peroxidation, the free radical AAPH was usedCitation10. The final solution in the UV cuvette consisted of 10 µl sodium linoleate solution (c = 16 mM) and 0.93 ml phosphate buffer pH 7.4 (c = 50 mM). Afterwards, 10 µl of the tested compound solution in DMSO (final concentration 0.1 mM) and 50 µl of AAPH solution (c = 40 mM) were added. The experiment was performed at 37 °C under air. Oxidation of linoleic acid sodium salt was monitored at 234 nm. The assay was repeated at least in triplicate and the standard deviation of absorbance was less than 10% of the mean. Trolox was used under the same experimental conditions as a reference compound.
In vitro haemolytic activity
Whole venous human blood (10 ml) was collected in a heparin-based container from a volunteer donor (45 years old, non-smoker) after obtaining his consent and washed three times with PBS pH 7.0 (centrifugation at 2500 rpm, 4 min, Centrifuge 5810 R, Eppendorf, Germany). Then, 4% (v/v) of fresh human erythrocyte solution (hErc) was prepared using a PBS pH 7.0 solution. A series of diluted compounds from 200 to 50 µg/ml were prepared in a total volume of 1 ml with hErc solution. After incubation for 1 h at 37 °C aerobically (without shaking), test tubes were centrifuged at room temperature (2500 rpm, 4 min). Absorbance of the supernatant (200 µl) was recorded at 540 nm using an iEMS microplate reader. The 0.1% (v/v) solution of Triton X100 served as a positive control (as total lysis of hErc), and intact hErc served as a negative control. Since compounds were diluted with 1.5% (v/v) DMSO, the same solution was used as a control. All tests were performed in triplicate and the results were expressed as haemolysis percentage (mean ± SD). Percentage of haemolysis was calculated using the following equation:
where Asample was absorbance of the tested compound, Anc was absorbance of the negative control (intact hErc) and Apc was absorbance of the positive control treated with 0.1% TritonX100. In statistical calculations, p values below 0.05 were considered significant throughout the test. For comparison of mean values, one-way ANOVA with Dunnet’s post-test was used.
Results and discussion
Chemistry
Preparation of PQ-ureidoamides 5a–f was complex and involved several reaction steps: preparation of Btc-amino acids 2a–c from BtcCl 1 and the corresponding amino acid, conversion of Btc-amino acid to Btc-amino acid chloride 3a–c, reaction of Btc-amino acid chlorides with amines or hydrazone and aminolysis of Btc-amino acid amides 4a–f with primaquine (Scheme 1).
Scheme 1. Synthesis of ureidoamides 5a–f and their precursors.
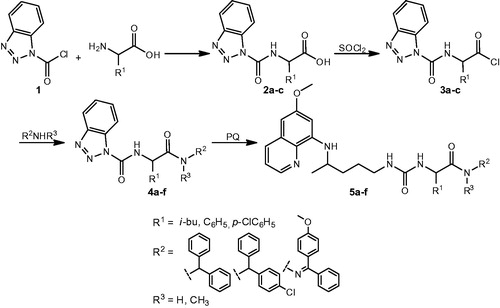
Three amino acids (L-leucine, D-phenylglycine, DL-p-chlorophenylglycine), three amines (diphenylmethanamine, (4-chlorophenyl)(phenyl)methanamine)), N-methyl-1,1-diphenylmethanamine and one hydrazone (4-methoxybenzophenone hydrazone) were used. Structures of PQ-ureidoamides 5a–f are given in .
Figure 1. Chemical structures of PQ-ureidoamides 5a–f.
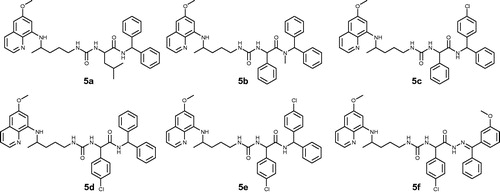
Compounds 5a–f are not fully in agreement with Lipinski’s and Gelovani’s rules for prospective small molecular drugs but show acceptable deviations from the rulesCitation21. The parameters were calculated with the Chemicalize.org programCitation22 and are presented in .
Table 1. Properties of PQ-ureidoamides 5a–f calculated with Chemicalize.org programme:Citation22 Lipinski's and Gelovani's parameters.
Biological evaluation
Based on previous findings on the antimicrobial effects of PQ-derivatives, we tested PQ-ureidoamides 5a–f toward several bacterial and fungal species. To assess this, we applied the broth dilution method to determine MIC of each compound towards a selected panel of microorganisms: Gram-positive bacteria (Staphylococcus aureus, Enterococcus faecalis, Enterococcus hyrae, Kocuria rhizophila, Bacillus cereus, Bacillus subtilis), Gram-negative bacteria (Escherichia coli, Pseudomonas aeruginosa, Burkholderia caepacia) and seven fungi (Candida albicans, Candida kefyr, Candida krusei, Candida parapsylosis, Candida tropicalis, Issatchenkia orientalis Kudrjanzev, Aspergillus niger). MICs of the tested compounds, together with positive controls (tetracycline and amphotericin B), were determined after 18- or 24-h incubation of the selected microbial species and the values are listed in Supplementary material.
Susceptibility of the tested microbial species towards PQ-derivatives 5a–f showed species-dependent and compound-dependent activity. All MICs lower than 100 µg/ml were set as a pronounced antimicrobial activity. Only compounds 5a and 5d showed antibacterial activity against the tested Staphylococcus spp. with the lowest MIC value of 6.5 µg/ml (MIC range 6.5–15 µg/ml for 5a and 12.5–25 µg/ml for 5d). Compound 5a showed weak activity towards K. rhizophila and P. aeruginosa (MIC = 25 µg/ml) and B. subtilis (MIC = 50 µg/ml). Furthermore, compounds 5c and 5d showed low antibacterial activity against two Gram-negative species (E. coli and P. aeruginosa, MIC range from 25 to 65 µg/ml). E. coli displayed a slight susceptibility towards compound 5e as well (MIC = 45–60 µg/ml). None of the tested compounds showed a wide spectrum of antifungal activity against yeasts and filamentous fungi.
Modern medicine uses a broad variety of medical devices and implants, the application of which is often associated with attachment of microorganisms and biofilm formation. Microorganisms within a biofilm are more resistant to different antimicrobial treatments; they can survive even under harsh environmental conditions and withstand the host’s immune systemCitation23. Therefore, search for novel compounds that will combat biofilms at different levels of formation and maturation is extremely important. Since the PQ-derivatives 5a–f showed, to some extent, antimicrobial potential, our interest was also to see whether they were effective against biofilm formation. The antibiofilm efficacy of compounds 5a–f was evaluated against Gram-positive (Staphylococcus aureus, Enterococcus faecalis) and Gram-negative bacteria (Escherichia coli, Pseudomonas aeruginosa) and several Candida species (C. albicans, C. kefyr, C. krusei, C. parapsylosis, C. tropicalis). The activity was compared to that of gentamycin, amphotericin B and the parent compound PQ ().
Table 2. Sensitivity of microbial strains to PQ-ureidoamides expressed as minimum biofilm eradication concentration (MBEC).
As shown in , antibiofilm potency of all compounds against the biofilm formed by E. coli (both strains) was prominent, with lower MBEC than the MBECs of gentamycin and PQ (25 µg/ml), especially for 5a and 5c. These two compounds were also very potent in eradicating the biofilm formed by P. aeruginosa. Compound 5a had very low MBEC (at the level of gentamycin) for S. aureus. In contrast, we found that none of the tested compounds was effective against biofilm formation by C. parapsylosis, E. faecalis, C. tropicalis and C. krusei. As already mentioned, compounds 5a–f had high efficiency against biofilm formation of Gram-negative bacteria E. coli (both tested strains). This finding is unexpected since Gram-negative bacteria have a more complex cell envelope, which is a relatively impermeable structure formed of an inner cytoplasmic membrane separated by a peptidoglycan layer from an outer envelope. One of the resistance mechanisms that differ Gram-negative from Gram-positive bacteria is the membraneCitation24. Since the exact mechanism underlying PQ activity is unclear, one of the suggested possibilities of such activity is the formation of reactive oxidative speciesCitation25, which could be the mechanism also involved in both antibacterial action and inhibition of biofilm formation. Tested PQ-derivatives, like the parent compound, might bind to specific sites within bacterial membranes and cause changes in membrane proteins, modulate their conformational status, and/or modify their lipid structureCitation26. Presumably, this action is not specific but may play an important role in the mechanism of antibacterial action. To extend this, it would be of interest to address drug–membrane interactions at different levels so as to approach molecular events that might underlie antibacterial action of PQ derivatives. In addition, it is known that PQ interferes with DNA synthesisCitation26. If this also occurs when we treat bacteria with PQ-derivatives, it could be possible that they depress the formation of an extracellular matrix that holds bacterial cells together in the biofilm, since the matrix is composed, among others, of protein and DNACitation27.
All compounds reduced biofilm formation of the yeast Candida albicans with MBEC values similar to or even lower than the standard antimycotic drug amphotericin B. It could be possible that PQ-ureidoamides, similar to the parent compound PQ, target Fe-S cluster proteins and affect yeast respiratory growthCitation28. Compounds 5e and 5f showed higher antibiofilm potential against yeast, especially against biofilm formation of C. kefyr with a rather low MBEC (6.25 µg/ml). This non-albicans Candida strain is one of the species identified as a common cause of candidiasis as a nosocomial infectionCitation29. These infections are often connected with frequent use of broad-spectrum antibiotics and different medical devices (central venous catheters, urinary catheters, prosthetic devices), and the finding of an efficient antibiofilm agent is of importance in this aspect as well. Since PQ induces morphological changes in membranes of P. fallax and inhibition of functional transport vesicle formation in the Golgi apparatusCitation30, one can speculate that a similar interaction may occur with the tested compounds. The study showed non-specific interactions of PQ (in the charged form) with the polar region of the phospholipidsCitation26. Thus, phospholipid–drug interactions may be the mechanism by which the tested compounds affect biofilm formation as well. It has been proposed that PQ-induced local changes of the lipid layer of the membrane may influence the protein function and start a signalling cascade that may lead to other biological events influencing the steady-state of a cell, in this case a bacterial or yeast cellCitation31.
Free radicals play an important role in the inflammatory process and several diseases, as well as in potential damage of cellular compounds such as DNA, proteins and lipids. Consequently, compounds with antioxidative properties could be expected to offer protection and therapeutic treatment. LOX inhibitors are potential agents for the treatment of inflammatory and allergic diseases, certain types of cancer, and cardiovascular diseasesCitation32. Thus, to evaluate the antioxidative potential of PQ-derivatives 5a–f, we have used three different antioxidant assays: (a) the interaction with the stable free radical DPPH, (b) the interaction with the water-soluble azo compound AAPH and (c) the inhibition of soybean lipoxygenase in vitro. DPPH interaction of the tested compounds was examined at a 100 µM concentration after 20 and 60 min. As shown in , DPPH-reducing ability of all tested compounds was very low, possibly due to stereochemical reasons.
Table 3. DPPH-reducing ability, IC50 values of soybean lipoxygenase (LOX) inhibition in vitro and anti-lipid peroxidation activity (LP) of compounds 5a–f.
In the AAPH assay, the highly reactive alkylperoxyl radicals are intercepted mainly through a hydrogen atom transfer (HAT) from the antioxidant. Therefore, particularly effective HAT agents are compounds with a high hydrogen atom donating ability, that is, compounds with low heteroatom-H bond dissociation energies and/or compounds from which hydrogen abstraction leads to sterically hindered radicals as well as compounds from which abstraction of hydrogen leads to C-centered radicals stabilised by resonance. LOX inhibition experiments showed that all PQ-ureidoamides, with the exception of compound 5a, were active, with IC50 values ranging from 40 to 69 µΜ. Again, derivative 5c was the most potent inhibitor (IC50 = 40 µM), followed by 5d (IC50 was slightly higher, e.g. 41.5 µM) and 5e (IC50 = 56 µM). The results showed that the presence of a chloro substituent was beneficial to activity, while position of the chlorine atom had no influence. Compounds 5a and 5b without a chloro substituent showed lower activity. However, the presence of the second Cl atom (5e) and a combination of chloro and methoxy substituents (5f) diminished the activity. Although these results are preliminary, they suggest a possible anti-inflammatory effect related to an antioxidative ability. More studies are needed to establish the anti-inflammatory activity and their mechanism of action.
In further study, the potential of compounds 5a–f to inhibit lipid peroxidation (LP) was evaluated as well. The results are given in . All derivatives inhibited lipid peroxidation significantly (80–99%), except for compound 5a (derivative with an aliphatic chain in the amino acid region). Phenylglycine derivative 5c with a chlorobenzhydryl substituent was the most potent (99%). Our results indicate that LOX inhibition was accompanied and correlated with LP inhibition.
PQ-ureidoamides 5a–f with potential antibacterial application could be intended for injectable use and thus in vitro haemolytic study was performed to evaluate haemoglobin release as an indicator of red blood cell lysis. Such studies are needed to address possible aspects of compound toxicity. Haemolytic activity of compounds 5a–f was evaluated using the fresh human erythrocyte assay, following the procedure described in experimental section ().
Table 4. Haemolytic activity of PQ-ureidoamides 5a–f.
Haemolytic activity tests showed no statistical differences between the tested PQ-derivatives and the parent compound PQ. The solvent DMSO (1.5%, v/v) did not cause significant haemolytic activity either. Overall, the tested compounds expressed no significant haemolytic activities in concentrations up to 200 µg/ml after 1 h of exposure to a 4% suspension of fresh human erythrocytes under aerobic conditions.
Conclusions
The occurrence of drug-resistant strains of bacteria is rising, and therefore, new approaches to treat bacterial infections are emerging, particularly for Gram-negative bacteria. Our findings suggest that the PQ-ureidoamides tested in this study may offer a new option for the treatment of biofilm-associated infections. They prevent bacterial adhesion to the surface and diminish the formation of a biofilm on abiotic surfaces at the concentration level of referent antimicrobial drugs or even at lower concentrations that are considered relevant for possible future use in vivo. Compounds 5a–f had high efficiency against biofilm formation of Gram-negative bacteria E. coli (MBEC from 6.25 to 50 µg/ml, four compounds had lower MBECs than gentamycin) and yeast C. albicans with MBEC values similar to or higher than the standard antimycotic drug amphotericin B. Compounds 5e and 5f showed higher antibiofilm potential against yeast, especially against biofilm formation of C. kefyr with a rather low MBEC (6.25 µg/ml). The favourable non-toxic profile of the PQ-ureidoamides tested on a series of human cancer cell lines and rat skeletal myoblastsCitation14 is in line with the guidelines for novel antibiofilm drugs. However, further studies are needed to clarify the mechanism of the antibiofilm effect observed. Some of our compounds kill bacteria at their planktonic stage as well: leucine derivative 5a and p-chlorophenylglycine PQ-ureidoamide 5d showed significant antibacterial activity against three S. aureus strains (MIC = 6.5 µg/ml), while compounds 5c and 5d showed weak antibacterial activity against E. coli and P. aeruginosa. Finally, compounds 5b–f significantly expressed anti-lipid peroxidation activity, whereas phenylglycine derivative 5c with a chlorobenzhydryl substituent exhibited antioxidant and anti-LOX profile.
Disclose statement
The research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
IENZ_1423067_Supplementary_Materials.pdf
Download PDF (2.2 MB)Additional information
Funding
Related Research Data
References
- Primaquine – FDA prescribing information, side effects and uses. [internet]. 2017. Sanofi-Aventis U.S. LLC. Available from: https://www.drugs.com/pro/primaquine.html, [last accessed 9 Jan 2018].
- van Huijsduijnen RH, Guy RK, Chibale K, et al. Anticancer properties of distinct antimalarial drug classes. PLoS One 2013;8:e82962.
- Liu F, Shang Y, Chen SZ. Chloroquine potentiates the anti-cancer effect of lidamycin on non-small cell lung cancer cells in vitro. Acta Pharmacol Sin 2014;35:645–52.
- Das AK. Anticancer effect of antimalarial artemisinin compounds. Ann Med Health Sci Res 2015;5:93–102.
- Pascolo S. Time to use a dose of chloroquine as an adjuvant to anti-cancer chemotherapies. Eur J Pharmacol 2016;771:139–44.
- Lin YC, Lin JF, Wen SI, et al. Chloroquine and hydroxychloroquine inhibit bladder cancer cell growth by targeting basal autophagy and enhancing apoptosis. Kaohsiung J Med Sci 2017;33:215–23.
- Džimbeg G, Zorc B, Kralj M, et al. The novel primaquine derivatives of N-alkyl, cycloalkyl or aryl urea: synthesis, cytostatic and antiviral activity evaluations. Eur. J Med Chem 2008;43:1180–7.
- Šimunović M, Perković I, Zorc B, et al. Urea and carbamate derivatives of primaquine: Synthesis, cytostatic and antioxidant activities. Bioorg Med Chem 2009;17:5605–13.
- Perković I, Tršinar S, Žanetić J, et al. Novel 1-acyl-4-substituted semicarbazide derivatives of primaquine − synthesis, cytostatic, antiviral and antioxidative studies. J Enzyme Inhib Med Chem 2013;28:601–10.
- Pavić K, Perković I, Cindrić M, et al. Novel semicarbazides and ureas of primaquine with bulky aryl or hydroxyalkyl substituents: synthesis, cytostatic and antioxidative activity. Eur J Med Chem 2014;86:502–14.
- Perković I, Antunović M, Marijanović I, et al. Novel urea and bis-urea primaquine derivatives with hydroxyphenyl and halogenphenyl substituents: synthesis and biological evaluation. Eur J Med Chem 2016;124:622–36.
- Pavić K, Perković I, Gilja P, et al. Design, synthesis and biological evaluation of novel primaquine-cinnamic acid conjugates of the amide and acylsemicarbazide type. Molecules 2016;21:1629–53.
- Pavić K, Perković I, Pospíšilová Š, et al. Primaquine hybrids as promising antimycobacterial and antimalarial agents. Eur J Med Chem 2018;143:769–79
- Pavić K, Supek F, Levatić J, et al. QSAR based synthesis of novel primaquine ureidoamides. In: 25th Croatian Meeting of Chemists and Chemical Engineers; 19–22 Apr 2017; Poreč, Croatia.
- Van Acker H, Van Dijck P, Coenye T. Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends Microbiol 2014;22:326–33.
- Hobley L, Harkins C, MacPhee CE, Stanley-Wall NR. Giving structure to the biofilm matrix: an overview of individual strategies and emerging common themes. FEMS Microbiol Rev 2015;39:649–69.
- Bjarnsholt T. The role of bacterial biofilms in chronic infections. APMIS Suppl 2013;136:1–51.
- Uppuluri P, Chaturvedi AK, Srinivasan A, et al. Dispersion as an important step in the Candida albicans biofilm developmental cycle. PLoS Pathog 2010;6:e1000828.
- Rabin N, Zheng Y, Opoku-Temeng C, et al. Agents that inhibit bacterial biofilm formation. Future Med Chem 2015;7:647–71.
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society for Clinical Microbiology and Infectious Diseases (ESCMID). EUCAST Discussion Document E. Dis 5.1: determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution, Clin. Microbiol. Infec. 2003, 9 (insert), 1.
- Luzina EL, Popov AV. Synthesis, evaluation of anticancer activity and COMPARE analysis of N-bis(tri fluoromethyl)alkyl-N’-substituted ureas with pharmacophoric moieties. Eur J Med Chem 2012;53:364–73.
- Chemicalize. ChemAxon Ltd. 2017. Available from: http://www.chemicalize.org
- Donlan RM, Costerton JW. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev 2002;15:167–93.
- Frirdich E, Whitfield C. Lipopolysaccharide inner core oligosaccharide structure and outer membrane stability in human pathogens belonging to the Enterobacteriaceae. J Endotoxin Res 2005;11:133–44.
- Bowman ZS, Morrow JD, Jollow DJ, McMillan DC. Primaquine-induced hemolytic anemia: role of membrane lipid peroxidation and cytoskeletal protein alterations in the hemotoxicity of 5-hydroxyprimaquine. J Pharmacol Exp Ther 2005;314:838–45.
- Basso LG, Rodrigues RZ, Naal RMZG, Costa-Filho AJ. Effects of the antimalarial drug primaquine on the dynamic structure of lipid model membranes. Biochim Biophys Acta 2011;1808:55–64.
- Branda SS, Vik S, Friedman L, Kolter R. Biofilms: the matrix revisited. Trends Microbiol 2005;13:20–6.
- Laleve A, Vallieres C, Golinelli-Cohen MP, et al. The antimalarial drug primaquine targets Fe-S cluster proteins and yeast respiratory growth. Redox Biol 2016;7:21–9.
- Cuéllar-Cruz M, López-Romero E, Villagómez-Castro JC, Ruiz-Baca E. Candida species: new insights into biofilm formation. Future Microbiol 2012;7:755–71.
- Hiebsch RR, Raub TJ, Wattenberg BW. Primaquine blocks transport by inhibiting the formation of functional transport vesicles. Studies in a cell-free assay of protein transport through the Golgi apparatus. J Biol Chem 1991;266:20323–8.
- Vale N, Moreira R, Gomes P. Primaquine revisited six decades after its discovery. Eur J Med Chem 2009;44:937–53.
- Pergola C, Werz O. 5-Lipoxygenase inhibitors: a review of recent developments and patents. Expert Opin Ther Pat 2010;20:355–75.