
Abstract
A set of compounds incorporating carbon-based zinc-binding groups (ZBGs), of the type PhX (X = COOH, CONH2, CONHNH2, CONHOH, CONHOMe), and the corresponding derivatives with sulphur(VI)-based ZBGs (X = SO3H, SO2NH2, SO2NHNH2, SO2NHOH, SO2NHOMe) were tested as inhibitors of all mammalian isoforms of carbonic anhydrase (CA, EC 4.2.1.1), CA I–XV. Three factors connected with the ZBG influenced the efficacy as CA inhibitor (CAI) of the investigated compounds: (i) the pKa of the ZBG; (ii) its geometry (tetrahedral, i.e. sulphur-based, versus trigonal, i.e. carbon-based ZBGs), and (iii) orientation of the organic scaffold induced by the nature of the ZBG. Benzenesulphonamide was the best inhibitor of all isoforms, but other ZBGs led to interesting inhibition profiles, although with an efficacy generally reduced when compared to the sulphonamide. The nature of the ZBG also influenced the CA inhibition mechanism. Most of these derivatives were zinc binders, but some of them (sulfonates, carboxylates) may interact with the enzyme by anchoring to the zinc-coordinated water molecule or by other inhibition mechanisms (occlusion of the active site entrance, out of the active site binding, etc.). Exploring structurally diverse ZBGs may lead to interesting new developments in the field of CAIs.
1. Introduction
Simple molecules/ions such as CO2, bicarbonate and protons are essential in many important physiologic processes in all life kingdoms (Bacteria, Archaea, and Eukarya) and for this reason, relatively high amounts of carbonic anhydrases (CAs, EC 4.2.1.1), the enzymes which use these molecules/ions as substrates, are present in most of the investigated organisms, all over the phylogenetic treeCitation1–13. There are seven genetically distinct CA families known to dateCitation4–7. The α-CAs are present in vertebrates, arthropods, sponges, corals, fungi, protozoa, algae, and cytoplasm of green plants but also in many Bacteria speciesCitation1,Citation7–13. β-CAs are predominantly found in Bacteria, algae, and chloroplasts of both mono- as well as dicotyledons, but also in many fungi and some ArchaeaCitation1–9. The γ-CAs were found in Archaea, Bacteria, and plantsCitation1–13, whereas the δ-, ζ-, and θ-CAs seem to be present only in marine diatomsCitation2. The η-CAs were found to date only in protozoa of the Plasmodium typeCitation5. In all these organisms, CAs catalyse the reversible hydration of carbon dioxide to bicarbonate and protons (hydronium ions), and are involved in crucial physiological processes connected with respiration and transport of CO2/bicarbonate, pH and CO2 homeostasis, electrolyte secretion in a variety of tissues/organs, biosynthetic reactions (e.g. gluconeogenesis, lipogenesis and ureagenesis), bone resorption, calcification, tumourigenicity, and many other physiologic or pathologic processes (thoroughly studied in vertebrates)Citation1–13. In algae, plants and some bacteria they play an important role in photosynthesis and other biosynthetic reactionsCitation10. In diatoms, CAs play a crucial role in carbon dioxide fixationCitation4,Citation10. Many such enzymes from vertebrates, protozoa, fungi, and bacteria are well-known drug targetsCitation1–13, and their inhibitors possess pharmacologic applications, being clinically used for the management of glaucomaCitation1,Citation14, edemaCitation1,Citation15, obesityCitation16, epilepsyCitation17, hypoxic tumorsCitation1,Citation2,Citation18,Citation19, idiopathic intracranial hypertensionCitation20, etc. Ultimately, some human (h) CA isoforms have also been validated as drug targets for cerebral ischemiaCitation21, neuropathic painCitation22, and arthritisCitation23, whereas many such enzymes present in pathogenic organisms may lead to the development of anti-infectives with a new mechanism of actionCitation24,Citation25.
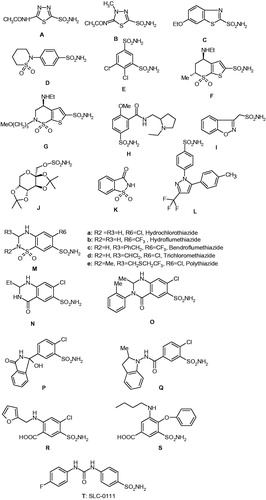
The main class of CA inhibitors (CAIs) is constituted by the primary sulphonamides and their isosteres (sulphamates, sulphamides)Citation1–6. They bind in deprotonated form, as sulphonamidate anions, to the metal ion from the enzyme active site, which is most frequently but not in all CA classes, a zinc ionCitation1–6,Citation12,Citation13. Sulphonamides are the most important class of CAIsCitation1,Citation2,Citation5,Citation6,Citation8,Citation16–19, since at least 20 such compounds are in clinical use for decades whereas one of them is in clinical development in the last period – Citation1–6,Citation14–25. They include acetazolamide A, methazolamide B, ethoxzolamide C, sulthiame D, dichlorophenamide E, dorzolamide F, brinzolamide G, sulpiride H, zonisamide I, topiramate J (a sulphamate), saccharin K, celecoxib L, chlorothiazide M and structurally related highceiling diuretics of types N, O, P, Q, R, and S, including the widely prescribed hydrochlorothiazide Ma, furosemide R, bumethanide S, all of them being compounds in clinical use for many years, as diuretics, antiglaucoma agents, or antiepilepticsCitation1–6,Citation14,Citation26–28. Compound T, SLC-0111, is in phase II clinical trials as an antitumor/antimetastatic agent, and was discovered in the author's laboratoryCitation18,Citation19,Citation27 (. Most of sulphonamides A–S act as potent CAIs and are in clinical use for decades, but their main problem is related to the fact that by inhibiting most of the catalytically active CA isoforms found in vertebrates (13 such isoforms are known to dateCitation1–13), they show a wide range of side effectsCitation1–6,Citation14–19. Newer generation inhibitors, among which T is a good example, were structurally-based drug designed in such a way as to act as isoform-selective inhibitors for the transmembrane, tumour-associated isoforms hCA IX and XII (h = human), and as thus, show less side effects compared to the classical CAIs of types A–S discussed aboveCitation27. X-ray crystallography of enzyme–inhibitor adducts was exhaustively been used for designing isoform-selective CAIs belonging to the sulphonamide, sulphamate, and sulphamide classesCitation12,Citation13,Citation26–28.
Figure 1. Clinically used sulphonamides/sulphamates with CA inhibitory action (A–S) and SLC-0111 (T) in phase II clinical trials as an antitumour/antimetastatic agentCitation18,Citation19.
However, in the last period, many other compounds with a similar mechanism of action were reported, among which dithiocarbamatesCitation29, xanthatesCitation30, monothiocarbamatesCitation31, some carboxylatesCitation32, hydroxamatesCitation33, phosphonatesCitation34, and borolsCitation35. All these CAIs are collectively termed as zinc binders, and their inhibition mechanism towards α-CAs is shown in . These CAIs bind in deprotonated form, as anions, to the Zn(II) ion from the enzyme active site, which is in a tetrahedral geometry, being coordinated by three His residues from the enzyme and by the zinc-binding group (ZBG) of the inhibitor (. There is overwhelming X-ray crystallographic evidence showing this type of binding for sulphonamides, sulphamates, sulphamides, dithiocarbamates, and their derivatives, hydroxamates, some carboxylates, one phosphonate and some borolsCitation12,Citation13,Citation26–35. The ZBG also interacts with two other conserved residues in all α-CAs, acting as “gate keepers”, i.e. Thr199 (hydrogen bonded through its OH group with the water molecule/hydroxide ion coordinated to the zinc in the uninhibited enzyme, and with the ZBG, as shown in , in the enzyme–inhibitor adducts) and Glu106, which is hydrogen bonded to Thr199 through its carboxylate moiety ()Citation21,Citation44–46,57–65. In other CA genetic families, the gate keepers are diverse amino acid residues, but the mechanism described above for α-class enzymes is probably valid (with small variations) for the remaining CA familiesCitation1–7,Citation12.
Figure 2. General scheme showing CAIs belonging to the zinc binders class (sulphonamides, sulphamates, sulphamides, carboxylates, hydroxamates, phosphonates, borols, etc.) in interaction with an α-CACitation1,Citation3. The ZBG is coordinated to the metal ion and makes hydrogen bonds with the gate keeper residues Thr199–Glu106, conserved in all α-CAsCitation1–3. The scaffold of the inhibitor may occupy either the hydrophylic or hydrophobic (or both) halves of the active siteCitation12, whereas the tails are orientated towards the exit of the cavity where the most variable amino acid residues among the different mammalian CAs are locatedCitation26–28. The interactions between the scaffold/tail with the enzyme are not shown.
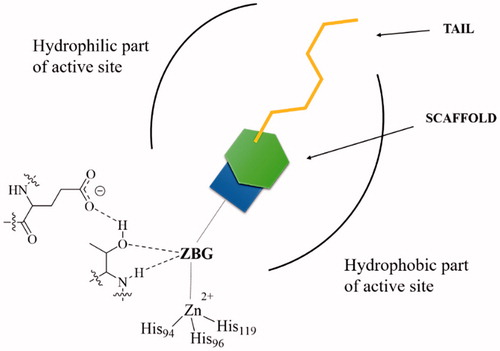
In addition to the ZBG, which is highly important in the design of CAIs, an inhibitor, as shown in , also possesses a scaffold and a tail. The scaffold is generally an aromatic, heterocyclic, aliphatic, sugar, or a combination of these moietiesCitation1–3. The tail started to be incorporated in the structure of CAIs starting with 1999, with the discovery of the tail approachCitation26–28, and has the role to assure isoform selectivity to the inhibitors, since these moieties, of a highly variable structural type, interact with the residues at the entrance of the active site cavity, which are the most variable regions among the various CA isoforms found in mammalsCitation1–3,Citation12,Citation26–28. Up until now, with few exceptions mentioned above (dithiocarbamatesCitation27–31, carboxylatesCitation32, hydroxamatesCitation33, phosphonatesCitation34, and borolsCitation35), the most successful strategies for obtaining potent and selective CAIs were based on ZBGs incorporating sulphur(VI) derivatives, basically variations on the sulphonamide themeCitation1–3,Citation12,Citation13,Citation26–28. In this article, I will present a comparison of sulphur-based versus carbon-based ZBGs for designing CAIs, and how the nature of this structural fragment from the inhibitor molecule is in fact the main factor influencing the inhibition profile, not only due to the coordination to the metal ion, but also due to structural constraints which the scaffolds experience when the ZBG is attached to the inhibitor molecule.
2. Materials and methods
2.1. Chemistry
Compounds 1–10 investigated here as CAIs were prepared as reported in the literatureCitation36–38, by reaction of phenylsulphonyl chloride or benzoyl chloride (from Sigma-Aldrich, Milan, Italy) with nucleophiles (water, ammonia, hydrazine hydrate, hydroxylamine hydrochloride, O-methyl-hydroxylamine, all commercially available from Sigma-Aldrich, Milan, Italy). These compounds are known in the literature and their physico-chemical and spectral data confirmed their structures (data not shown)Citation36–38.
2.2. Carbonic anhydrase inhibition assay
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activityCitation39. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled-deionised water and dilutions up to 0.1 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the enzyme–inhibitor (E–I) complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation17,Citation22–28 and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation22–28.
3. Results and discussion
In order to investigate how the ZBGs based on two different elements, i.e. sulphur and carbon, influence the potency as CAI of the compounds incorporating them, five sulphur(VI) derivatives based on the benzenesulphonic acid 1 and the same number of carbon(IV) derivatives, based on benzoic acid 6, were investigated as inhibitors of all catalytically active mammalian isoforms, CA I–XV. For both series, the amide (compounds 2 and 7, respectively), hydrazide (3 and 8), hydroxylamide (4 and 9) and O-methoxylated amide (5 and 10) were considered as possible CAIs, and were investigated for their interaction with the human (h) hCA I-XI, and hCA XIV, as well as the murine mCA XIII and mCA XV ().
Table 1. Inhibition of CA isozymes I–XV (of human = h, and murine = m origin) with compounds 1–10.
It may be observed that the scaffold of all these compounds 1–10 was identical, i.e. the simple phenyl moiety, whereas the ZBGs incorporate the central element in tetrahedral geometry for the sulphur derivatives 1–5, and in trigonal, planar geometry for the carbon ones 6–10 (as the central carbon atom has the sp2 hybridization). Thus, both the geometry and the acid-base properties of the ZBGs differ considerably among these compounds, due to these diverse hybridizations and electronic properties of the central element. Indeed, sulphonic acids such as 1 are very strong acids, with pKa in the negative range (1 has a pKa of –2.8), whereas benzoic acid has a pKa of 4.2Citation40. The same differences exist for their derivatives, with the aromatic sulphonamides behaving as much stronger acids (pKa of 2 is of 10.1Citation41) than the corresponding amides (pKa of 6 is of around 14.5Citation40). Thus, the main differences between the sulphur-based versus the carbon based ZBGs are: (i) geometry of the ZBG due to the diverse hybridization of the main element found in it; (ii) pKa of the ZBG. Both these two factors are known to be relevant for compounds to efficiently bind to Zn(II) in CAs and act as CAIsCitation1–13.
The following structure–activity relationship (SAR) was observed from the data of :
hCA I was efficiently inhibited by the primary sulphonamide 2, with an inhibition constant of 86 nM, whereas the remaining derivatives showed a much weaker inhibitory action. For the sulphur-based ZBGs, the N-hydroxy sulphonamide 4 and the sulphonic acid 1 were the next best inhibitors after 2, with KIs of 2.73–8.65 µM, whereas the N-amino and N-methoxy sulphonamides 3 and 5 were much less efficient CAIs compared to 2 (KIs of 69.8 to >1000 µM). Benzoic acid 6 was a very weak inhibitor (KI of 730 µM) but all its derivatives 7–10 showed an enhanced activity (KIs of 72.5–324 µM). In fact, the X-ray crystal structure of the hydroxamic acid 9 bound to hCA IICitation38 (the adduct with isoform hCA I was not yet characterised by means of crystallography) showed that the hydroxamate ZBG is bidentate, unlike the sulphonamide or carboxylate ones, with both oxygens present in the CONHOH fragment being coordinated to the catalytic zinc ion, confirming thus the possibility to use this ZBG for designing efficient CAIs.
hCA II was also best inhibited by the primary sulphonamide 2, with an inhibition constant of 101 nM, whereas all the remaining sulphur-based derivatives showed micromolar inhibition, with KIs in the range of 5.31–9.30 µM. For this isoform, there are not relevant differences of activity between the NHOH and NHNH2 substituted derivatives 3 and 4, which showed very similar inhibitory action. The N-methoxysulphonamide 5 was on the other hand very similar in its inhibitory behaviour to the sulphonic acid 1 (KIs of 8.96–9.30 µM). In fact, the X-ray crystal structures of 2, 3, and 4 bound to this isoform are availableCitation37,Citation38,Citation42, showing that although the interaction between the ZBG and the metal ion and amino acid residues in its neighbourhood are different for the three compounds, all of them lead to efficient inhibitors due to various factors which will be discussed in detail shortly. For the carbon-based ZBGs, the best inhibition was observed for the carboxylate 6 (KI of 30.1 µM), followed by the carboxamide 7 (KI of 93.2 µM), whereas the remaining derivatives were much weaker CAIs (KIs of 179–341 µM, ). Thus, for hCA II (as for hCA I), there is a net loss of CAI activity in the carbon-based ZBGs compared to the corresponding sulphur-based ones.
hCA III was inhibited in the low micromolar range by the sulphur-containing compounds 1–5 (KIs of 2.26–6.63 µM) and in the high micromolar range by the carbon-based derivatives 6–10 (KIs of 35.7–44.8 µM). It is interesting to note the rather limited range of variation for the KIs for both subseries, meaning that the nature of the ZBG does not influence inhibitory potential so strongly for this isoform compared to hCA I and II discussed above. Indeed, the sulphonic acid 1 and the sulphonamide 2 only differ by a factor of 3 in their inhibition against hCA III (whereas for hCA I the sulphonamide 2 was 100 times a better inhibitor compared to the sulphonic acid 1, see ). Even more striking were the results for the carbon based ZBG, with the carboxylic acid 6 and the carboxamide 7 having practically the same inhibition constant.
hCA IV, a membrane-bound isoform, was also better inhibited by the sulphur-based than the carbon-based derivatives. For the first subseries, again the sulphonamide and the sulphonic acid were the most efficient inhibitors, with KIs of 7.96–9.10 µM, whereas the N-amino, N-hydroxy-, and N-methoxy sulphonamides were less effective, with KIs of 24.6–78.4 µM. For the second subseries, the hydroxamate 9 was the best inhibitor (KI of 84.7 µM) whereas the remaining derivatives were much less effective (KIs of 111–434 µM). As for hCA I and II, there are net differences of activity between the sulphur-based and the carbon-based inhibitors.
For the first mitochondrial isoform, hCA VA, the most efficient inhibitors were the benzenesulphonamide 2 and its N-substituted derivatives 3–5, with KIs of 1.68–7.75 µM, whereas the sulphonic acid 1 was a much weaker inhibitor (KI of 80.9 µM). For the carbon-based compounds, the carboxylate 6 was a micromolar inhibitor (KI of 7.12 µM), thus almost as active as some of the sulphur-containing compounds (derivatives 2–5). On the other hand, derivatives 7–10 were around one order of magnitude less efficient hCA VA inhibitors compared to 6, with KIs of 46.8–89.1 µM.
The second mitochondrial isoform hCA VB had a different inhibition profile compared to hCA VA, with which it has a rather high homologyCitation1–4. Thus, the primary sulphonamide 2, its N-methoxy-derivative 5 and benzoic acid 6 showed the best inhibitory action, with KIs of 8.82–10.7 µM, whereas all the remaining derivatives were less efficient inhibitors, with KIs ranging between 53.6 and 95.2 µM. In this case, a similar behaviour of some of the sulphur-based and carbon-based ZBG-containing compounds was observed, such as, for example, the SO2NHNH2 and CONHNH2 pairs (3 and 8), or SO2NHOH and CONHOH derivatives 4 and 9, which possess similar inhibitory action ().
The secreted isoform hCA VI was potently inhibited by sulphonamide 2 (KI of 97 nM), with the next most efficient inhibitor being the sulphonic acid 1 (KI of 8.80 µM). All other compounds were much weaker inhibitors, with KIs in the range of 27.0–91.6 µM ().
The brain-associated cytosolic isoform hCA VII was also efficiently inhibited by the sulphonamide 2 (KI of 95 nM), whereas the remaining compounds with sulphur-based ZBGs showed a very similar behaviour of low micromolar inhibitors (KIs of 7.93–9.56 µM). All the carbon-based compounds were on the other hand high micromolar inhibitors (KIs of 70.7–92.2 µM).
The tumour-associated transmembrane isoform hCA IX was efficiently inhibited by sulphonamide 2 (KI of 97 nM), and surprisingly, the next most efficient inhibitor was the carboxamide 7, with a KI of 3.67 µM. Apart the sulphonic acid 1, which was a low micromolar inhibitor, the remaining derivatives showed inhibition constants in the high micromolar range, of 45.9–66.5 µM (). Many of the carbon-based ZBGs were thus more efficient in producing tighter binders for hCA IX compared to the sulphur-based ZBGs, which is a quite surprising result.
The second transmembrane, tumour-associated enzyme hCA XII was again best inhibited by the sulphonamide 2 (KI of 90 nM), whereas all the remaining sulphur-containing compounds were medium potency inhibitors, with inhibition constants in the range of 1.53–8.32 µM. Among the carbon-based derivatives, the best inhibitor was the hydroxamate 9 (KI of 9.51 µM) and the amide 7 (KI of 11.2 µM), with the remaining compounds acting as less efficient inhibitors (inhibition constants in the range of 20.4–45.0 µM).
mCA XIII, a cytosolic isoform quite similar to the corresponding human enzymeCitation1–4, was best inhibited by the sulphonamide 2 (KI of 100 nM), with the remaining sulphur derivatives 1, 3–5 acting as low micromolar inhibitors (KIs of 4.97–8.72 µM). The carbon-based inhibitors 6–10 were less effective for inhibiting mCA XIII, with KIs in the range of 23.0–63.3 µM.
hCA XIV, another transmembrane isoform, was also efficiently inhibited by the primary sulphonamide 2 (KI of 92 nM), but many of the carbon-based compounds also showed submicromolar inhibitory action. Indeed, benzoic acid 6, its hydrazide 8 and hydroxamate 9, showed KIs in the range of 0.94–0.99 µM. However, the amide 7 and the methylated hydroxamate 10 were weaker inhibitors, with inhibition constants in the range of 10.0–27.0 µM. The compounds with sulphur-containing ZBGs (except the sulphonamide discussed above) were low micromolar inhibitors, with KIs in the range of 1.57–8.79 µM.
mCA XV (the isoform XV is not present in primates, where it is encoded by a pseudogene, but is present in other vertebrates, including rodentsCitation1–4) was efficiently inhibited by the sulphonamide 2 (KI of 100 nM), whereas the remaining sulphur-based derivatives were micromolar inhibitors (KIs in the range of 5.16–8.03 µM). The carbon-based compounds were much less efficient inhibitors, with KIs in the range of 72.8–106 µM ().
The inhibition profiles of the investigated mammalian isoforms with both types of inhibitors were very different, with the same compound showing a range of inhibitory potencies. For example, although the primary sulphonamide was the best inhibitor of all isoforms, its efficacy ranged between 86 nM (for the most sensitive isoform, hCA I) and 8.82 µM (for the least inhibited isoform, hCA VB), i.e. differing by a factor of >100. This situation was in fact observed for most other compounds investigated here, showing thus that a series of different factors, not only pKa and geometry of the ZBG may influence the binding efficacy of the compound to the active site.
4. Conclusions
By analysing the inhibitory action of the 10 structurally related derivatives towards the 13 diverse mammalian CA isoforms (), one can observe that the inhibition efficacy is a multifactorial process, not easy to rationalise, even for these relatively simple compounds for which the scaffold was constant (a phenyl moiety). However, the X-ray crystal structures for the adducts of four of the considered compounds discussed here bound to hCA II, the sulphonamide 2Citation42 (PDB code 2WEJ), the N-hydroxysulphonamide 4 (PDB code 3T5UCitation37), the N-methoxy-sulphonamide 5 (PDB code 3T5ZCitation37) and the hydroxamate 9 (PDB code 4FL7Citation38) are known and they were crucial for rationalising the results presented here. Considering these data, it appears that at least three factors connected with the ZBG influence the efficacy as CAI of the investigated compounds:
(i) The pKa of the ZBG, which as mentioned above, varied on almost 20 pKa units, from the highly acidic sulphonic acid 1 to the poorly acidic amide 7. The way in which this parameter influences CAI properties was in fact highly debated already from the first phases of research in the CAIs fieldCitation1–4, as it is not so straightforward how this parameter influences activity. Indeed, a zinc binder must be deprotonated for efficiently binding the zinc ion (. However, the highly acidic sulphonic acid 1 (pKa of –2.8) is for example a much weaker CAI compared to the poorly acidic sulphonamide 2 (pKa of 10.1). Thus, pKa is not the only parameter influencing CAI effects, or probably it is not the most influential factor. An effective zinc binding inhibitor must dissociate easily to form the anion, but it should not be a too strong or a too weak acid. This is why probably both the sulphonic acids and the amides are weaker CAIs compared to the sulphonamides.
(ii) The geometry of the ZBG. The tetrahedral, i.e. sulphur-based, and the trigonal, i.e. carbon-based ZBGs, are able to bind in a diverse manner to the amino acid residues in the neighbourhood of the metal ion, such as Thr199 (and Thr200) and to the metal ion itselfCitation1–4,Citation37,Citation38. Indeed, the sulphur-based ZBGs seem to always bind in a monodentate fashion to the metal ion, probably also due to the fact that the sulphonamide and its N-substituted derivatives are more acidic than the corresponding N-substituted carbon-derived ZBGs of the carboxamide type (except the COOH, which is of course more acidic than the sulphonamide moiety). For example, the hydroxamate 9, as mentioned earlier, is one of the few bidentate ZBGs known to date among CAIs, and binds in deprotonated form to the zinc ion by means of both of its oxygen atoms from the CONHO– moietyCitation38. On the other hand, both sulphur-based and carbon-based ZBGs also participate in a strong hydrogen bond with Thr199, an amino acid residue conserved in all α-CAsCitation1–4. As seen from , the four ZBGs present in these inhibitors (except for the bulky SO2NHOMe one) are rather superimposable. However, the way in which they interact with Zn(II) and residues at the bottom of the active site is rather diverse, as seen from . For the three sulphonamide-based ZBGs, the metal ion is coordinated monodentately by the deprotonated N– atom of the ZBG, and a hydrogen bond with the OH of Thr199 further stabilises the adduct. The hydroxamate ZBG, as mentioned earlier, is also deprotonated but acts as a bidentate ligand (.
Figure 3. Superimposition of the hCA II – 2 adduct (blue, PDB code 2WEJCitation42) with the hCA II – 4 adduct (violet, PDB code 3T5UCitation37), the hCA II – 5 adduct (silver, PDB code 3T5ZCitation37) and the hCA II – hydroxamate 9 adduct (green, PDB code 4FL7Citation38).
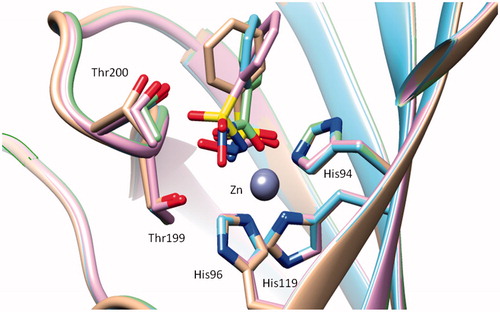
(iii) The orientation of the organic scaffold induced by the nature of the ZBG, is the last factor influencing activity, and is probably the most unexpected one. This is obvious from the superimposition shown in . It may be seen that the nature of the ZBG present in these four inhibitors leads to a rather diverse orientation of the phenyl scaffold in some of them. Thus, the phenyl scaffolds of the four inhibitors shown in are not at all superimposable, and this is mainly due to the influence that the ZBG has on the scaffold. It may be seen that a dramatic tilting of the phenyl group in 5 and 9 (compared to 2 and 4) leads to different orientations of the scaffold in these CAIs. But can we exploit these facts for designing other types of CAIs? I will present some examples which demonstrate that these findings can indeed lead to new developments in the field. In fact, the presence of a heteroatom (or a small functionality) between the organic scaffold and the ZBG may induce a diverse orientation of the scaffold, with profound consequences for the activity of the CAI. This is well illustrated for the examples given below in . Introduction of an oxygen atom or a NH moiety between the ZBG and the phenyl ring in sulphonamide 2 leads to the sulphamate 2a and sulphamide 2b. These two derivatives were tested for the inhibition of 4 CA isoformsCitation43a,b, and as seen from data of , they are much more potent CAIs compared to the lead 2. The sulphamate 2a is a low nanomolar inhibitor of the four CA isoforms, and the sulphamide 2b, although slightly less effective than the sulphamate is also a very potent CAI. Although there are no X-ray crystal structures of 2a and 2b bound to CA II (to be compared with the hCA II – 2 adduct), presumably the extra O and NH moieties present in these compounds lead to a diverse orientation of the scaffold when the compounds are bound to the enzyme. This was in fact well documented for the sulphamide analogue of topiramate J (possessing the NHSO2NH2 instead of the OSO2NH2 ZBG)Citation43c which is a much weaker CAI compared to J, and its X-ray crystal structure in adduct with hCA II showed a completely different binding modeCitation43c compared to topiramate in its hCA II adductCitation43d.
Table 2. hCA I, II, IX, and XII inhibition data with compounds 2, 2a, 2b, 7, and 7a.
On the other hand, introduction of the oxygen atom between the scaffold and the ZBG in amide 7 led to the carbamate 7a, which was also tested as CAI against four CA isoforms (). As seen from data of , in this case only hCA XII was better inhibited by the carbamate 7a compared to the carboxamide 7, whereas for the remaining isoforms the carboxamide was a better inhibitor compared to the carbamate. However, both derivatives act as rather weak CAIs compared to the sulphonamide, sulphamate, and sulphamide 2, 2a, and 2b (), but probably their activity may be optimised by choosing scaffolds which interact in a favourable manner with the enzyme active site.
Another aspect, which has not been contemplated in detail here due to lack of X-ray crystallographic data, is the fact that the nature of the ZBG may even induce a diverse inhibition mechanism. In fact, the zinc binders are just one and the best studied example of CAIs, but work in the last 10 years led to the discovery of several non-classical inhibition mechanisms, such as the anchoring of the inhibitor to the zinc-coordinated water molecule/hydroxide ionCitation44–46, occlusion of the active site cavity entranceCitation47 and out of the active site bindingCitation32f. Not to mention that there are many classes of potent CAIs (e.g. tertiary and secondary sulphonamides are the most investigated ones) for which the inhibition mechanism is not yet elucidatedCitation48,Citation49. For example, a sulphonic acid derivative obtained by the CA mediated hydrolysis of a sulphocoumarin, was shown to anchor to the zinc-coordinated water moleculeCitation44, opening the possibility that the sulphonic acid 1 investigated here may have a similar inhibition mechanism. The carboxylates are probably the most complicated family of CAIs since a variety of inhibition mechanisms were reported for many of them. In fact, some carboxylates coordinate to the metal ion in a monodentate fashionCitation32, others were observed anchored to the zinc coordinated waterCitation32, others were found at the entrance of the active siteCitation47, and one such derivative even outside the active site cavityCitation32f. Thus, it is unknown at this moment whether compound 6 (and also its derivatives 7, 8, or 10) inhibits CAs by the classical mechanism shown in or by alternative mechanisms. It is even possible that different isoforms are inhibited by different mechanisms with the same compound, as recently exemplified for acesulfame potassium salt, which is coordinated to the zinc ion in its complex with CA IX and anchored to the water coordinated to the metal ion in the CA II adductCitation50.
The conclusion of all these data is that although the sulphonamide seems to be the best ZBG for generating effective inhibitors of all CA isoforms, other ZBGs investigated here afforded interesting inhibition profiles, with efficacies reduced compared to the primary sulphonamide, but probably prone to be optimised, if an adequate scaffold is attached to them. Furthermore, as discussed above, the nature of the ZBG also influences the CA inhibition mechanism, with most of the derivatives investigated here probably being zinc binders. However, some of them (sulfonates, carboxylates) may interact with the enzyme by anchoring to the zinc-coordinated water molecule or by other inhibition mechanisms (occlusion of the active site entrance, out of the active site binding, etc.) which are less well investigated for the moment. Thus, exploring structurally diverse ZBGs may lead to many other interesting new developments in the design of isoform-selective CAIs, with the present findings attempting to share more light in this field.
Acknowledgements
I am particularly grateful to Dr. Alfonso Maresca for skilful technical assistance and to Dr. Marta Ferraroni for .
Disclosure statement
The author declares no conflict of interest.
Additional information
Funding
Related Research Data
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- (a) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (b) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- (a) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72. (b) Del Prete S, Vullo D, De Luca V, et al. Biochemical characterization of the δ-carbonic anhydrase from the marine diatom Thalassiosira weissflogii, TweCA. J Enzyme Inhib Med Chem 2014;29:906–11. (c) Del Prete S, Vullo D, De Luca V, et al. Biochemical characterization of recombinant beta-carbonic anhydrase (PgiCAb) identified in the genome of the oral pathogenic bacterium Porphyromonas gingivalis. J Enzyme Inhib Med Chem 2015;30:366–70.
- (a) Supuran CT, Capasso C. The eta-class carbonic anhydrases as drug targets for antimalarial agents. Expert Opin Ther Targets 2015;1:551–63. (b) Del Prete S, De Luca V, De Simone G, et al. Cloning, expression and purification of the complete domain of the η-carbonic anhydrase from Plasmodium falciparum. J Enzyme Inhib Med Chem 2016;31(Suppl.4):54–9. (c) Vullo D, Del Prete S, Fisher GM, et al. Sulfonamide inhibition studies of the η-class carbonic anhydrase from the malaria pathogen Plasmodium falciparum. Bioorg Med Chem 2015;23:526–31.
- (a) Zimmerman S, Innocenti A, Casini A, et al. Carbonic anhydrase inhibitors. Inhibition of the prokariotic beta and gamma-class enzymes from Archaea with sulfonamides. Bioorg Med Chem Lett 2004;1:6001–6. (b) Supuran CT, Capasso C. An overview of the bacterial carbonic anhydrases. Metabolites 2017;7:E56. (c) Nocentini A, Vullo D, Del Prete S, et al. Inhibition of the β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa with monothiocarbamates. J Enzyme Inhib Med Chem 2017;32:1064–70.
- (a) Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;3:325–32. (b) Supuran CT, Capasso C. Carbonic anhydrase from Porphyromonas gingivalis as a drug target. Pathogens 2017;6:E30. (c) Supuran CT, Capasso C. New light on bacterial carbonic anhydrases phylogeny based on the analysis of signal peptide sequences. J Enzyme Inhib Med Chem 2016;3:1254–60. (d) Ferraroni M, Del Prete S, Vullo D, et al. Crystal structure and kinetic studies of a tetrameric type II β-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Acta Crystallogr D Biol Crystallogr 2015;71:2449–56.
- (a) Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74. (b) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94.
- (a) Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;1:199–229. (b) Supuran CT. Drug interaction considerations in the therapeutic use of carbonic anhydrase inhibitors. Expert Opin Drug Metab Toxicol 2016;12:423–31.
- (a) Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnologic use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30. (b) Alterio V, Langella E, Viparelli F, et al. Structural and inhibition insights into carbonic anhydrase CDCA1 from the marine diatom Thalassiosira weissflogii. Biochimie 2012;94:1232–41. (c) Moya A, Tambutté S, Bertucci A, et al. Carbonic anhydrase in the scleractinian coral Stylophora pistillata: characterization, localization, and role in biomineralization. J Biol Chem 2008;283:25475–84. (d) Bertucci A, Moya A, Tambutté S, et al. Carbonic anhydrases in anthozoan corals—a review. Bioorg Med Chem 2013;21:1437–50. (e) Dathan NA, Alterio V, Troiano E, et al. Biochemical characterization of the chloroplastic β-carbonic anhydrase from Flaveria bidentis (L.) “Kuntze”. J Enzyme Inhib Med Chem 2014;29:500–4.
- (a) Vullo D, De Luca V, Del Prete S, et al. Sulfonamide inhibition studies of the γ-carbonic anhydrase from the Antarctic cyanobacterium Nostoc commune. Bioorg Med Chem 2015;23:1728–34. (b) Supuran CT. Bacterial carbonic anhydrases as drug targets: towards novel antibiotics? Front Pharmacol 2011;2:34. (c) Maresca A, Vullo D, Scozzafava A, et al. Inhibition of the beta-class carbonic anhydrases from Mycobacterium tuberculosis with carboxylic acids. J Enzyme Inhib Med Chem 2013;28:392–6.
- (a) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;1:4421–68. (b) De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810. (c) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11. (d) Di Fiore A, De Simone G, Alterio V, et al. The anticonvulsant sulfamide JNJ-26990990 and its S,S-dioxide analog strongly inhibit carbonic anhydrases: solution and X-ray crystallographic studies. Org Biomol Chem 2016;14:4853–8.
- (a) Winum JY, Supuran CT. Recent advances in the discovery of zinc binding motifs for the development of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:321–4. (b) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- (a) Carta F, Supuran CT, Scozzafava A. Novel therapies for glaucoma: a patent review 2007–2011. Expert Opin Ther Pat 2012;22:79–88. (b) Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16. (c) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47.
- Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91.
- (a) Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;2:725–35. (b) Supuran CT. Carbonic anhydrase inhibitors as emerging drugs for the treatment of obesity. Expert Opin Emerg Drugs 2012;17:11–5.
- (a) Bibi D, Mawasi H, Nocentini A, et al. Design and comparative evaluation of the anticonvulsant profile, carbonic-anhydrate inhibition and teratogenicity of novel carbamate derivatives of branched aliphatic carboxylic acids with 4-aminobenzensulfonamide. Neurochem Res 2017;4:1972–82. (b) Carta F, Birkmann A, Pfaff T, et al. Lead development of thiazolylsulfonamides with carbonic anhydrase inhibitory action. J Med Chem 2017;6:3154–64. (c) Supuran CT, Kalinin S, Tanç M, et al. Isoform-selective inhibitory profile of 2-imidazoline-substituted benzene sulfonamides against a panel of human carbonic anhydrases. J Enzyme Inhib Med Chem 2016;31(Supp1.1):197–202. (d) Aggarwal M, Kondeti B, McKenna R. Anticonvulsant/antiepileptic carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:717–24.
- (a) Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008–2013). Expert Opin Ther Pat 2013;2:737–49. (b) Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res 2011;7:3364–76. (c) Pettersen EO, Ebbesen P, Gieling RG, et al. Targeting tumour hypoxia to prevent cancer metastasis. From biology, biosensing and technology to drug development: the METOXIA Consortium. J Enzyme Inhib Med Chem 2015;30:689–721.
- (a) Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48. (b) Federici C, Lugini L, Marino ML, et al. Lansoprazole and carbonic anhydrase IX inhibitors sinergize against human melanoma cells. J Enzyme Inhib Med Chem 2016;31(Suppl.1):119–25. (c) Kusuzaki K, Matsubara T, Murata H, et al. Natural extracellular nanovesicles and photodynamic molecules: is there a future for drug delivery? J Enzyme Inhib Med Chem 2017;3:908–16. (d) Bragagni M, Carta F, Osman SM, et al. Synthesis of an acridine orange sulfonamide derivative with potent carbonic anhydrase IX inhibitory action. J Enzyme Inhib Med Chem 2017;32:701–6.
- Supuran CT. The safety and clinical efficacy of acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6.
- (a) Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. J Enzyme Inhib Med Chem 2016;3:894–9. (b) Bejaoui M, Panatzi E, De Luca V, et al. Acetazolamide protects steatotic liver grafts against cold ischemia reperfusion injury. J Pharmacol Exp Ther 2015;355:191–8.
- (a) Carta F, Di Cesare Mannelli L, Pinard M, et al. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg Med Chem 2015;2:1828–40. (b) Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother 2016;16:961–8. (c) Fragai M, Comito G, Di Cesare Mannelli L, et al. Lipoyl-homotaurine derivative (ADM_12) reverts oxaliplatin-induced neuropathy and reduces cancer cells malignancy by inhibiting carbonic anhydrase IX (CAIX). J Med Chem 2017;60:9003–11.
- (a) Margheri F, Ceruso M, Carta F, et al. Overexpression of the transmembrane carbonic anhydrase isoforms IX and XII in the inflamed synovium. J Enzyme Inhib Med Chem 2016;31(Suppl.4):60–3. (b) Bua S, Di Cesare Mannelli L, Vullo D, et al. Design and synthesis of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the treatment of rheumatoid arthritis. J Med Chem 2017;6:1159–70. (c) Angeli A, Tanini D, Capperucci A, Supuran CT. Synthesis of novel selenides bearing benzenesulfonamide moieties as carbonic anhydrase I, II, IV, VII, and IX inhibitors. ACS Med Chem Lett 2017;8:1213–7.
- (a) Capasso C, Supuran CT. Anti-infective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin Ther Pat 2013;2:693–704. (b) Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs – antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzyme Inhib Med Chem 2014;2:379–87. (c) Schlicker C, Hall RA, Vullo D, et al. Structure and inhibition of the CO2-sensing carbonic anhydrase Can2 from the pathogenic fungus Cryptococcus neoformans. J Mol Biol 2009;3:1207–20. (d) Aspatwar A, Hammarén M, Koskinen S, et al. β-CA-specific inhibitor dithiocarbamate Fc14-584B: a novel antimycobacterial agent with potential to treat drug-resistant tuberculosis. J Enzyme Inhib Med Chem 2017;32:832–40.
- (a) Maresca A, Carta F, Vullo D, Supuran CT. Dithiocarbamates strongly inhibit the beta-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;2:407–11. (b) Vermelho AB, da Silva Cardoso V, Ricci Junior E, et al. Nanoemulsions of sulfonamide carbonic anhydrase inhibitors strongly inhibit the growth of Trypanosoma cruzi. J Enzyme Inhib Med Chem 2018;33:139–46. (c) Nocentini A, Cadoni R, Del Prete S, et al. Benzoxaboroles as efficient inhibitors of the β-carbonic anhydrases from pathogenic fungi: activity and modeling study. ACS Med Chem Lett 2017;8:1194–8. (d) Vullo D, Kumar RSS, Scozzafava A, et al. Sulfonamide inhibition studies of the β-carbonic anhydrase from the bacterial pathogen Clostridium perfringens. J Enzyme Inhib Med Chem 2018;33:31–6. (e) De Vita D, Angeli A, Pandolfi F, et al. Inhibition of the α-carbonic anhydrase from Vibrio cholerae with amides and sulfonamides incorporating imidazole moieties. J Enzyme Inhib Med Chem 2017;32:798–804.
- (a) Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed Engl 2007;4:7697–9. (b) Scozzafava A, Menabuoni L, Mincione F, Supuran CT. Carbonic anhydrase inhibitors. A general approach for the preparation of water soluble sulfonamides incorporating polyamino-polycarboxylate tails and of their metal complexes possessing long lasting, topical intraocular pressure lowering properties. J Med Chem 2002;4:1466–76. (c) Carta F, Garaj V, Maresca A, et al. Sulfonamides incorporating 1,3,5-triazine moieties selectively and potently inhibit carbonic anhydrase transmembrane isoforms IX, XII and XIV over cytosolic isoforms I and II: solution and X-ray crystallographic studies. Bioorg Med Chem 2011;1:3105–19. (d) Vomasta D, Innocenti A, König B, Supuran CT. Carbonic anhydrase inhibitors: two-prong versus mono-prong inhibitors of isoforms I, II, IX, and XII exemplified by photochromic cis-1,2-α-dithienylethene derivatives. Bioorg Med Chem Lett 2009;19:1283–6.
- (a) Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902. (b) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun 2010;46:8371–3.
- (a) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II and IX with sulfonamides incorporating 1,2,4-triazine moieties. Bioorg Med Chem Lett 2004;1:5427–33. (b) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors. Novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumor-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;1:3102–8. (c) Carta F, Scozzafava A, Supuran CT. Sulfonamides (RSO2NH2): a patent review 2008–2012. Expert Opin Ther Pat 2012;22:747–58. (d) Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors. Part 74. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring? J Med Chem 1999;4:2641–50. (e) Borras J, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors. Part 73. Synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406. (f) Abbate F, Winum JY, Potter BV, et al. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with EMATE, a dual inhibitor of carbonic anhydrases and steroid sulfatase. Bioorg Med Chem Lett 2004;14:231–4.
- (a) Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates strongly inhibit carbonic anhydrases and show antiglaucoma action in vivo. J Med Chem 2012;55:1721–30. (b) Monti SM, Maresca A, Viparelli F, et al. Dithiocarbamates strongly inhibit the beta-class fungal carbonic anhydrases from Cryptococcus neoformans, Candida albicans and Candida glabrata. Bioorg Med Chem Lett 2012;22:859–62. (c) Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun 2012;4:1868–70. (d) Bozdag M, Carta F, Vullo D, et al. Synthesis of a new series of dithiocarbamates with effective human carbonic anhydrase inhibitory activity and antiglaucoma action. Bioorg Med Chem 2015;23:2368–76.
- Carta F, Akdemir A, Scozzafava A, et al. Xanthates and trithiocarbonates strongly inhibit carbonic anhydrases and show antiglaucoma effects in vivo. J Med Chem 2013;56:4691–700.
- (a) Vullo D, Durante M, Di Leva FS, et al. Monothiocarbamates strongly inhibit carbonic anhydrases in vitro and possess intraocular pressure lowering activity in an animal model of glaucoma. J Med Chem 2016;5:5857–67. (b) Nocentini A, Vullo D, Del Prete S, et al. Inhibition of the β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa with monothiocarbamates. J Enzyme Inhib Med Chem 2017;32:1064–70.
- (a) Carta F, Innocenti A, Hall RA, et al. Carbonic anhydrase inhibitors. Inhibition of the β-class enzymes from the fungal pathogens Candida albicans and Cryptococcus neoformans with branched aliphatic-/aromatic carboxylates and their derivatives. Bioorg Med Chem Lett 2011;21:2521–6. (b) Innocenti A, Vullo D, Scozzafava A, et al. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2005;15:573–8. Interaction of isozymes I, II, IV, V and IX with carboxylates. (c) Maresca A, Vullo D, Scozzafava A, et al. Inhibition of the beta-class carbonic anhydrases from Mycobacterium tuberculosis with carboxylic acids. J Enzyme Inhib Med Chem 2013;28:392–6. (d) Mori M, Cau Y, Vignaroli G, et al. Hit recycling: discovery of a potent carbonic anhydrase inhibitor by in silico target fishing. ACS Chem Biol 2015;10:1964–9. (e) Langella E, D’Ambrosio K, D’Ascenzio M, et al. A combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes within carbonic anhydrase active site. Chemistry 2016;22:97–100. (f) D’Ambrosio K, Carradori S, Monti SM, et al. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem Commun 2015;51:302–5. (g) Cau Y, Vullo D, Mori M, et al. Potent and selective carboxylic acid inhibitors of tumor-associated carbonic anhydrases IX and XII. Molecules 2018;23:E17. (h) Cadoni R, Pala N, Lomelino C, et al. Exploring heteroaryl-pyrazole carboxylic acids as human carbonic anhydrase XII inhibitors. ACS Med Chem Lett 2017;8:941–6.
- (a) Scozzafava A, Supuran CT. Carbonic anhydrase and matrix metalloproteinase inhibitors. Sulfonylated amino acid hydroxamates with MMP inhibitory properties act as efficient inhibitors of carbonic anhydrase isozymes I, II and IV, and N-hydroxysulfonamides inhibit both these zinc enzymes. J Med Chem 2000;43:3677–87. (b) de Menezes Dda R, Calvet CM, Rodrigues GC, et al. Hydroxamic acid derivatives: a promising scaffold for rational compound optimization in Chagas disease. J Enzyme Inhib Med Chem 2016;31:964–73. (c) Scozzafava A, Supuran CT. Hydroxyurea is a carbonic anhydrase inhibitor. Bioorg Med Chem 2003;11:2241–6. (d) Nuti E, Orlandini E, Nencetti S, et al. Carbonic anhydrase and matrix metalloproteinase inhibitors. Inhibition of human tumor-associated isozymes IX and cytosolic isozyme I and II with sulfonylatedhydroxamates. Bioorg Med Chem 2007;15:2298–311. (e) Santos MA, Marques S, Vullo D, et al. Carbonic anhydrase inhibitors. Inhibition of cytosolic/tumor-associated isoforms I, II and IX with iminodiacetic carboxylates/hydroxamates also incorporating benzenesulfonamide moieties. Bioorg Med Chem Lett 2007;17:1538–43. (f) Supuran CT. Hydroxamates as carbonic anhydrase inhibitors. In: Gupta SP, ed. Hydroxamic acids: a unique family of chemicals with multiple biological activities. Heidelberg: Springer Verlag; 2013:55–69.
- (a) Reich R, Hoffman A, Veerendhar A, et al. Carbamoylphosphonates control tumor cell proliferation and dissemination by simultaneously inhibiting carbonic anhydrase IX and matrix metalloproteinase-2. Toward nontoxic chemotherapy targeting tumor microenvironment. J Med Chem 2012;55:7875–82. (b) Tauro M, Loiodice F, Ceruso M, et al. Dual carbonic anhydrase/matrix metalloproteinase inhibitors incorporating bisphosphonic acid moieties targeting bone tumors. Bioorg Med Chem Lett 2014;2:2617–20. (c) Temperini C, Innocenti A, Guerri A, et al. Phosph(on)ate as a zinc binding group in metalloenzyme inhibitors: X-ray crystal structure of the antiviral drug foscarnet complexed to human carbonic anhydrase I. Bioorg Med Chem Lett 2007;1:2210–15. (d) Luisi G, Angelini G, Gasbarri C, et al. Dual targeting of cancer-related human matrix metalloproteinases and carbonic anhydrases by chiral N-(biarylsulfonyl)-phosphonic acids. J Enzyme Inhib Med Chem 2017;32:1260–4.
- (a) Alterio V, Cadoni R, Esposito D, et al. Benzoxaborole as a new chemotype for carbonic anhydrase inhibition. Chem Commun (Camb) 2016;5:11983–6. (b) Nocentini A, Cadoni R, Dumy P, et al. Carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani chagasi are inhibited by benzoxaboroles. J Enzyme Inhib Med Chem 2018;33:286–9.
- Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996;31:1001–10.
- Di Fiore A, Maresca A, Alterio V, et al. Carbonic anhydrase inhibitors: X-ray crystallographic studies for the binding of N-substituted benzenesulfonamides to human isoform II. Chem Commun (Camb) 2011;47:11636–8.
- Di Fiore A, Maresca A, Supuran CT, De Simone G. Hydroxamate represents a versatile zinc binding group for the development of new carbonic anhydrase inhibitors. Chem Commun (Camb) 2012;48:8838–40.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Guthrie JP. Hydrolysis of esters of oxy acids: pKa values for strong acids. Can J Chem 1978;56:2342–54.
- Remko M. Molecular structure, pKa, lipophilicity, solubility and absorption of biologically active aromatic and heterocyclic sulfonamides. J Mol Struct 2010;944:34–42.
- Scott AD, Phillips C, Alex A, et al. Thermodynamic optimisation in drug discovery: a case study using carbonic anhydrase inhibitors. ChemMedChem 2009;4:1985–9.
- (a) Winum JY, Vullo D, Casini A, et al. Carbonic anhydrase inhibitors. Inhibition of cytosolic isozymes I and II and transmembrane, tumor-associated isozyme IX with sulfamates including EMATE also acting as steroid sulfatase inhibitors. J Med Chem 2003;4:2197–204. (b) Casini A, Winum JY, Montero JL, et al. Carbonic anhydrase inhibitors: inhibition of cytosolic isozymes I and II with sulfamide derivatives. Bioorg Med Chem Lett 2003;1:837–40. (c) Winum JY, Temperini C, El Cheikh K, et al. Carbonic anhydrase inhibitors: clash with Ala65 as a means for designing inhibitors with low affinity for the ubiquitous isozyme II, exemplified by the crystal structure of the topiramate sulfamide analogue. J Med Chem 2006;4:7024–31. (d) Casini A, Antel J, Abbate F, et al. Carbonic anhydrase inhibitors: SAR and X-ray crystallographic study for the interaction of sugar sulfamates/sulfamides with isozymes I, II and IV. Bioorg Med Chem Lett 2003;13:841–5.
- (a) Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;5:293–300. (b) Grandane A, Tanc M, Di Cesare Mannelli L, et al. 6-Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J Med Chem 2015;58:3975–83.
- (a) Ferraroni M, Carta F, Scozzafava A, et al. Thioxocoumarins inhibit carbonic anhydrases through a different mechanism compared to coumarins. J Med Chem 2016;59:462–73. (b) Carta F, Temperini C, Innocenti A, et al. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 2010;53:5511–22.
- (a) Bayram E, Senturk M, Kufrevioglu OI, Supuran CT. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg Med Chem 2008;16:9101–5. (b) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68. (c) Sentürk M, Gülçin I, Beydemir S, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9. (d) Oztürk Sarikaya SB, Topal F, Sentürk M, et al. In vitro inhibition of α-carbonic anhydrase isozymes by some phenolic compounds. Bioorg Med Chem Lett 2011;21:4259–62. (e) Innocenti A, Gülçin I, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Antioxidant polyphenols effectively inhibit mammalian isoforms I–XV. Bioorg Med Chem Lett 2010;20:5050–3. (f) Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: inhibition of mammalian isoforms I–XIV with a series of substituted phenols including paracetamol and salicylic acid. Bioorg Med Chem 2008;16:7424–8.
- (a) Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;1:3057–62. (b) Küçükbay FZ, Küçükbay H, Tanc M, et al. Synthesis and carbonic anhydrase inhibitory properties of amino acid - coumarin/quinolinone conjugates incorporating glycine, alanine and phenylalanine moieties. J Enzyme Inhib Med Chem 2016;31:1198–202. (c) Vullo D, Isik S, Bozdag M, et al. 7-Amino-3,4-dihydro-1H-quinolin-2-one, a compound similar to the substituted coumarins, inhibits α-carbonic anhydrases without hydrolysis of the lactam ring. J Enzyme Inhib Med Chem 2015;3:773–7. (d) Maresca A, Scozzafava A, Supuran CT. 7,8-Disubstituted – but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8.
- (a) Scozzafava A, Carta F, Supuran CT. Secondary and tertiary sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2013;2:203–13. (b) Alp C, Ozsoy S, Alp NA, et al. Sulfapyridine-like benzenesulfonamide derivatives as inhibitors of carbonic anhydrase isoenzymes I, II and VI. J Enzyme Inhib Med Chem 2012;2:818–24. (c) Le Darz A, Mingot A, Bouazza F, et al. Fluorinated pyrrolidines and piperidines containing tertiary benzenesulfonamides: selective carbonic anhydrase II inhibitors. J Enzyme Inhib Med Chem 2015;30:737–45.
- (a) De Monte C, Carradori S, Secci D, et al. Cyclic tertiary sulfamates: selective inhibition of the tumor-associated carbonic anhydrases IX and XII by N- and O-substituted acesulfame derivatives. Eur J Med Chem 2014;8:240–6. (b) D’Ascenzio M, Carradori S, Secci D, et al. Selective inhibition of human carbonic anhydrases by novel amide derivatives of probenecid: synthesis, biological evaluation and molecular modelling studies. Bioorg Med Chem 2014;22:3982–8. (c) Carradori S, Mollica A, Ceruso M, et al. New amide derivatives of probenecid as selective inhibitors of carbonic anhydrase IX and XII: biological evaluation and molecular modelling studies. Bioorg Med Chem 2015;23:2975–81.
- Murray AB, Lomelino CL, Supuran CT, McKenna R. “Seriously Sweet”: acesulfame K exhibits selective inhibition using alternative binding modes in carbonic anhydrase isoforms. J Med Chem 2018; in press. doi: 10.1021/acs.jmedchem.7b01470.