
Abstract
Xanthohumol (XN), a prenylated chalcone unique to hops (Humulus lupulus) and two derived prenylflavanones, isoxanthohumol (IX) and 8-prenylnaringenin (8-PN) gained increasing attention as potential anti-diabetic and cancer preventive compounds. Two enzymes of the aldo-keto reductase (AKR) superfamily are notable pharmacological targets in cancer therapy (AKR1B10) and in the treatment of diabetic complications (AKR1B1). Our results show that XN, IX and 8-PN are potent uncompetitive, tight-binding inhibitors of human aldose reductase AKR1B1 (Ki = 15.08 μM, 0.34 μM, 0.71 μM) and of human AKR1B10 (Ki = 20.11 μM, 2.25 μM, 1.95 μM). The activity of the related enzyme AKR1A1 was left unaffected by all three compounds. This is the first time these three substances have been tested on AKRs. The results of this study may provide a basis for further quantitative structure–activity relationship models and promising scaffolds for future anti-diabetic or carcinopreventive drugs.
1. Introduction
The female inflorescences of hops (Humulus lupulus) have long been used in traditional Chinese and Ayurvedic medicine mainly to treat sleep disturbancesCitation1 and are also known for their antibiotic propertiesCitation2. Among the secondary plant compounds that occur in the resinous inflorescences of H. lupulus are prenylated chalcones and other flavonoidsCitation3. These compounds are well known for their bittering and preserving qualities in the brewing process of beerCitation3,Citation4, as well as for their bioactivity (antibiotic, anti-viral and antioxidant properties)Citation5–7. Recently, the prenylated chalcone xanthohumol (XN) has come into focus of biomedical research, due to its versatile bioactive characteristicsCitation8. Since XN is a unique prenylflavonoid occurring in hops, beer is the only noteworthy dietary source for XN in central Europe. In addition to XN, hops inflorescences also contain flavanones like isoxanthohumol (IX) and 8-prenylnaringenin (8-PN)Citation9 (), but at 10- to 100-fold lower concentrations than XN. The main metabolic routes for XN, IX and 8-PN have been characterised extensively, both, in vitro by using human and rat liver microsomes Citation3,Citation10–12 and in vivo, in rat modelsCitation13,Citation14. XN is subject to spontaneous conversion into IX via intramolecular Michael additionCitation15. IX, in contrast, undergoes an enzymatic O-demethylation via hepatic CYP1A2 to 8-PN, which has been described as the most potent phytoestrogen found in natureCitation5,Citation16. All three substances are currently under investigation due to their anti-diabeticCitation17,Citation18, anti-carcinogenicCitation3,Citation8,Citation19 and antioxidant propertiesCitation20. Moreover, XN has been shown to have anti-HIV traitsCitation21,Citation22.
Figure 1. Chemical structures of xanthohumol (XN), isoxanthohumol (IX) and 8-prenylnaringenin (8-PN).
The pathogenesis of several inflammatory conditions, such as diabetes, cancer and sepsis, have in many cases been linked to enzymes of the aldo-keto reductase (AKR) superfamilyCitation23–29. Among them, AKR1A1, also known as human aldehyde reductase, is involved in the reduction of biogenic and xenobiotic carbonyl group containing compounds, such as the cytotoxic lipid peroxidation-derived aldehydes 4-ONE and 4-HNECitation30,Citation31 and the chemotherapeutic drugs doxorubicin (DOX) or daunorubicin (DAUN)Citation32. It is widely expressed in most tissues, with particularly high levels reported in the cerebrum, liver and kidneysCitation33.
The same applies for the homologous enzyme AKR1B1 (60% sequence identity to AKR1A1), commonly known as human aldose reductaseCitation34. AKR1B1 mediates the first step of the “polyol pathway” by reducing glucose to sorbitol through a NADPH-dependent reaction. Further, sorbitol is oxidised to fructose by sorbitol dehydrogenase, using NAD+ as a co-factorCitation35,Citation36. Under normoglycemic conditions only 3% of total glucose is converted into sorbitol through the polyol pathway. Under hyperglycemic conditions, however, the flux of glucose through this metabolic pathway may increase up to 10-fold (30%), which might finally lead to accumulation of excess sorbitolCitation35,Citation37–39. The resulting osmotic stress and imbalances of the pyridine nucleotide redox status decrease the cell’s antioxidative capabilities. In diabetes mellitus, this promotes the formation of advanced glycation end products (AGEs)Citation34,Citation40, which themselves lead to diabetic complications, such as microangiopathies, nephropathies, retinopathies, peripheral neuropathies and cataractCitation41–43. Consequently, aldose reductase inhibitors (ARIs) are at the focus of exploratory pharmaceutical research, as they yield the potential to prevent or control the onset of these diabetic complications. In vitro, AKR1B1 displays poor affinity to glucose (KM = 50–100 mM) and hydrophobicity of the putative substrate-binding domain essentially precludes efficient carbohydrate reductionCitation44. Therefore, lipid peroxidation-derived hydrophobic aldehydes, such as 4-hydroxynonenal (HNE) and other related nephro- and hepatotoxic compounds are more likely to be physiological substrates of AKR1B1. Finally, high catalytic efficiencies of AKR1B1 towards these compounds suggest an important metabolic role in the detoxification of lipid-derived aldehydesCitation27,Citation44,Citation45. Physiologically and despite the relatively high KM for glucose, however, selective AKR1B1 inhibition remains an effective treatment option for diabetic complications derived from glucose-related adductsCitation44,Citation46.
Overlapping substrate spectra are considered a major pitfall of many ARIs, as they often inhibit AKR1A1 and AKR1B1 simultaneously, thereby interfering with their central role in detoxification processes. This has raised the demand for selective inhibitors that preferentially bind to AKR1B1 and leave the activity of AKR1A1 unaffectedCitation47–49.
Additionally, the AKR1B subclass contains the AKR1B1 homologue AKR1B10 (71% sequence identity to AKR1B1), a NADPH-dependent oxidoreductase that converts both endo- and exogenous carbonyl group containing compounds to their corresponding alcoholsCitation50–52. In contrast to AKR1B1, AKR1B10 exhibits a more restricted substrate specificity towards isoprenyl aldehydes including farnesal and geranylgeranial, which are reduced to their corresponding alcohol metabolitesCitation52.
These metabolites are involved in protein prenylation, a process suspected of being crucial for carcinogenesisCitation28. Moreover, AKR1B10 participates in the reduction of retinal to retinol, and thereby balances the homeostasis of retinoic acid, which is able to modulate cell proliferation and differentiationCitation53,Citation54, as well as the reductive metabolism of carbonyl group containing exogenous compoundsCitation50–52.
AKR1B10 is physiologically expressed in the small and large intestine and in the adrenal glandsCitation55,Citation56. AKR1B10 overexpression has been reported in several types of cancer, including lungCitation57,Citation58, pancreaticCitation26,Citation28,Citation59 and hepatocellular tumorsCitation24. At the same time, AKR1B10 fulfills a regulatory role in cell proliferation and differentiation by modulating the metabolism of retinoids and prenylation of oncoproteins, including KrasCitation26,Citation59. Since AKR1B10 overexpression is already detectable in precancerous lesionsCitation50,Citation60, metabolic events mediated by this enzyme could play a crucial role in the development of cancer. In particular, membrane proteins like Ras and Ras-related GTP-binding proteins rely on prenylation to exert their functions in processes of cellular growth and differentiationCitation28,Citation61,Citation62. Within the metabolism of farnesal and geranylgeranial whose reduced metabolites are important intermediates of cholesterol synthesis and protein prenylation, AKR1B10 performs key reactions by reducing farnesyl and geranylgeranial to farnesol and geranylgeraniol. The latter are further phosphorylated to farnesyl and geranylgeranyl pyrophosphates, two main intermediates of cholesterol synthesis involved in protein prenylationCitation28. Increased levels of AKR1B10 expression in neoplastic cells alterate prenylation processes and post-translational modifications of the aforementioned protooncoproteins and, hence, accelerate tumor formationCitation28,Citation53.
Thus, the finding of new potent and selective inhibitors for both AKR1B10 and AKR1B1 are current challenges in biomedical research, in order to prevent uncontrolled cell proliferation and to reduce microangiopathies, as found in diabetic complications. In this article, we investigated the inhibitory potential of three hop-derived substances on three members of the AKR superfamily (namely AKR1A1, AKR1B1 and AKR1B10). Our results provide evidence that XN, IX and 8-PN are potent inhibitors of AKR1B1 and AKR1B10, while not affecting the activity of the closely related enzyme AKR1A1.
2. Material and methods
2.1. Materials
2.1.1. Chemicals and reagents
XN was a friendly gift from Dr. Klaus Kammhuber, Lfl Hop Research Center Huell (Huell, Germany). IX and 8-PN were purchased from Biomol (Hamburg, Germany). NADPH was obtained from Carl Roth GmbH+Co. (Karlsruhe, Germany). Glucose, glyceraldehyde and farnesal were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2. Methods
2.2.1. Preparation of recombinant proteins
The recombinant enzymes AKR1A1, AKR1B1 and AKR1B10 were prepared in an Escherichia coli expression system according to previously published methods: AKR1A1 was a gentle gift from Prof. Dr. Vladimir WsolCitation63; AKR1B1 was a friendly gift from Dr. Nina Kassner; information about production and purification of AKR1B10 has been published beforeCitation64. Genetic information on the specific inserts of all obtained plasmids was verified by sequencing (MWG Eurofins). The plasmids were then transformed into E. coli BL21 (DE3) cells. For over-expression of 6 × His-tagged enzymes, a 400-ml culture (containing the appropriate antibiotic; plasmid dependent) was grown to an optical density of 0.6 at 600 nm at 37 °C. Protein over-expression was induced by adding isopropyl-1-thio-galactopyranoside (IPTG) to the culture medium (final concentration of 1 mM). After 3 h, cells were harvested by centrifugation (6000 g, 15 min) and re-suspended in 20 ml PBS-I buffer (20 mM NaH2PO4, 500 mM NaCl, 10 mM imidazole, pH 7.4). Cell disruption was performed by ultrasonication with cooling on ice, to avoid heating. The sample was subsequently centrifuged at 100,000 g at 4 °C for 1 h. The obtained supernatants, containing the target protein were purified using Ni-affinity chromatography on an ÄKTA-Purifier System (Amersham Pharmacia Biotech, Uppsala, Sweden). Purification progress was monitored by SDS-PAGE of the obtained fractions (not shown). Enzyme concentrations were determined using a Qubit 2.0 fluorometric quantitation system (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions.
2.2.2. Determination of inhibition parameters
Catalytic properties were determined by measuring the decrease in absorbance at 340 nm at 37 °C (Cary 100 scan photometer, Varian, Pal Alto, CA, USA). A reaction mixture without inhibitor consisted of different concentrations of substrate (see for details), 200 μM NADPH, 0.1 M NaH2PO4 buffer (pH 7.4) and an appropriate amount of enzyme in a total assay volume of 0.8 ml. Final enzyme concentrations in the assay ranged from 583 nM (AKR1B10) to 712 nM (AKR1B1). For inhibitor selectivity studies on AKR1A1, AKR1B1 and AKR1B10 stock solutions of the inhibitors XN, XI and 8-PN were prepared in dimethyl sulfoxide (DMSO). The final concentration of DMSO in the assay was ≤0.5%. Activity measurements were started without pre-incubation by adding an appropriate amount of enzyme. When collecting data for dose–response curves, initial velocities of the glyceraldehyde reduction (concentration at KM; enzyme specific) in the presence of inhibitors were assayed as described above. The percentage of inhibition was calculated considering the activity in the absence of inhibitor to be 100%.
Table 1. IC50 and Ki values of the AKR1B1 and AKR1B10-catalysed GA reduction in the presence of the inhibitors XN, IX and 8-PN.
Initially, the half maximal inhibitory concentrations (IC50 values) were determined for each inhibitor in the presence of each enzyme, using the shared substrate glyceraldehyde (set to their specific Km; 3.6 mM, 50 µM and 4 mM for AKR1A1, AKR1B1 and AKR1B10, respectively) to assess specificity among the structurally similar members of the AKR-superfamily. For IC50 determination, experimental data were normalised and fitted to a sigmoidal curve as implemented in GraphPad6 (GraphPad Software Inc., La Jolla, CA, USA). Whenever tight-binding inhibition was observed, the inhibition constant Ki was determined by fitting inhibition data to the Morrison equation as implemented in GraphPad Prism6 (GraphPad Software Inc., La Jolla, CA, USA)Citation65, using non-linear regression.
In order to verify the inhibitory potency, enzyme-specific physiological substrates for AKR1B1 (glucose, KM = 32 mM) and AKR1B10 (farnesal; KM = 5 µM) were used to determine inhibition parameters. Enzyme inhibition parameters were assayed as described above. The inhibition mechanism of each compound for the respective enzymes was analysed by plotting IC50 values at different substrate concentrations (at least five inhibitor and substrate concentrations)Citation65,Citation66. All data obtained were plotted and analysed using GraphPad Prism6 (GraphPad Software Inc., La Jolla, CA, USA).
3. Results
3.1. Determination of inhibitor selectivity
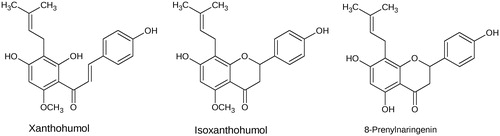
Initially, dose–response curves for XN, IX and 8-PN with AKR1A1, AKR1B10 and AKR1B1, using glyceraldehyde, were calculated (IC50- and Ki-values are summarised in ). exemplarily shows the determination of IC50- and Ki-values for IX with AKR1B1. IX turned out to be the most effective inhibitor among the three substances for both AKR1B1 and AKR1B10 (IC50 = 0.57 and 1.09 µM, respectively). The IC50 for IX is 6 to 15 times lower than compared to XN (). Interestingly, the activity of AKR1A1 was unaffected by all three substances (IC50 > 50 µM).
Figure 2. Dose–response (a) and inhibition curve of the AKR1B1-catalysed glyceraldehyde reduction by isoxanthohumol (b). Enzymatic activity is expressed as the ratio of inhibited vs. non-inhibited reaction rate. Data were fitted to the Morrison equation for tight-binding inhibitors. All data are presented as mean ± standard deviation from at least three experiments.
3.2. Determination of inhibition parameters for physiological substrates
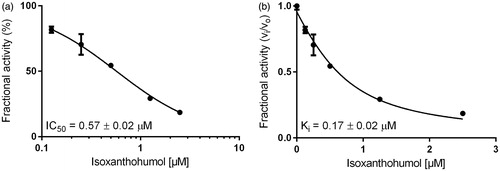
Inhibition constants, as well as the mode of inhibition on the reduction of two specific physiological substrates, were further calculated by using glucose for AKR1B1 and farnesal for AKR1B10, at concentrations equal to their corresponding KM values (as determined before: 32 mM and 5 µM, respectively–data not shown). and show the exemplified determination of the IC50- and Ki-values (calculated using the Morrison equationCitation65), in presence of IX, for AKR1B1 and AKR1B10, respectively. and summarise the obtained IC50 and the inhibition constants for the respective enzymes.
Table 2. IC50 and Ki values of the AKR1B1-catalysed glucose reduction in the presence of the inhibitors XN, IX and 8-PN.
Figure 3. Dose–response (a) and inhibition curve of the AKR1B1-catalysed glucose reduction by isoxanthohumol (b). Enzymatic activity is expressed as the ratio of inhibited vs. non-inhibited reaction rate. Data were fitted to the Morrison equation for tight-binding inhibitors. All data are presented as mean ± standard deviation from at least three experiments.
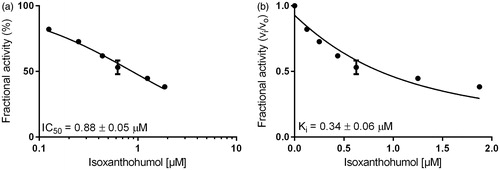
Figure 4. Dose–response (a) and inhibition curve of the AKR1B10-catalysed farnesal reduction by isoxanthohumol (b). Enzymatic activity is expressed as the ratio of inhibited vs. non-inhibited reaction rate. Data were fitted to the Morrison equation for tight-binding inhibitors. All data are presented as mean ± standard deviation from at least three experiments.
Table 3. IC50 and Ki values of the AKR1B10-catalysed farnesal reduction in the presence of the inhibitors XN, IX and 8-PN.
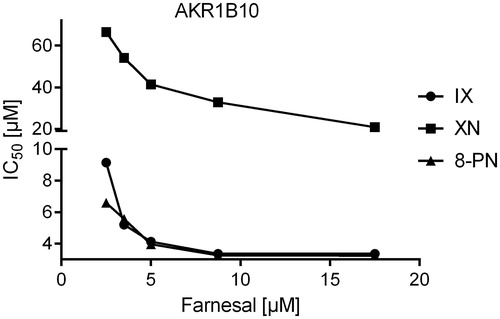
Since all three substances turned out to be tight-binding inhibitors of AKR1B1 and AKR1B10 (ratio [Inhibitor]:[Enzyme] ranging from 0.9 to 8.2), IC50-values were determined at five different substrate concentrations, including KM to analyse the mode of inhibition. All inhibitors exhibited an uncompetitive mode of inhibition for both, AKR1B1 and AKR1B10, in the presence of their physiological substrates ( and ).
Figure 5. IC50-values of the AKR1B1-catalysed glucose reduction as a function of substrate concentration in the presence of the inhibitors xanthohumol (squares), isoxanthohumol (circles) and 8-prenylnaringenin (triangles).
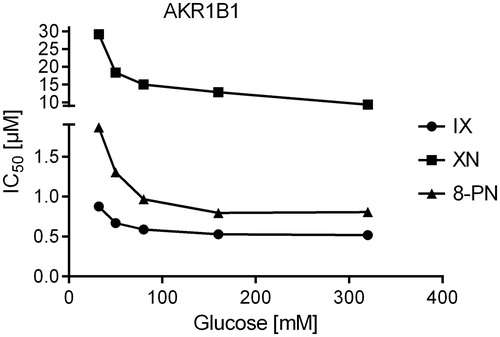
Figure 6. IC50-values of the AKR1B10-catalysed farnesal reduction as a function of substrate concentration in the presence of the inhibitors xanthohumol (squares), isoxanthohumol (circles) and 8-prenylnaringenin (triangles).
A closer examination of the binding mechanism of the three inhibitors was carried out by investigating their mode with respect to the co-factor binding site for NADPH. Interestingly, all the three substances behave as uncompetitive inhibitors of AKR1B10, while only IX displays the typical pattern of an uncompetitive inhibitor on AKR1B1, whereas XN and 8-PN inhibit in a non-competitive fashion (see .
Figure 7. IC50-values of the AKR1B1-(a) and AKR1B10-(b) catalysed glyceraldehyde reduction as a function of co-substrate (NADPH) concentration in the presence of the inhibitors isoxanthohumol (squares), xanthohumol (circles) and 8-prenylnaringenin (triangles).

4. Discussion
Aldose reductase (AKR1B1) and AKR1B10 are involved in numerous pathologies and have therefore been proposed as suitable targets for drug development. Under hyperglycemic conditions, AKR1B1 promotes osmotic imbalance and modifies physiological ratios of the redox couples of both NAD+ and NADP+, with decreasing antioxidant defense abilities. Finally, it induces a potential increase in protein glycation phenomena due to an increase in AGE levels. This has led to the identification of AKR1B1 as a primary target to prevent the onset of secondary diabetic effects. AKR1B1’s homologue AKR1B10 is overexpressed in multiple cancer types, including malignancies of breast, prostate and lungs, and is deemed suitable as a target in cancer treatment. Recent studies have shown that plant/hops specific XN, IX and 8-PN act as anti-diabetic, anti-carcinogenic and antioxidative agents. Hitherto, it has not been explored how this mode of action relates to the role of AKRs in the pathogenesis of the complications described above.
Our results show that XN, IX and 8-PN are potent inhibitors of the AKR1B’s subfamily members B1 and B10 with IC50 and Ki values ranging at low µM or nM concentrations, using d,l-glyceraldehyde as a generic substrate (see ). In the following, glucose (AKR1B1) and farnesal (AKR1B10) were used as corresponding physiological substrates (see ). After all, IX turned out to be the most potent inhibitor on both AKR1B1 and AKR1B10, with all substrates tested (Ki = 0.17 μM and 0.52 μM with AKR1B1 and AKR1B10 in presence of GA as substrate and Ki = 0.34 μM and 2.25 μM with AKR1B1 and AKR1B10 in presence of glucose and farnesal, respectively).
Interestingly, all three inhibitors exhibit a 4′-OH group and a 2-benzyl substituent in their structure (see . In a previous work, Rastelli et al.Citation67 have shown that a 4′-OH group is crucial for the inhibition of AKR1B1 by 7-hydroxy-2–(4′-hydroxybenzil)-4H-1-benzopyran-4-one, a modified form of the potent ARI quercetin, and 2′,4,4′-trihydroxychalcone, and that it specifically binds to Thr113. The same work further showed that the 2-benzyl substituent, owing to its hydrophobic aromatic nature and conformation, optimally fits an additional hydrophobic pocket of the enzyme, lined by Trp111 and Leu300. In particular, the binding of this substituent to this additional pocket provides a selective kind of inhibition for AKR1B1 with respect to the closely related enzyme AKR1A1Citation67.
There is also an evidence of prenylated flavonoids, that possess additional hydrophobic and anionic characteristic moieties (prenyl groups) at the C-8 position of their flavonoid skeletons, playing important roles in aldose reductase inhibitionCitation68,Citation69. In this sense, a comparison can be made between 8-PN (AKR1B1 IC50 = 0.81 μM obtained in this study) and its isomer 6-prenylnaringenin (6-PN), which is also a phytoestrogenic prenylflavanone occurring in hops (AKR1B1 IC50 = 6.2 μMCitation70). In fact, 8-PN exhibits an inhibitory potency that is 7.6 times greater as compared to that of 6-PN, applying glyceraldehyde as substrate. This might as well stress the importance of prenyl moieties at the C-8 position in terms of AKR1B1 inhibition. Although no IC50 data for 6-PN in presence of AKR1B10 were available at the time of this study, the low IC50 value obtained for 8-PN, might as well indicate a similar effect.
We further demonstrated that the three substances have no inhibitory effect on the closely related AKR member AKR1A1. This is an important result since many candidate inhibitors failed in clinical trials due to their undesired property of inhibiting both enzymes, AKR1B1 and AKR1A1Citation25,Citation44. Nevertheless, none of the inhibitors characterised in this study showed a selective inhibition, neither for AKR1B1 nor for AKR1B10. However, our studies may provide a basis for further modification of the molecules’ scaffolds, in order to design novel selective inhibitors for the respective enzymes. In fact, plenty of current studies aimed to find selective inhibitors for AKR1B1 over AKR1B10, and vice versa, in order to exclusively treat complications of diabetes or cancer. Yet, only a few selective inhibitors have been identified so far, including Androst-3β,5α,6β,19-tetrol (IC50 = 0.86 µM) or oleanolic acid (IC50 = 0.09 µM)Citation71–75. So far, numerous tested phytogenic inhibitors have shown only moderate degrees of selectivityCitation71,Citation76.
Our results show that IX (IC50 = 0.57 μM for AKR1B1 and IC50 = 1.09 μM for AKR1B10) and 8-PN (IC50 = 0.81 μM for AKR1B1 and IC50 = 0.99 μM for AKR1B10) inhibit both enzymes with an efficacy 6 to 15 times greater than XN when applying GA as substrate ().
In presence of the physiological substrates, glucose and farnesal, the efficacy of IX (IC50 = 0.88 μM for AKR1B1 and IC50 = 0.63 μM for AKR1B10) and 8-PN (IC50 = 1.87 μM for AKR1B1 and IC50 = 3.96 μM for AKR1B10) is 10–30 times greater than that of XN ( and ).
IX and 8-PN are strongly related prenylated flavonoids, whereas XN is a prenylated chalcone that exhibits an open ring in its core structure, which might serve as a possible explanation for its lower inhibitory potency. Our enzyme inhibition experiments showed that all compounds under investigation exhibit an uncompetitive mode of inhibition on both, AKR1B1 and AKR1B10. Uncompetitive inhibitors exclusively bind to the enzyme-substrate complex (ES), thus circumventing competition with the substrate for the active site. For AKR1B1, this constitutes a promising mechanism of inhibition, since the affinity of uncompetitive inhibitors is greatest at saturating concentrations of the substrate, as observed under hyperglycemic conditions (>7 mM, diabetic range)Citation74,Citation77. Theoretically, under the aforementioned pathological conditions, a drastic inhibition of AKR1B1 would occur even at low inhibitor concentrations.
In order to closely examine the binding mechanism of the substances, further experiments were performed to investigate whether XN, IX and 8-PN compete with NADPH at the co-factor binding site. However, our work revealed no competition between the three tested substances and NADPH, neither for AKR1B1, nor for AKR1B10. All substances tested exhibited an uncompetitive mode of inhibition for AKR1B10 with respect to NADPH, while only IX showed an uncompetitive inhibition pattern for AKR1B1, with respect to the co-factor binding site. XN and 8-PN exhibited a non-competitive mode of inhibition on AKR1B1.
Interestingly, the abovementioned substances show an uncompetitive mode of action with respect to both, the substrate and the co-factor binding site. Hypothetically speaking, this would mean that the inhibitor does not bind to the enzyme–NADPH binary complex as expected, but, instead, to the subsequent and transient enzyme conformation after the product release but before the release of NADP+. This mechanism was previously hypothesised by Harris and Kozarich, 1997Citation78 and Copeland 2005Citation79 for the binding of epristeride to the NADPH-dependent steroid-5α reductase (M. mulatta).
Hence, XN, 8-PN, IX for AKR1B10, and IX for AKR1B1 might act as uncompetitive inhibitors that bind to an enzyme species, following the formation of the initial ES complex. More challenging to explain is the behavior of 8-PN and XN, which both have been showing a non-competitive mode of inhibition with respect to AKR1B1 at the NADPH binding site.
Since the models obtained from molecular docking experiments (data not shown) did not clarify the binding mechanism of the three compounds, further investigations by X-ray diffraction are required in future studies.
Overall, we could show that the hop compounds tested have a selective inhibitory effect on the AKR1B subfamily, with respect to AKR1A1. All substances exhibit an uncompetitive mode of inhibition on both, AKR1B1 and AKR1B10. This fact might be promising when it comes to designing novel drugs and molecular therapies with high efficacy even at low concentrations. The results of this study may also provide a basis for further quantitative structure–activity relationship models and favorable scaffolds for new selective inhibitors of AKR1B1 or AKR1B10.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Zanoli P, Zavatti M. Pharmacognostic and pharmacological profile of Humulus lupulus L. J Ethnopharmacol 2008;116:383–96.
- Van Cleemput M, Heyerick A, Libert C, et al. Hop bitter acids efficiently block inflammation independent of GRalpha, PPARalpha, or PPARgamma. Mol Nutr Food Res 2009;53:1143–55.
- Stevens JF, Page JE. Xanthohumol and related prenylflavonoids from hops and beer: to your good health! Phytochemistry 2004;65:1317–30.
- Anioł M, Świderska A, Stompor M, et al. Antiproliferative activity and synthesis of 8-prenylnaringenin derivatives by demethylation of 7-O- and 4′-O-substituted isoxanthohumols. Med Chem Res 2012;21:4230–8.
- Adegbola P, Aderibigbe I, Hammed W, et al. Antioxidant and anti-inflammatory medicinal plants have potential role in the treatment of cardiovascular disease: a review. Am J Cardiovasc Dis 2017;7:19–32.
- Samarghandian S, Farkhondeh T, Samini F. Honey and health: a review of recent clinical research. Pharmacognosy Res 2017;9:121–7.
- Zakaryan H, Arabyan E, Oo A, et al. Flavonoids: promising natural compounds against viral infections. Arch Virol 2017;162:2539–51.
- Liu M, Hansen P, Wang G, et al. Pharmacological profile of xanthohumol, a prenylated flavonoid from hops (Humulus lupulus). Molecules 2015;20:754–79.
- Stevens JF, Taylor AW, Nickerson GB, et al. Prenylflavonoid variation in Humulus lupulus: distribution and taxonomic significance of xanthogalenol and 4’-O-methylxanthohumol. Phytochemistry 2000;53:759–75.
- Nikolic D, Li Y, Chadwick LR, et al. Metabolism of 8-prenylnaringenin, a potent phytoestrogen from hops (Humulus lupulus l.), by human liver microsomes. Drug Metab Dispos 2004;32:272–9.
- Yilmazer M, Stevens JF, Buhler DR. In vitro glucuronidation of xanthohumol, a flavonoid in hop and beer, by rat and human liver microsomes. FEBS Lett 2001;491:252–6.
- Yilmazer M, Stevens JF, Deinzer ML, et al. In vitro biotransformation of xanthohumol, a flavonoid from hops (Humulus lupulus), by rat liver microsomes. Drug Metab Dispos Biol Fate Chem 2001;29:223–31.
- Nookandeh A, Frank N, Steiner F, et al. Xanthohumol metabolites in faeces of rats. Phytochemistry 2004;65:561–70.
- Martinez SE, Davies NM. Enantiospecific pharmacokinetics of isoxanthohumol and its metabolite 8-prenylnaringenin in the rat. Mol Nutr Food Res 2015;59:1674–89.
- Legette L, Karnpracha C, Reed RL, et al. Human pharmacokinetics of xanthohumol, an antihyperglycemic flavonoid from hops. Mol Nutr Food Res 2014;58:248–55.
- Guo J, Nikolic D, Chadwick LR, et al. Identification of human hepatic cytochrome P450 enzymes involved in the metabolism of 8-Prenylnaringenin and isoxanthohumol from hopf (Humulus lupulus l.). Drug Metab Dispos 2006;34:1152–9.
- Costa R, Rodrigues I, Guardão L, et al. Modulation of VEGF signaling in a mouse model of diabetes by xanthohumol and 8-prenylnaringenin: unveiling the angiogenic paradox and metabolism interplay. Mol Nutr Food Res 2017;61:1600488.
- Costa R, Rodrigues I, Guardão L, et al. Xanthohumol and 8-prenylnaringenin ameliorate diabetic-related metabolic dysfunctions in mice. J Nutr Biochem 2017;45:39–47.
- Colgate EC, Miranda CL, Stevens JF, et al. Xanthohumol, a prenylflavonoid derived from hops induces apoptosis and inhibits NF-kappaB activation in prostate epithelial cells. Cancer Lett 2007;246:201–9.
- Negrão R, Duarte D, Costa R, et al. Isoxanthohumol modulates angiogenesis and inflammation via vascular endothelial growth factor receptor, tumor necrosis factor alpha and nuclear factor kappa B pathways: isoxanthohumol modulates angiogenesis and inflammation. BioFactors 2013;39:608–22.
- Wang Q, Ding Z, Liu J, et al. Xanthohumol, a novel anti-HIV-1 agent purified from Hops. Antiviral Res 2004;64:189–94.
- Cos P, Maes L, Vlietinck A, et al. Plant-derived leading compounds for chemotherapy of human immunodefiency virus (HIV) infection – an update (1998–2007). Planta Med 2008;74:1323–37.
- Ramana KV, Srivastava SK. Aldose reductase: a novel therapeutic target for inflammatory pathologies. Int J Biochem Cell Biol 2010;42:17–20.
- Jin J, Liao W, Yao W, et al. Aldo-keto Reductase Family 1 member B 10 mediates liver cancer cell proliferation through sphingosine-1-phosphate. Sci Rep 2016;6:22746.
- Alexiou P, Pegklidou K, Chatzopoulou M, et al. Aldose reductase enzyme and its implication to major health problems of the 21(st) century. Curr Med Chem 2009;16:734–52.
- Zhang W, Li H, Yang Y, et al. Knockdown or inhibition of aldo-keto reductase 1B10 inhibits pancreatic carcinoma growth via modulating Kras-E-cadherin pathway. Cancer Lett 2014;355:273–80.
- Balestri F, Cappiello M, Moschini R, et al. Modulation of aldose reductase activity by aldose hemiacetals. Biochim Biophys Acta BBA - Gen Subj 2015;1850:2329–39.
- Chung YT, Matkowskyj KA, Li H, et al. Overexpression and oncogenic function of aldo-keto reductase family 1B10 (AKR1B10) in pancreatic carcinoma. Mod Pathol 2012;25:758–66.
- Kurahashi T, Kwon M, Homma T, et al. Reductive detoxification of acrolein as a potential role for aldehyde reductase (AKR1A) in mammals. Biochem Biophys Res Commun 2014;452:136–41.
- Ellis EM. Reactive carbonyls and oxidative stress: potential for therapeutic intervention. Pharmacol Ther 2007;115:13–24.
- Rotondo R, Moschini R, Renzone G, et al. Human carbonyl reductase 1 as efficient catalyst for the reduction of glutathionylated aldehydes derived from lipid peroxidation. Free Radic Biol Med 2016;99:323–32.
- Bains OS, Takahashi RH, Pfeifer TA, et al. Two allelic variants of aldo-keto reductase 1A1 exhibit reduced in vitro metabolism of daunorubicin. Drug Metab Dispos 2008;36:904–10.
- O’connor T, Ireland LS, Harrison DJ, et al. Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members. Biochem J 1999;343(Pt 2):487–504.
- Srivastava SK, Ramana KV, Bhatnagar A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr Rev 2005;26:380–92.
- Yabe-Nishimura C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol Rev 1998;50:21–33.
- El-Kabbani O, Ruiz F, Darmanin C, et al. Aldose reductase structures: implications for mechanism and inhibition. Cell Mol Life Sci CMLS 2004;61:750–62.
- González RG, Barnett P, Aguayo J, et al. Direct measurement of polyol pathway activity in the ocular lens. Diabetes 1984;33:196–9.
- Tang WH, Martin KA, Hwa J. Aldose reductase, oxidative stress, and diabetic mellitus. Front Pharmacol 2012;3:87.
- Hwang JJ, Jiang L, Hamza M, et al. The human brain produces fructose from glucose. JCI Insight 2017;2:e90508.
- Brings S, Fleming T, Freichel M, et al. Dicarbonyls and advanced glycation end-products in the development of diabetic complications and targets for intervention. Int J Mol Sci 2017;18:984.
- Del Corso A, Cappiello M, Mura U. From a dull enzyme to something else: facts and perspectives regarding aldose reductase. Curr Med Chem 2008;15:1452–61.
- Kanchan D, Kale S, Somani G, et al. Thymol, a monoterpene, inhibits aldose reductase and high-glucose-induced cataract on isolated goat lens. J Pharm Bioallied Sci 2016;8:277.
- El Gamal H, Eid AH, Munusamy S. Renoprotective effects of aldose reductase inhibitor epalrestat against high glucose-induced cellular injury. Bio Med Res Int 2017;2017:1–11.
- Ramana KV. Aldose reductase: new insights for an old enzyme. Biomol Concepts 2011;2:103–114.
- Barski OA, Tipparaju SM, Bhatnagar A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev 2008;40:553–624.
- Suryanarayana P, Kumar PA, Saraswat M, et al. Inhibition of aldose reductase by tannoid principles of Emblica officinalis: implications for the prevention of sugar cataract. Mol Vis 2004;10:148–54.
- Kato A, Kobayashi K, Narukawa K, et al. 6,7-Dihydroxy-4-phenylcoumarin as inhibitor of aldose reductase 2. Bioorg Med Chem Lett 2010;20:5630–3.
- Ibrar A, Tehseen Y, Khan I, et al. Coumarin-thiazole and -oxadiazole derivatives: synthesis, bioactivity and docking studies for aldose/aldehyde reductase inhibitors. Bioorganic Chem 2016;68:177–86.
- Carbone V, Zhao H-T, Chung R, et al. Correlation of binding constants and molecular modelling of inhibitors in the active sites of aldose reductase and aldehyde reductase. Bioorg Med Chem 2009;17:1244–50.
- Gallego O, Ruiz FX, Ardevol A, et al. Structural basis for the high all-trans-retinaldehyde reductase activity of the tumor marker AKR1B10. Proc Natl Acad Sci 2007;104:20764–9.
- Matsunaga T, Shintani S, Hara A. Multiplicity of mammalian reductases for xenobiotic carbonyl compounds. Drug Metab Pharmacokinet 2006;21:1–18.
- Jin Y, Penning TM. Aldo-keto reductases and bioactivation/detoxication. Annu Rev Pharmacol Toxicol 2007;47:263–92.
- Endo S, Matsunaga T, Mamiya H, et al. Kinetic studies of AKR1B10, human aldose reductase-like protein: endogenous substrates and inhibition by steroids. Arch Biochem Biophys 2009;487:1–9.
- Yang ZN, Davis GJ, Hurley TD, et al. Catalytic efficiency of human alcohol dehydrogenases for retinol oxidation and retinal reduction. Alcohol Clin Exp Res 1994;18:587–91.
- Hyndman DJ, Flynn TG. Sequence and expression levels in human tissues of a new member of the aldo-keto reductase family. Biochim Biophys Acta 1998;1399:198–202.
- Cao D, Fan ST, Chung SS. Identification and characterization of a novel human aldose reductase-like gene. J Biol Chem 1998;273:11429–35.
- Fukumoto S, Yamauchi N, Moriguchi H, et al. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers’ non-small cell lung carcinomas. Clin Cancer Res 2005;11:1776–85.
- Jung Y-J, Lee EH, Lee CG, et al. AKR1B10-inhibitory Selaginella tamariscina extract and amentoflavone decrease the growth of A549 human lung cancer cells in vitro and in vivo. J Ethnopharmacol 2017;202:78–84.
- Li H, Yang AL, Chung YT, et al. Sulindac inhibits pancreatic carcinogenesis in LSL-KrasG12D-LSL-Trp53R172H-Pdx-1-Cre mice via suppressing aldo-keto reductase family 1B10 (AKR1B10). Carcinogenesis 2013;34:2090–8.
- Matkowskyj KA, Bai H, Liao J, et al. Aldoketoreductase family 1B10 (AKR1B10) as a biomarker to distinguish hepatocellular carcinoma from benign liver lesions. Hum Pathol 2014;45:834–43.
- Riou P, Kjaer S, Garg R, et al. 14-3-3 proteins interact with a hybrid prenyl-phosphorylation motif to inhibit G proteins. Cell 2013;153:640–53.
- Gao J, Liao J, Yang G-Y. CAAX-box protein, prenylation process and carcinogenesis. Am J Transl Res 2009;1:312–25.
- Skarydova L, Tomanova R, Havlikova L, et al. Deeper insight into the reducing biotransformation of bupropion in the human liver. Drug Metab Pharmacokinet 2014;29:177–84.
- Martin H-J, Breyer-Pfaff U, Wsol V, et al. Purification and characterization of AKR1B10 from human liver: role in carbonyl reduction of xenobiotics. Drug Metab Dispos 2005;34:464–70.
- Copeland RA. Enzymes: a practical introduction to structure, mechanism, and data analysis. 2nd ed. New York, NY: Wiley; 2000.
- Bisswanger H. Enzyme kinetics. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2008.
- Rastelli G, Antolini L, Benvenuti S, et al. Structural bases for the inhibition of aldose reductase by phenolic compounds. Bioorg Med Chem 2000;8:1151–8.
- Jung HA, Moon HE, Oh SH, et al. Kinetics and molecular docking studies of kaempferol and its prenylated derivatives as aldose reductase inhibitors. Chem Biol Interact 2012;197:110–8.
- Jung HA, Yoon NY, Kang SS, et al. Inhibitory activities of prenylated flavonoids from Sophora flavescens against aldose reductase and generation of advanced glycation endproducts. J Pharm Pharmacol 2008;60:1227–36.
- Shim SH, Kim Y, Lee JY, et al. Aldose reductase inhibitory activity of the compounds from the seed of Psoralea corylifolia. J Korean Soc Appl Biol Chem 2009;52:568–72.
- Huang L, He R, Luo W, et al. Aldo-keto reductase family 1 member B10 inhibitors: potential drugs for cancer treatment. Recent Pat Anticancer Drug Discov 2016;11:184–96.
- Cousido-Siah A, Ruiz FX, Fanfrlík J, et al. IDD388 polyhalogenated derivatives as probes for an improved structure-based selectivity of AKR1B10 inhibitors. ACS Chem Biol 2016;11:2693–705.
- Kabir A, Endo S, Toyooka N, et al. Evaluation of compound selectivity of aldo-keto reductases using differential scanning fluorimetry. J Biochem (Tokyo) 2016;161:215–222.
- Chen W, Chen X, Zhou S, et al. Design and synthesis of polyhydroxy steroids as selective inhibitors against AKR1B10 and molecular docking. Steroids 2016;110:1–8.
- Takemura M, Endo S, Matsunaga T, et al. Selective inhibition of the tumor marker aldo-keto reductase family member 1B10 by oleanolic acid. J Nat Prod 2011;74:1201–6.
- Zemanova L, Hofman J, Novotna E, et al. Flavones inhibit the activity of AKR1B10, a promising therapeutic target for cancer treatment. J Nat Prod 2015;78:2666–74.
- Mohamadi-Nejad A, Moosavi-Movahedi AA, Hakimelahi GH, et al. Thermodynamic analysis of human serum albumin interactions with glucose: insights into the diabetic range of glucose concentration. Int J Biochem Cell Biol 2002;34:1115–24.
- Harris GS, Kozarich JW. Steroid 5alpha-reductase inhibitors in androgen-dependent disorders. Curr Opin Chem Biol 1997;1:254–9.
- Copeland RA. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem Anal 2005;46:1–265.