
Abstract
An efficient synthesis of substituted pyrido[2,3-d]pyrimidines was carried out and evaluated for in vitro anticancer activity against five cancer cell lines, namely hepatic cancer (HepG-2), prostate cancer (PC-3), colon cancer (HCT-116), breast cancer (MCF-7), and lung cancer (A-549) cell lines. Regarding HepG-2, PC-3, HCT-116 cancer cell lines, 7-(4-chlorophenyl)-2-(3-methyl-5-oxo-2,3-dihydro-1H-pyrazol-1-yl)-5-(p-tolyl)- pyrido[2,3-d]pyrimidin-4(3H)-one (5a) exhibited strong, more potent anticancer (IC50: 0.3, 6.6 and 7 µM) relative to the standard doxorubicin (IC50: 0.6, 6.8 and 12.8 µM), respectively. Kinase inhibitory assessment of 5a showed promising inhibitory activity against three kinases namely PDGFR β, EGFR, and CDK4/cyclin D1 at two concentrations 50 and 100 µM in single measurements. Further, a molecular docking study for compound 5a was performed to verify the binding mode towards the EGFR and CDK4/cyclin D1 kinases.
Graphical Abstract
Introduction
Cancer is one of the leading causes of death in the world, characterized by the loss of control of cell proliferation, leading almost invariably to death, in untreated patientsCitation1,Citation2. Chemotherapy, alone or in combination with surgery, is commonly the most efficient anti-cancer remedy. However, the use of available chemotherapeutics is limited mainly due to drug resistance and toxicitiesCitation3. Developing resistance to chemotherapy belongs to many reasons like poor uptake of the drug, alternative metabolic paths, and increased production of the target protein, mutations that block the drug binding to its target or efflux systems that expel drugs from the cellCitation4–8. So, combination of chemotherapies with different targets increases efficiency, antagonizes the resistance, and decreases toxicity as well. Traditional anticancer drugs work by disrupting the function of DNA. Some of these drugs may affect DNA directly or inhibit the enzymes controlling DNA synthesis. These drugs are mostly nonselective and having cytotoxicity to both cancer and normal cellsCitation9,Citation10. The advances in molecular biology and genetics improve identification of molecular targets that are unique to cancer cells or overexpressed on them. The design of agents affecting these targets promises the development of more selective anticancer drugs with less toxic side effectsCitation11. Pyrido[2,3-d]pyrimidines were reported to display antitumor properties, which may be attributed to inhibition of different enzymes that involved in carcinogenesis cases. The prominent examples were pyrido[2,3-d]pyrimidines A–E that exhibited a potent inhibitory activity against various kinases, e.g. TKs, PI3K, and CDK4/6Citation12,Citation13 (). Based on the structural features of the previous pyrido[2,3-d]pyrimidines, we aimed to prepare a new group of pyrido[2,3-d]pyrimidinone congeners, which were screened for their inhibitory activity against TKs, CDK4/6, and PI3K enzymes. Simultaneously, they were tested for their anticancer activity against cancer cells expressing the previous enzymes. Further, molecular modeling study was performed to explore the most appropriate binding modes of the most potent target compounds that matched ligands binding modes. The applied modeling program was Molecular Operating Environment (MOE®) 2008.10.
Figure 1. Reported and proposed pyrido[2,3-d]pyrimidine conjugates with anticancer and tyrosine kinase inhibitory activity.
![Figure 1. Reported and proposed pyrido[2,3-d]pyrimidine conjugates with anticancer and tyrosine kinase inhibitory activity.](/cms/asset/bd5277e9-3cfa-4a20-bf4e-990fe3dd7ecf/ienz_a_1437729_f0001_c.jpg)
Materials and methods
Chemistry
Melting points by using electro thermal apparatus on open capillary tubes were recorded. IR spectra (KBr) were performed on a Shimadzu 435 IR Spectrophotometer (Shimadzu Bruker, Tokyo, Japan) (ν cm−1). 1H and 13 C NMR NMR spectra were recorded on Varian Mercury VX-300 NMR 300 MHz spectrophotometer (Stoughton, USA) or on Agilent Technologies 400 MHz NMR spectrophotometer (Santa Clara, CA, USA) using DMSO-d6 as a solvent 300 MHz oron Agilent Technologies 400 MHz NMR spectrophotometer and the chemical shifts were estimated in ppm, relative to TMS as an internal standard. Physical data (C, H, and N) agreed with the proposed structures and obtained by using a Vario Elemental analyzer (Yamashitacho, Yokohama, Japan) (within ±0.4% of the theoretical values). Mass spectra were recorded on a DI-50 unit of Shimadzu GC/MS-QP 2010 plus Spectrometer (Livingston, West Lothian, UK) or on single quadrupole Mass Spectrometer ISQ LT (Thermo Scientific) (Austin, TX, USA).
General procedure for the synthesis of 2,3-dihydro-2-thioxo-5,7-diarylpyrido[2,3-d]pyrimidin-4(1H)-one (3a–c, e–g)
2-Thioxopyrimidine 1 (1.43 g, 0.01 mol) and a series of α,β-unsaturated ketones 2a–g (0.01 mol) in dry DMF (20 ml) was refluxed for 10–15 h and the reaction progress was monitored by TLC. The solid mass created on cooling was filtered and crystallized from (DMF) to give the title products.
5-(2,4-Dichlorophenyl)-2,3-dihydro-2-thioxo-7-p-tolylpyrido[2,3-d]pyrimidin-4(1H)-one (3a)
Yield: 67%; mp: 309–311 °C; IR (KBr, cm−1) ν: 3402 (2NH), 1697 (C=O); 1HNMR (400 MHz, DMSO-d6): δ, 12.13 & 13.02 (2s, 2H, 2NH), 8.12 (d, 2H, J = 8.4 Hz), 7.92 (s, 1H, C-6 pyridine), 7.41 (s, 1H), 7.38 (d, 2H, J = 8.4 Hz), 7.33 (d, 2H, J = 8.4 Hz), 2.35 (s, 3H, CH3); MS: [m/z (%), 417 (0.84, M++4), 415 (1.96, M++2), 413 (2.28, M+), 119 (100)].
7-(4-Bromophenyl)-2,3-dihydro-2-thioxo-5-p-tolylpyrido[2,3-d]pyrimidin-4(1H)-one (3b)
Yield: 88%; mp: 310–312 °C; IR (KBr, cm−1) ν: 3410 (2NH), 1701 (C=O); 1HNMR (400 MHz, DMSO-d6): δ12.13, 13.02 (2s, 2H, 2NH), 8.17 (d, 2H, J = 8 Hz), 7.92 (s, 1H, C-6 pyridine), 7.72 (d, 2H, J = 8.8 Hz), 7.41 (d, 2H, J = 8.8 Hz), 6.95 (d, 2H, J = 8.8 Hz), 2.06 (s, 3H, CH3); MS: [m/z (%), 425 (2.57, M++2), 423 (2.04, M+), 180 (100)].
7-(4-Chlorophenyl)-2,3-dihydro-2-thioxo-5-p-tolylpyrido[2,3-d]pyrimidin-4(1H)-one (3c)
Yield: 75%; mp: 302–304 °C; IR (KBr, cm−1) ν: 3394 (2NH), 1705 (C=O); 1HNMR (300 MHz, DMSO-d6): δ 12.25, 13.00 (2s, 2H, 2NH), 8.25 (d, 2H, J = 8.7 Hz), 7.94 (s, 1H, C-6 pyridine), 7.59 (d, 2H, J = 8.7 Hz), 7.34 (d, 2H, J = 9 Hz), 7.22 (d, 2H, J = 8.1 Hz), 2.37 (s, 3H, CH3); MS: [m/z (%), 381 (7.05, M+ +2), 379 (14.24, M+), 40 (100)].
2,3-Dihydro-5-(4-methoxyphenyl)-2-thioxo-7-p-tolylpyrido[2,3-d]pyrimidin-4(1H)-one (3e)
Yield: 75%; mp: 361–363 °C; IR (KBr, cm−1) ν: 3421 (2NH), 1678 (C=O); 1HNMR (400 MHz, DMSO-d6): δ 12.28, 12.95 (2s, 2H, 2NH), 8.11 (d, 2H, J = 8 Hz), 7.55 (s, 1H, C-6 pyridine), 7.39 (d, 2H, J = 8.4 Hz), 7.38 (d, 2H, J = 8.8 Hz), 6.95 (d, 2H, J = 8.8 Hz), 3.82 (s, 3H, OCH3), 2.36 (s, 3H, CH3); 13CNMR (100 MHz, DMSO-d6): δ 175.71, 159.97, 153.79, 153.21, 141.19, 134.27, 130.88, 130.84, 129.93, 127.94, 119.23, 113.31, 108.02, 55.64, 21.42; MS: [m/z (%), 375 (14.85, M+), 91 (100)].
7-(4-Chlorophenyl)-2,3-dihydro-5-(3,4,5-trimethoxyphenyl)-2-thioxopyrido[2,3-d]pyrimidin-4(1H)-one (3f)
Yield: 74%; mp: 306–308 °C; IR (KBr, cm−1) ν: 3402 (2NH), 1708 (C=O); 1HNMR (400 MHz, DMSO-d6): δ 12.31, 13.22 (2s, 2H, 2NH), 8.26 (d, 2H, J = 8 Hz), 7.93 (s, 1H, C-6 pyridine), 7.60 (d, 2H, J = 8.8 Hz), 6.76 (s, 2H), 3.70, 3.76 (2s, 9H, 3OCH3); MS: [m/z (%), 457 (0.1, M+ +2), 455 (1.6, M+), 95 (100)].
2,3-Dihydro-5-(3,4,5-trimethoxyphenyl)-2-thioxo-7-p-tolylpyrido[2,3-d]pyrimidin-4-(1H)-one (3g)
Yield: 55%; mp: 318–320 °C; IR (KBr, cm−1) ν: 3421 (2NH), 1701 (C=O); 1HNMR (300 MHz, DMSO-d6): δ12.13, 12.98 (2s, 2H, 2NH), 8.15 (d, 2H, J = 8.4 Hz), 7.94 (s, 1H, C-6 pyridine), 7.34 (d, 2H, J = 8.1 Hz), 6.76 (s, 2H), 3.77, 3.72 (2s, 9H, 3OCH3), 2.38 (s, 3H, CH3); MS: [m/z (%), 435 (12.70, M+), 77.07 (100)].
General procedure for the synthesis of 2-hydrazinyl -5,7-diarylpyrido[2,3-d]pyrimidin-4(3H)-one (4a–f)
Derivatives 3a,c–g (0.004 mol) and hydrazine reagent 99% (3 ml, 0.006 mol) was refluxed in absolute ethyl alcohol (20 ml) for 10–15 h. On cooling, the residue created was filtered and purified from (DMF).
5-(2,4-Dichlorophenyl)-2-hydrazinyl-7-p-tolylpyrido[2,3-d]pyrimidin-4(3H)-one (4a)
Yield: 27%; mp: 167–169 °C; IR (KBr, cm−1) ν: 3410, 3190 (2NH, NH2), 1678 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 10.13 (1s, 1H, NH), 7.98 (s, 1H, C-6 pyridine), 7.55 (d, 2H, J = 8 Hz), 7.42 (d, 1H, J = 6.8 Hz), 7.38 (d, 2H, J = 10 Hz), 7.34 (d, 2H, J = 8 Hz), 6.17 (s, 1H, NH), 4.32 (s, 2H, NH2), 2.36 (s, 3H, CH3); MS: [m/z (%), 415 (1.69, M++4), 413 (2.00, M++2), 411 (4.86, M+)].
7-(4-Chlorophenyl)-2-hydrazinyl-5-p-tolylpyrido[2,3-d]pyrimidin-4(3H)-one (4b)
Yield: 54%; mp: 233–234 °C; IR (KBr, cm−1) ν: 3421, 3190 (2NH, NH2), 1716 (C=O); 1HNMR (400 MHz, DMSO-d6): δ8.29, 8.28 (2s, 3H, NH, NH2), 8.19 (d, 2H, J = 8.8 Hz) , 8.08 (s, 1H, NH), 7.93 (s, 1H, C-6 pyridine), 7.53 (d, 2H, J = 8 Hz), 7.25 (d, 2H, J = 8 Hz), 7.17 (d, 2H, J = 8 Hz), 2.30 (s, 3H, CH3); MS: [m/z (%), 379 (2.32, M+ +2), 367 (100)].
2-Hydrazinyl-5-(4-methoxyphenyl)-7-p-tolylpyrido[2,3-d]pyrimidin-4(3H)-one (4d)
Yield: 73%; mp: 260–262 °C; IR (KBr, cm−1) ν: 3425, 3290 (2NH, NH2), 1728 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 10.17 (s, 1H, NH), 8.26 (s,1H, NH), 8.06 (d, 2H, J = 8 Hz), 7.30 (s, 1H, C-6 pyridine), 7.33 (d, 2H, J = 8.4 Hz), 7.29 (d, 2H, J= 8.8 Hz), 6.92 (d, 2H, J = 8.8 Hz), 4.20 (s, 2H, NH2), 3.79 (s, 3H, OCH3), 2.38 (s, 3H, CH3); MS: [m/z (%), 373 (100, M+)].
7-(4-Chlorophenyl)-2-hydrazinyl-5-(3,4,5-trimethoxyphenyl)pyrido[2,3-d]pyrimidin-4-(3H)-one (4e)
Yield: 37%; mp: 279–281 °C; IR (KBr, cm−1) ν: 3421, 3111 (2NH, NH2), 1716 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 10.03 (s, 2H, NH), 8.30 (d, 2H, J = 8 Hz), 7.93 (s, 1H, C-6 pyridine), 6.79 (d, 2H, J = 8 Hz), 7.62 (d, 2H, J = 8 Hz), 6.16 (s, 2H, NH2), 3.77, 3.75 (2s, 9H, 3OCH3); MS: [m/z (%), 454 (1.33, M++1), 453 (3.52, M+), 77 (100)].
2-Hydrazinyl-5-(3,4,5-trimethoxyphenyl)-7-p-tolylpyrido[2,3-d]pyrimidin-4(3H)-one (4f)
Yield: 68%; mp: 301–303 °C; IR (KBr, cm−1) ν: 3348, 3103 (2NH, NH2), 1716 (C=O); 1HNMR (400 MHz, DMSO-d6): δ 10.12 (s, 2H, 2NH), 8.19 (d, 2H, J = 8 Hz), 7.99 (s, 1H, C-6 pyridine), 7.36 (d, 2H, J = 8 Hz), 6.76 (d, 2H, J = 8 Hz), 6.12 (s, 2H, NH2), 3.76, 3.77 (2s, 9H, 3OCH3), 2.36 (s, 3H, CH3); MS: [m/z (%), 433 (1.49, M+), 115 (100)].
General procedure for the synthesis of2–(3-methyl-5-oxo-2,3-dihydro-1H-pyrazol-1-yl)-5,7-diarylpyrido[2,3-d]pyrimidin-4(3H)-one (5a–d), 2-(3-amino-5-oxo-2,3-dihydro-1H-pyrazol-1-yl)-5,7-diarylpyrido[2,3-d]pyrimidin-4(3H)-one (6a–d) and 1-(4-oxo-5,7-diaryl-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)pyrazolidine-3,5-dione (7a–c)
2-Hydrazinyl derivatives (0.01 mol) and active methylene (ethyl acetoacetate, ethyl cyanoacetate, or diethylmalonate) (0.01 mol) was refluxed in glacial acetic acid for 4–6 h. After completion of the reaction, the precipitate created on pouring into ice-water was filtered and purified from (acetic acid) to give the title compounds 5a–d, 6a–d and 7a–c, respectively.
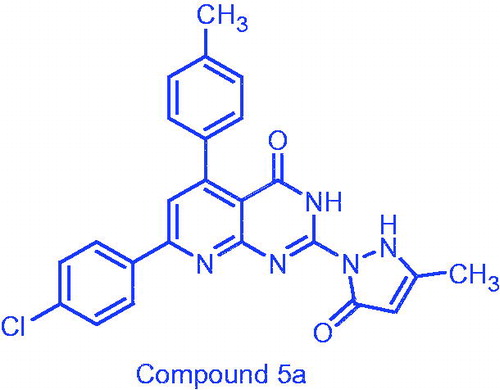
7-(4-Chlorophenyl)-2-(3-methyl-5-oxo-2,3-dihydro-1H-pyrazol-1-yl)-5-(p-tolyl)pyrido-[2,3-d]pyrimidin-4(3H)-one (5a)
Yield: 67%; mp: 385–387 °C; IR (KBr, cm−1) ν: 3422 (2NH), 1716 (2C=O); 1HNMR (400 MHz, DMSO-d6): δ 11.16, 11.66 (2s, 2H, 2NH), 8.21 (d, 2H), 8.18 (s, 1H, C-4 pyrazolone), 7.57 (d, 2H, J = 8 Hz), 7.50 (s, 1H, C-6 pyridine), 7.30 (d, 2H, J = 8 Hz), 7.19 (d, 2H, J = 8 Hz), 2.35 (s, 3H, CH3), 1.89 (s, 3H, CH3); 13CNMR (100 MHz, DMSO-d6): δ 172.44, 161.77, 157.81, 154.58, 153.98, 137.86, 136.26, 136.07,135.88, 129.63, 129.34, 129.09, 128.42, 118.39, 106.54, 21.50, 21.32; MS: [m/z (%), 446 (22, M++2), 444 (50, M+), 186 (100)].
2-(3-Methyl-5-oxo-2,3-dihydro-1H-pyrazol-1-yl)-5,7-di-p-tolylpyrido[2,3-d]pyrimidin-4-(3H)-one (5b)
Yield: 53%; mp: 342–344 °C; IR (KBr, cm−1) ν: 3429 (2NH), 1727, 1651 (2C=O); 1H NMR (400 MHz, DMSO-d6): δ 11.93, 13.59 (2s, 2H, 2NH), 8.28 (d, 2H, J = 8 Hz), 8.17 (s, 1H, C4-pyrazolone), 7.68 (s, 1H, C6-pyridine), 7.59 (d, 2H, J = 8 Hz), 7.37 (d, 2H, J = 8 Hz), 7.24 (d, 2H, J = 8 Hz), 2.35, 2.39 (2s, 6H, 2CH3), 1.88 (s, 3H, CH3); MS: [m/z (%), 423 (0.79, M+), 367 (100)].
7-(4-Chlorophenyl)-2-(3-methyl-5-oxo-2,3-dihydro-1H-pyrazol-1-yl)-5-(3,4,5-trimethoxyphenyl)pyrido[2,3-d]pyrimidin-4(3H)-one (5c)
Yield: 42%; mp: 323–325 °C; IR (KBr, cm−1) ν: 3421(2NH), 1719 (2C=O); 1HNMR (400 MHz, DMSO-d6): δ 11.97, 13.71 (2s, 2H, 2NH), 8.31 (d, 2H, J = 8 Hz), 8.20 (s, 1H, C-4 pyrazolone), 7.78 (s, 1H, C-6 pyridine), 7.62 (d, 2H, J = 8 Hz), 6.78 (s, 2H), 3.73, 3.77 (2s, 9H, 3OCH3), 1.88 (s, 3H, CH3); 13CNMR (100 MHz, DMSO-d6): δ175.81, 162.13, 158.37, 155.58, 154.61, 152.70, 137.94, 136.27, 135.92, 134.87, 129.94, 129.49, 118.54, 106.85, 105.95, 60.50, 56.49; MS: [m/z (%), 521 (0.83, M++2), 519 (3.84, M+), 376 (100)].
2-(3-Methyl-5-oxo-2,3-dihydro-1H-pyrazol-1-yl)-7-(p-tolyl)-5-(3,4,5-trimethoxy phenyl)pyrido[2,3-d]pyrimidin-4(3H)-one (5d)
Yield: 43%; mp: 357–359 °C; IR (KBr, cm−1) ν: 3435 (2NH), 1719 (2C=O)); 1H NMR (400 MHz, DMSO-d6): δ 11.94, 13.67 (2s, 2H, 2NH), 8.20 (d, 2H), 8.18 (s,1H, C4-pyrazolone), 7.36 (d, 2H, J = 8 Hz), 7.72 (s, 1H, C6-pyridine), 6.76 (d, 2H, J = 8 Hz), 3.73, 3.77 (2s, 9H, 3OCH3), 2.38 (s, 3H, CH3), 1.88 (s, 3H, CH3); MS: [m/z (%), 500 (1.59, M++1), 106 (100)].
2-(3-Amino-5-oxo-2,3-dihydro-1H-pyrazol-1-yl)-7-(4-chlorophenyl)-5-(p-tolyl)pyrido [2,3-d]pyrimidin-4(3H)-one (6a)
Yield: 65%; mp: 382–384 °C; IR (KBr, cm−1) ν: 3430 (br, 2NH, NH2), 1726, 1644 (2C=O); 1HNMR (400 MHz, DMSO-d6): δ 13.90 (s, 2H, 2NH), 8.30 (d, 2H, J = 8 Hz), 8.18 (s, 1H, C-4 pyrazolone), 7.69 (s, 1H, C-6 pyridine), 7.61 (d, 2H, J = 8 Hz), 7.37 (d, 2H, J = 8 Hz), 7.25 (d, 2H, J = 8 Hz), 3.96 (s, 2H, NH2), 2.38 (s, 3H, CH3); MS: [m/z (%), 446 (22, M++2), 444 (50, M+), 186 (100)].
2-(3-Amino-5-oxo-2,3-dihydro-1H-pyrazol-1-yl)-5,7-di-p-tolylpyrido[2,3-d]pyrimidin-4-(3H)-one (6b)
Yield: 42%; mp: 380–382 °C; IR (KBr, cm−1) ν: 3429 (br, 2NH, NH2), 1727, 1680 (2C=O); 1H NMR (400 MHz, DMSO-d6): δ 12.48 (s, 2H, 2NH), 8.16 (d,2H, J = 8 Hz), 8.13 (s, 1H, C-4 pyrazolone), 7.80 (s, 1H, C-6 pyridine), 7.36 (d, 2H, J = 8 Hz), 7.32 (d, 2H, J = 8 Hz), 7.22 (d, 2H, J = 8 Hz), 3.97 (s, 2H, NH2), 2.37, 2.38 (2s, 6H, 2CH3); MS: [m/z (%), 424 (2, M+), 175 (100)].
2-(3-Amino-5-oxo-2,3-dihydro-1H-pyrazol-1-yl)-7-(4-chlorophenyl)-5-(3,4,5-trimethoxyphenyl)pyrido[2,3-d]pyrimidin-4(3H)-one (6c)
Yield: 68%; mp: 339–341 °C; IR (KBr, cm−1) ν: 3433 (br, 2NH, NH2), 1726, 1678 (2C=O); 1HNMR (400 MHz, DMSO-d6): δ 13.68 (s, 2H, 2NH), 8.31 (d,2H), 8.20 (s, 1H, C-4 pyrazolone), 7.79 (s, 1H, C-6 pyridine), 7.60 (d, 2H), 6.78 (d, 2H), 3.96 (s, 2H, NH2), 3.79, 3.77 (2s, 9H, 3OCH3); MS: [m/z (%), 424 (2, M+), 175 (100)].
2-(3-Amino-5-oxo-2,3-dihydro-1H-pyrazol-1-yl)-7-(p-tolyl)-5-(3,4,5-trimethoxy phenyl)pyrido[2,3-d]pyrimidin-4(3H)-one (6d)
Yield: 52%; mp: 350–352 °C; IR (KBr, cm−1) ν: 3390 (br, 2NH, NH2), 1716, 1675 (2C=O); 1H NMR (400 MHz, DMSO-d6): δ 11.46, 13.61 (2s, 2H, 2NH), 8.19 (d, 2H), 8.17 (s, 1H, C-4 pyrazolone), 7.72 (s, 1H, C-6 pyridine), 7.36 (d, 2H, J = 8 Hz), 6.78 (s, 2H), 3.96 (s, 2H, NH2), 3.73, 3.77 (2s, 9H, 3OCH3), 2.38 (s, 3H, CH3); 13 C NMR (100 MHz, DMSO-d6): δ 171.74, 163.73, 159.72, 155.23, 154.45, 153.36, 152.68, 150.89, 144.52, 141.52, 141.35, 137.86, 135.06, 134.46, 130.03, 128.11, 118.25, 106.80, 60.49, 56.47, 21.44; MS: [m/z (%), 500 (M+, 0.7); 107 (100)].
1-(7-(4-Chlorophenyl)-4-oxo-5-(p-tolyl)-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl) pyrazolidine-3,5-dione (7a)
Yield: 23%; mp: 358–360 °C; IR (KBr, cm−1) ν: 3421 (2NH), 1716, 1730 (3C=O); 1HNMR (400 MHz, DMSO-d6): δ 12.39 (s, 2H, 2NH), 8.29 (d, 2H, J = 8 Hz), 7.89 (s, 1H, C-6 pyridine), 7.61 (d, 2H, J = 8 Hz), 7.33 (d, 2H, J = 8 Hz), 7.22 (d, 2H, J = 8 Hz), 2.95 (s, 2H, C-4 pyrazolidinone), 2.37 (s, 3H, CH3); MS: [m/z (%), 447 (14,M++2), 445 (2,M+), 77 (100)].
1-(7-(4-Chlorophenyl)-4-oxo-5-(3,4,5-trimethoxyphenyl)-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)pyrazolidine-3,5-dione (7b)
Yield: 66%; mp: 370–372 °C; IR (KBr, cm−1) ν: 3421 (2NH), 1732, 1660 (3C=O); 1HNMR (400 MHz, DMSO-d6): δ 13.95 (s, 2H, 2NH), 8.20 (d, 2H, J = 8 Hz), 7.70 (s, 1H, C-6 pyridine), 7.33 (d, 2H, J = 8 Hz), 6.65 (d, 2H, J = 8 Hz), 3.73, 3.77 (2s, 9H, 3OCH3), 2.95 (s, 2H, C4-pyrazolidinone); MS: [m/z (%), 521 (4, M++2), 523 (0.8, M+), 95 (100)].
1-(4-Oxo-7-(p-tolyl)-5-(3,4,5-trimethoxyphenyl)-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)pyrazolidine-3,5-dione (7c)
Yield: 34%; mp: 352–354 °C; IR (KBr, cm−1) ν: 3448 (2NH), 1732, 1690 (3C=O); 1HNMR (400 MHz, DMSO-d6): δ 12.92 (br s, 2H, 2NH), 8.31 (d, 2H, J = 8 Hz), 7.93 (s, 1H, C6-pyridine), 7.37 (d, 2H, J = 8 Hz), 6.78 (d, 2H, J = 8 Hz), 3.72, 3.76 (2s, 9H, OCH3), 2.94 (s, 2H, C-4 pyrazolidinone), 2.36 (s, 3H, CH3); 13CNMR (100 MHz, DMSO-d6): δ 170.45, 167.54, 167.16, 151.59, 147.78, 141.47, 134.15, 130.22, 128.01, 121.34, 106.85, 90.72, 60.47, 56.47; MS: [m/z (%), 501 (20, M+), 55 (100)].
General procedure for the synthesis of2-(2-(aryl-2-ylmethylene)hydrazinyl)-5,7-diarylpyrido[2,3-d]pyrimidin-4(3H)-one (8a–f)
2-Hydrazinylpyrido[2,3-d]pyrimidines 4a,d (0.01 mol) and aromatic aldehydes namely, benzaldehyde, 4-chlorobenzaldehyde, 4-anisaldehyde, or thiophene-2-carbaldehyde (0.02 mol) in acetic acid (10 ml) was refluxed for 4 h. The mixture was poured onto ice-water and the residue formed was filtered and purified from (acetic acid).
7-(4-Chlorophenyl)-2-(2-(thiophen-2-ylmethylene)hydrazinyl)-5-(p-tolyl)pyrido[2,3-d]pyrimidin-4(3H)-one (8a)
Yield: 65%; mp: 360–362 °C; IR (KBr, cm−1) ν: 3421 (2NH), 1728 (C=O); 1HNMR (400 MHz, DMSO-d6): δ 13.5 (s, 2H, 2NH), 8.29 (d, 2H), 8.18 (s, 1H, N=CH), 7.70 (s, 1H, C-6 pyridine), 7.61–7.65 (m, 3H), 7.45 (d, 2H, J = 8 Hz), 7.32 (d, 2H, J = 12.3 Hz), 7.00 (d, 2H, J = 8 Hz), 2.38 (s, 3H, CH3); MS: [m/z (%) 473 (15, M++2), 471 (10, M+), 43 (100)].
2-(4-Chlorobenzylidene)hydrazinyl)-7-(4-chlorophenyl)-5-p-tolylpyrido[2,3-d]pyrimidin-4(3H)-one (8b)
Yield: 23%; mp: 366–368 °C; IR (KBr, cm−1) ν: 3423 (2NH), 1719 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 13.71 (s, 2H, 2NH), 8.29 (d, 2H, J = 8 Hz), 8. 18 (s,1H, N=CH), 7.91 (d, 2H, J = 8 Hz), 7.59 (d, 2H, J = 8 Hz), 7.50 (s, 1H, C-6 pyridine), 7.39–7.34 (m, 4H), 7.25 (d, 2H, J = 8 Hz), 2.38 (s, 3H, CH3); MS: [m/z (%) 503 (2.14, M++2), 499 (0.75, M+), 214 (100)].
7-(4-Chlorophenyl)-2-(2-(4-methylbenzylidene)hydrazinyl)-5-(p-tolyl)pyrido[2,3-d]pyrimidin-4(3H)-one (8c)
Yield: 58%; mp: 375–377 °C; IR (KBr, cm−1) ν: 3427 (2NH), 1728 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 13.5 (br s, 2NH), 8.28 (d, 2H, J = 8 Hz) , 8.18 (s, 1H, N=CH), 7.80 (d, 2H, J = 8 Hz), 7.60 (d, 2H, J = 8 Hz), 7.49 (s, 1H, C6-pyridine), 7.37 (d, 2H, J = 8 Hz), 7.33–7.23 (m, 4H), 2.28, 2.25 (2s, 6H, 2CH3); MS: [m/z (%) 481 (12, M++2), 64 (100)].
2-(2-Benzylidenehydrazinyl)-5–(4-methoxyphenyl)-7-(p-tolyl)pyrido[2,3-d]pyrimidin-4-(3H)-one (8d)
Yield: 53%; mp: 351–353 °C; IR (KBr, cm−1) ν: 3427 (2NH), 1728 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 13.60 (s, 2H, 2NH), 8.13–8.18 (m, 3H), 7.64 (s, 1H, C-6 pyridine), 7.41–7.45 (m, 4H), 7.35 (d, 2H, J = 8 Hz), 6.97 (d, 2H, J = 8.8 Hz), 3.82 (s, 3H, OCH3), 2.37 (s, 3H, CH3); MS: [m/z (%) 461 (4, M+), 43 (100)].
2-(4-Chlorobenzylidene)hydrazinyl)-5-(4-methoxyphenyl)-7-p-tolylquinazolin-4(3H)-one (8e)
Yield: 33%; mp: 361–363 °C; IR (KBr, cm−1) ν: 3422 (2NH), 1670 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 11.72, 11.28 (2s, 2H, 2NH), 8.25 (s, 1H, N=CH), 8.11 (d, 2H, J = 8 Hz), 8.00 (d, 2H, J = 8 Hz), 7.43 (s, 1H, C-6 pyridine), 7.41–7.50 (m, 4H), 7.30 (d, 2H, J = 8 Hz), 6.94 (d, 2H, J = 8 Hz), 3.81 (s, 3H, OCH3), 2.36 (s, 3H, CH3); MS: [m/z (%) 497 (26, M++2), 495 (84, M+), 384 (100)].
5-(4-Methoxyphenyl)-2-(2-(4-methylbenzylidene)hydrazinyl)-7-(p-tolyl)pyrido[2,3-d]pyrimidin-4(3H)-one (8f)
Yield: 27%; mp: 380–382 °C; IR (KBr, cm−1) ν: 3421 (2NH), 1670 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 11.82,11.73 (2s, 2H, 2NH), 8.10 (s, 1H, N=CH), 7.84 (d, 2H, J = 8 Hz), 7.37–7.45 (m, 4H), 7.33 (d, 2H, J = 8 Hz), 7.21 (d, 2H, J = 8 Hz), 6.95 (d, 2H, J = 8 Hz), 3.81 (s, 3H, OCH3), 2.35, 2.33 (s, 6H, 2CH3); 13 C NMR (100 MHz, DMSO-d6): 161.20, 159.91, 140.73, 140.44, 135.38, 134.64, 133.87, 132.21, 130.66, 129.56, 129.85, 128.24, 127.98, 127.90, 127.70, 127.21, 117.49, 113.46, 113.24, 55.61, 21.51, 21.38; MS: [m/z (%) 475 (9, M+), 42 (100)].
Biological evaluation
In-vitro antitumor assay
Anticancer screening (In vitro bioassay on human cancer cell lines) was determined by the Bioassay-Cell Culture Laboratory, National Research Centre, Cairo, Egypt. It was adopted against five cancer cell lines (HepG2, PC-3, HCT116, MCF-7, and A549); doxorubicin was used as a reference standard according to a previously reported methodsCitation14–16.
Kinase inhibition assay
The in-vitro enzyme inhibition determination for compound 5a (which showed promising anticancer activity against HePG-2, PC-3, HCT-116 cancer cell lines in comparison with doxorubicin was carried out in KINEXUS Corporation, Vancouver, British Columbia, Canada. Kinexus has developed an open-access, on-line resource called DrugKiNET, www.drugkinet.ca. The evaluation performed profiling of the compound 5a against a range of five protein kinases [(PDGFR beta, EGFR, CDK4/Cyclin D1, PI3K (p100b/p85a, PI3K (p100a/p85a)] according to a previously reported methodCitation16.
Molecular modeling study
All the molecular modeling calculations and docking simulation studies were performed utilizing Molecular Operating Environment (MOE®) 2008.10Citation17. The three-dimensional X-ray structures of EGFR (PDB code: 1M17)Citation18,Citation19 and CDK6 was used instead of CDK4 (PDB code: 2EUF)Citation20,Citation21 were obtained from the Protein Data Bank through the internet.
Results and discussion
Chemistry
New group of 5,7-diaryl pyrido[2,3-d]pyrimidinones 3a–g were created through reaction of the starting precursor 2-mercapto-4-hydroxy-6-aminopyrimidine 1 with α, β unsaturated ketones 2a–g in dry DMF according to the applied method reported for analogue 3dCitation22–24. Compound 3c as an example showed absorption band at 3394 cm−1 assigned for NH group in IR spectrum and three singlet signals at δ 7.94, 12.25, and 13.00 ppm attributed to C6 proton of pyridopyrimidine ring and 2NH protons in 1HNMR spectrum. Further, the MS revealed molecular ion peak at m/z 379 (M+) agreed with the molecular weight of the assigned structure. Also, 13 C-NMR spectrum of 3e analogue showed signal at 175.71 ppm for C=S group.
Nucleophilic attackCitation22–25 of hydrazine hydrate on thioxo derivatives 3a,c–g yielded the corresponding 2-hydrazinopyrido[2,3-d]pyrimidin-4(3H)-ones 4a–f (Scheme 1). IR spectrum of compound 4a showed existence of three absorption bands at 3410, 3190, and 1678 cm−1 corresponding to NH2, NH, and C=O groups, respectively. 1H NMR spectrum of 4d revealed singlet signals at δ2.38, 3.79, 4.20, 8.26, and 10.17 ppm assignable for CH3, OCH3, NH2, and two NH protons, respectively.
Scheme 1. Synthesis of Pyrido[2,3-d]pyrimidine derivatives.
![Scheme 1. Synthesis of Pyrido[2,3-d]pyrimidine derivatives.](/cms/asset/bc9cf3be-5070-40e0-b89e-91a13a318f85/ienz_a_1437729_sch0001.jpg)
Derivatives 4 b,c,e,f were condensed with active methylene compounds (ethylacetoacetate, ethylcyanoacetate, or diethylmalonate) in glacial acetic acidCitation26 to give the corresponding three-substituted pyrazolone derivatives 5a–d, 6a–d, 7a–c, respectively. Compounds 5a–d were confirmed by the existence of an additional band at 1651 cm−1 for C=O group of the newly created pyrazolone nucleus in IR spectrum of compound 5b. Additionally, the 1HNMR spectrum of 5a showed signals at δ 1.89 and 8.18 ppm assignable for CH3 group and C4 proton of pyrazole moiety. In addition, 13 C NMR spectra of 5a revealed signals at δ 21.32, 106.54, 161.77, and 172.44 ppm assigned for CH3 group, C-4 of pyazole ring, C=O of pyridine ring and C=O of pyrazolone moiety, respectively.
On the other hand, 6a–d derivatives displayed broad band at 3429 cm−1 for NH2 and two NH groups in IR spectrum of compound 6b. Additionally, 1H NMR spectrum showed three singlet signals at δ 3.97, 8.13, and 12.48 ppm related to NH2 group, C4 of pyrazole moiety, and two NH groups, respectively. 13CNMR spectrum for compound 6d showed signal at δ 21.44, 56.47, 60.49, and 106.80 ppm assigned for the carbon of the CH3, OCH3, and C4 of the pyrazole moiety. The carbonyl signals were recorded at δ 163.37 and 171.74 ppm.
Compounds 7a–c showed strong band for C=O group in pyrazole moiety at 1690 cm−1 in the IR spectrum of 7c and presence of singlet signal equivalent to one proton at δ 2.94 ppm represents the CH2 protons of the pyrazolyl moiety in 1H NMR spectrum. 13 C NMR spectra of compound 7c showed signals at δ 56.47, 60.47, 90.72, 167.16, 167.54 and 170.45 ppm corresponding to the carbon of OCH3, C4 of pyrazole ring and three C=O carbons, respectively.
Condensation of the hydrazinyl derivatives 4 b,d with various aromatic aldehydesCitation22,Citation25 afforded the corresponding 2-(2-(Aryl-2-ylmethylene) hydrazinyl)-5,7-diarylpyrido[2,3-d]pyrimidin-4(3H)-ones 8a–f. Structures of compounds 8a–f were confirmed by the presence of signals corresponding to the –N=CH proton at δ 8.10–8.25 ppm. 13CNMR spectrum for 8f revealed signals at δ 21.38, 21.51, 55.61, 159.91, and 161.20 ppm assigned for CH3, OCH3, –NH = CH and C=O carbons, respectively. MS of compound 8 b showed the molecular ion peak [M+] at m/z 499 and m/z 503 corresponding for the chlorine isotopes.
Biological evaluation
Invitro antitumor assay
All the synthesized compounds 3–8 were tested for their anticancer activity against hepatic cancer (HepG-2), prostate cancer (PC-3), colon cancer (HCT-116), breast cancer (MCF-7), and lung cancer (A-549) cell lines. Preliminary screening against the cancer cell lines was performed, using doxorubicin as a reference drug at doses of 100 μM. Variable results were recorded for the test compounds 3–8 (). Pyridopyrimidine derivatives that exhibited inhibitory activity >90% compared to doxorubicin were selected for IC50 and IC90 screening ( and ).
Table 1. Percentage of growth inhibition activity of compounds 3a–c,e–g, 4a,b, d–f, 5a–d, 6a–d, 7a–c, 8a–f against HepG-2, PC-3, HCT-116, MCF-7, and A-549 cell lines at (100 μM) dose.
Table 2. IC50 and IC90 of the test compounds 3a,f,g, 4a,b,d,e, 5a,d, 6a–d,f against HepG-2, PC-3 and HCT-116 cell lines.
Table 3. IC50 and IC90 of the test compounds 3a,f,g, 4a,b,d,e, 5a,d, 7a, 8a–d,f against MCF-7and A549 cell lines.
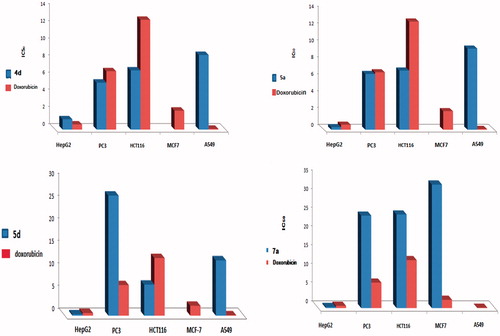
Almost, hepatic cancer cell line (HepG2) showed remarkable sensitivity towards the test compounds 3–8 compared to doxorubicin. Pyrazolylpyrido[2,3-d]pyrimidines 5a and 7a were equipotent, possessing twice activity relative to doxorubicin (IC50 of 0.3 μM each and 0.6 μM respectively). Pyrazolyl analogue 5d exhibited remarkable activity of IC50 0.9 μM. Hydrazino precursor 4a was almost equipotent to doxorubcin of IC50 0.7 μM. Compounds 3a, 4d and 8c displayed moderate activity of IC50 1.7 μM, 1.2 μM, and 1 μM, respectively. Noticeably, the test compounds showed lower anticancer activity than doxorubicin, concerning IC90 measurement ().
Regarding the PC-3 cell line, hydrazino precursor 4d and pyrazolyl analogue 5a were more potent than doxorubicin as a reference standard (IC505.47 μM, 6.6 μM, and 6.8 μM respectively). Also, pyrazolyl analogue 6d as well as the hydrazone derivative 8a displayed remarkable activity of IC50 7.9 μM and 7.97 μM respectively. On the other hand, pyridopyrimidine derivatives 3a, 4b, 8 b–d exhibited moderate activity with IC50 ranged from 9.2 μM to 12 μM. Interestingly, IC90 showed promising anti-prostate cancer activity for pyrazolyl analogue 5a versus doxorubicin (IC90 14.1 μM and 13.8 μM, respectively). The rest of the test compounds showed IC90 ranged from moderate to poor activity compared to doxorubicin ().
Potent activity against HCT-116 cell line was recorded for three of the test compounds, e.g. 2-hydraino derivative 4d and 3-methyl-5-oxo-pyrazolyl 5a,d. They were almost twice the activity of doxorubicin exerting IC50 6.9 μM, 7 μM, and 5.9 μM vs. 12.8 μM, respectively. Excepting pyridopyrimidines 4d and 5a,d, none of the test compounds showed high potency against HCT-116 colon cancer. Preferentially, the hydrazide derivative 4d was also more potent than doxorubicin by two folds, exerting IC90 21 μM vs. 51.7 μM, respectively. Simultaneously, compounds 3a and 5a exhibited higher activity than doxorubicin of IC90 48 μM and 36 μM, respectively (). In contrary, breast and lung cancer cell lines exhibited remarkable resistance towards all of the test compounds 3–8 ().
Structure-activity relationship
In general, the structure–activity relationships of the screened products indicated that, existence of pyrazolyl moiety at C-2 of pyrido[2,3-d]pyrimidines 5a,d, 6d, and 7a derivatives afforded the maximum potency of anticancer activity. Noticeably, 5a and 7a shared the same pyridopyrimidine scaffold with (4-CH3-phenyl) and (4-chlorophenyl) at C-5 and C-7 respectively. Compound 5a linked 3-methyl-5-oxopyrazolyl moiety showed broad anticancer effect against hepatic, prostate, and colon cancers. Replacing the methyl group of pyrazole moiety in 5a with carbonyl group in 7a afforded 3,5-dioxopyrazole where the activity profile was changed. Comparatively, it was retained against hepatic cancer but diminished against prostate and colon cancers in 7a. Moreover, compounds 5d and 6d shared pyridopyrimidine scaffold with (3,4,5-trimethoxyphenyl) and (4-CH3-phenyl) at C-5 and C-7, respectively. Replacing methypyrazolone moiety in 5d with aminopyrazolone in 6d shifted the anticancer activity from anti-colon to anti-prostate cancer, respectively. Noticeably, the steric factor potentially affected the anticancer activity in 5a–d. The data recorded remarkable decrease in the activity when (4-CH3-phenyl) group in 5a was exchanged by (3,4,5-trimethoxyphenyl) in 5c. Similarly, introduction of steric bulky group in 7b–c diminished the anticancer activity. In contrary, steric factor did not affect the anticancer activity in 6a–d, except 6d derivative that carried the bulky (3,4,5-trimethoxyphenyl) group at C-5 ().
Figure 2. IC50 of compounds 4d, 5a, 5d, 7a against HepG-2, PC-3, HCT-116, MCF-7, and A-549 cell lines.
Thioxo precursors 3a–c,e–g displayed poor anticancer activity against all cancer cell lines. Anti-hepatic cancer effect was greatly increased upon converting thioxo group in 3c to hydrazide moiety in 4b (equipotent to doxorubicin). Also, great records were displayed upon adopting the same replacement in 3e to afford hydrazide analogue 4d. The later was more potent than doxorubicin against prostate and colon cancers. Hydrophilic electron rich nature of the hydrazide moiety enabled the electronic factor to afford positive impact on the anticancer activity. Compounds 4b and 4d lacked their activity by extending the hydrazide moiety to benzylidene hydrazide in 8b,d. Obviously, 8a–f failed to record any potent activity against all cancer cell lines.
Kinase inhibition assay
Upon cellular screening on HepG-2, PC-3, and HCT-116, compound 5a exhibited higher anticancer activity in comparison with doxorubicin. So, it was subjected for in vitro inhibition assessment to measure its inhibitory activity against a panel of five different protein and lipid kinases. Two concentrations of 5a were applied (50 μM and 100 μM) in single measurement compared to blank control.
Three of the tested enzymes are protein kinases, e.g. platelet-derived growth factor receptor (PDGFR) and epidermal growth factor receptor (EGFR) are tyrosine kinases. The third enzyme was cyclin-dependent kinase (CDK4/CyclinD) that phosphorylates serine/threonine residues. The remaining tested enzymes are lipid kinases that belong to phosphoinositide 3-kinases (PI3Ks), e.g. PI3K (p100b/p85a) and PI3K (p100a/p85a).
Maximum inhibitory percentage of 5a was recorded () against PDGFR beta of 82% and 94% at concentrations of 50 μM and 100 μM, respectively. Also, remarkable inhibitory percentage was exhibited against EGFR of 81% and 86% at the previously mentioned concentrations. On the other hand, 5a at 50 μM recorded low inhibition percent against CDK4/Cyclin D1 of 35%. The inhibition of the previous enzyme was retrieved upon applying 100 μM of 5a to 75%. As a conclusion, the inhibitory effect of 5a against both PDGFR beta and EGFR was remarkable and directly proportional to the concentration. Interestingly, the inhibitory effect of 5a toward PDGFR beta was more prominent and more concentration sensitive than EGFR. For example, increase the concentration of 5a by two-fold from 50 μM to 100 μM afforded 10% increase of the activity against PDGFR. Concerning EGFR kinase, applying twice concentration of 5a showed 5% increase of the activity. Although the effect of 5a against CDK4/CyclinD1 was less prominent, but it was more concentration dependent. So, doubling the concentration of 5a enhanced the inhibitory effect by almost two-folds () (). Unexpectedly, compound 5a showed activation effect toward the two lipid kinases, PI3K (p100b/p85a) and PI3K (p100a/p85a) above the control experimental. The activation percentages were recorded in (). So, the potent anticancer activity of 5a against HepG-2, PC-3, and HCT-116 could be attributed to its remarkable inhibitory activity against protein kinase enzymes.
Figure 3. Percentage of kinases inhibition of compound 5a against EGFR, CDK4 at (50 and 100 μM).
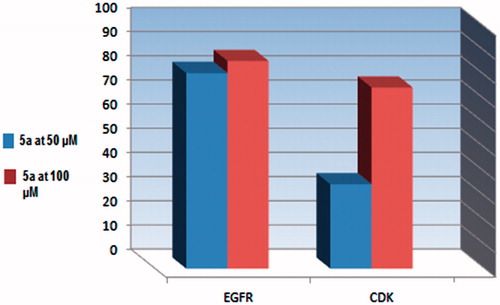
Table 4. Percentage of kinases inhibition of compound 5a at (50 and 100 μM).
Molecular docking results
Molecular docking technique represents the pattern of the interaction between a small molecule and a protein at the atomic level. This approach can explore the behavior of small molecules in the binding site of the target proteins. So, docking simulation was performed in this work using Molecular Operating Environment (MOE®)Citation17 2008.10. All the interaction energies and different calculations were automatically calculated.
Kinase inhibitory screening assay promoted compound 5a that showed promising inhibitory activity against three kinases, namely PDGFR β, EGFR, and CDK4/Cyclin D1. The X-ray crystallography of PDGFR β structure was not fully resolvedCitation27. On the other hand, X-ray crystallography structure was reported for EGFR (pdb code: 1M17)Citation18,Citation19 with erlotinib. Simultaneously, the structure of CDK4/CyclinD1 with PD0332991 was reported (PDB ID: 2EUF)Citation20,Citation21. So the docking study was achieved for both EGFR and CDK4/Cyclin D1 kinases to predict the binding modes, affinities, and orientations of compound 5a at the active sites of them.
Docking study on EGFR
Docking of 5a into the active site of EGFR explored minimum binding energy comparable to the reference, erlotinib (Docking score= −7.95 and −5.35 Kcal/mol, respectively). The orientation of pyrido[2,3-d]pyrimidinone scaffold of 5a was parallel to the hinge region due to the two bulky aryl rings. The scaffold posed in the adenine binding region of the ATP binding site. The pyrazolyl C=O carbon of 5a formed a hydrogen bond acceptor (distance: 2.44 Ao) with the backbone NH of Met769 in the hinge region. Para-tolyl moiety shared hydrophobic interaction with Gly695 and para-chlorophenyl ring formed arene-arene interaction with Phe699 ().
Figure 4. The proposed binding mode of compound 5a docked in the active site of EGFR. A and B showing 2D and 3D ligand-receptor interactions (hydrogen bonds are illustrated as arrows; C atoms are colored gray, N blue, and O red).
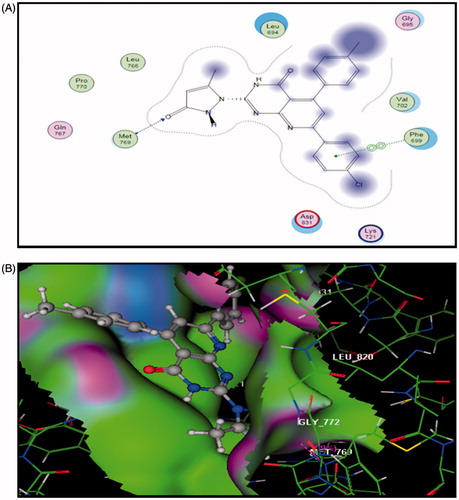
Figure 5. The proposed binding mode of compound 5a docked in the active site of CDK6. A and B showing 2D and 3D ligand-receptor interactions (hydrogen bonds are illustrated as arrows; C atoms are colored gray, N blue, and O red.
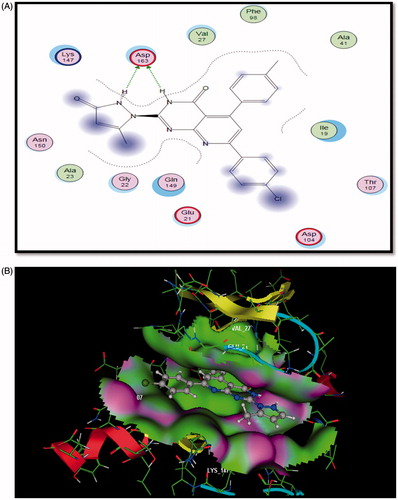
Docking study on CDK6
Compound 5a bound in ATP binding pocket of CDK6 displayed good fitting and minimum binding energy as well as the reference PD0332991 (Docking score= -5.187 and -4.145, respectively). Also, it formed bidentate hydrogen bonding between N3 of pyridopyrimidine ring and N2 of pyrazolone moiety to the main chain amide of Asp163 (distance =1.44, 1.43 Ao). Moreover, para-chlorophenyl moiety projected on the opposite side, sequestered into a hydrophobic region ().
So, docking simulation of compound 5a into kinase domain of EGFR and CDK6 postulated the vital role of both pyrido[2,3-d]pyrimidinone scaffold and the side chain substituent. Both moieties involved in binding mode interaction. Binding pose covered different macromolecular interactions, e.g. H-bonding, arene-arene, and hydrophobic interactions. Compound 5a could be anticipated to bind efficiently to the ATP binding site of EGFR compared to erlotinib. Furthermore, similar results were predicted for the binding pose of CDK6 with both PD0332991 and compound 5a.
Conclusions
A novel group of substituted pyrido[2,3-d]pyrimidinones 3–8, were synthesized and evaluated for in vitro anticancer activity. There was potent growth inhibitory activity against HepG-2, PC-3, and HCT-116 cell lines and weak activity against MCF-7 and A-549 cancer cells in comparison to doxorubicin. Regarding HepG-2 cell line, compounds 5a, 7a were more potent (IC50= 0.3 µM) than doxorubicin. Also, compound 4 b was nearly equipotent (IC50= 0.7 µM) to doxorubicin (IC50= 0.6 µM). Upon cellular screening on PC-3, compounds 4d, 5a exhibited higher activity than doxorubicin (IC50= 5.47, 6.6, 6.8 µM respectively). For HCT-116 cell line, 4d, 5a, and 5d derivatives showed two-folds the activity of doxorubicin (IC50= 6.9, 7, 5.9, and 12.8 µM, respectively). The highly potent anticancer compound 5a exhibited promising inhibitory activity against PDGFR β, EGFR, and CDK4/Cyclin D1 kinases at two concentrations of 50 μM and 100 μM in single measurement. Furthermore, the molecular docking study for compound 5a into the ATP binding site of EGFR and CDK6 explored an efficient binding as that of erlotinib and PD0332991, respectively.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Wu H-C, Chang D-K, Huang CT. Targeted therapy for cancer. J Cancer Mol 2006;2:57–66.
- Varmus H. The new era in cancer research. Science 2006;312:1162–5.
- Zheng LW, Li Y, Ge D, et al. Synthesis of novel oxime-containing pyrazole derivatives and discovery of regulators for apoptosis and autophagy in A549 lung cancer cells. Bioorg Med Chem Lett 2010;20:4766–70.
- Eckford PDW, Sharom FJ. ABC efflux pump-based resistance to chemotherapy drugs. Chem Rev 2009;109:2989–3011.
- Lage L. An overview of cancer multidrug resistance: a still unsolved problem. Cell Mol Life Sci 2008;65:3145–67.
- Luqmani YA. Mechanisms of drug resistance in cancer chemotherapy. Cancer Treat Rev 2003;29:297–307.
- Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nature Reviews/Drug Discovery 2006;5:219–34.
- Chorawala MR, Oza PM, Shah GB. Mechanisms of anticancer drugs resistance: an overview. IJPSDR 2012;4:1–9.
- Denny WA. DNA minor groove alkylating agents. Exp Opin Ther Patents 2000;10:459–74.
- Denny WA, Atwell GJ, Baguley BC, Wakelin LPG. Potential antitumor agents. Synthesis and antitumor activity of new classes of diacridines: importance of linker chain rigidity for DNA binding kinetics and biological activity. J Med Chem 1985;28:1568–74.
- Patrick GL. An introduction to medicinal chemistry. 3rd ed. New York: Oxford University Press Inc.; 2005: 489–553.
- Abou-Seri FSM, Abdel-Aziz HA, Abbas SE, et al. Synthesis and antitumor activity of pyrido[2,3-d]pyrimidine and pyrido[2,3-d] [1,2,4]triazolo[4,3-a]pyrimidine derivatives that induce apoptosis through G1 cell-cycle arrest. Eur J Med Chem 2014;83:155–66.
- Rodriguez R, Meuth M. Chk1 and p21 cooperate to prevent apoptosis during DNA replication fork stress. Mol Biol Cell 2006;17:402–12.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Nossier ES, El-Hallouty SM, Zaki ER. Synthesis, anticancer evaluation and molecular modeling of some substituted thiazolidinonyl and thiazolyl pyrazole derivatives. Int J Pharm Sci 2015;7:353–9.
- El-serwy WS, Mohamed NA, El-serwy WS, et al. Synthesis, molecular modeling studies and biological evaluation of novel pyrazole as antitumor and EGFR inhibitors. Int J Pharm Tech 2016;8:25192–209.
- Molecular Operating Environment (MOE), 2008.10; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7.
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain, alone and in complex with a 4 anilinoquinazoline inhibitor. J BioChem 2002;277:46265–72.
- Traxler P, Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol Ther 1999;82:195–206.
- Gahmen US, HouKim S. Structure basis for CDK6 activation by a virus encoded cyclin. Nature Struc Bio 2002;9:177–81.
- Tadesse S, Yu M, Kumarasiri M, et al. Targeting CDK6 in cancer: state of the art and new insights. Cell Cycle 2015;14:3220–30.
- Abdallaha MA, Gomha SM, Morad MA, Elaasser MM. Synthesis of pyridotriazolopyrimidines as antitumor agents. J Het Chem 2016;24:1242–51.
- Farghaly TA, Hassaneen HME. Synthesis of pyrido[2,3-d][1,2,4]triazolo[4,3-a]pyrimidin-5-ones as potential antimicrobial agents. Arch Pharm Res 2013;36:564–72.
- El-Nassan HB. Synthesis and antitumor activity of novel pyrido[2,3-d][1,2,4]triazolo[4,3-a]pyrimidin-5-one derivatives. Eur Med Chem 2011;46:2031–6.
- El-Gazzar AB, Gaafar AM, Aly AS. Synthesis of some new thiazolo[3,2-a]pyrido[2,3-d]pyrimidinones and Isoxazolo[5′,4′:4,5]thiazolo[3,2-a]pyrido[2,3-d]pyrimidinone. Phosphorus Sulfur Silicon 2002;177:45–58.
- Ismail MMF, Rateb HS, Hussein MM. Synthesis and docking studies of novel benzopyran-2-ones with anticancer activity. Eur J Med Chem 2010;45:3950–9.
- Conconi MT, Marzaro G, Urbani L, et al. Quinazoline-based multi-tyrosine kinase inhibitors: synthesis, modeling, antitumor and antiangiogenic properties. Eur J Med Chem 2013;67:373–83.