
Abstract
Alzheimer’s disease is debilitating neurodegenerative disorder in the elderly. Current therapy relies on administration of acetylcholinesterase inhibitors (AChEIs) -donepezil, rivastigmine, galantamine, and N-methyl-d-aspartate receptor antagonist memantine. However, their therapeutic effect is only short-term and stabilizes cognitive functions for up to 2 years. Given this drawback together with other pathological hallmarks of the disease taken into consideration, novel approaches have recently emerged to better cope with AD onset or its progression. One such strategy implies broadening the biological profile of AChEIs into so-called multi-target directed ligands (MTDLs). In this review article, we made comprehensive literature survey emphasising on donepezil template which was structurally converted into plethora of MTLDs preserving anti-cholinesterase effect and, at the same time, escalating the anti-oxidant potential, which was reported as a crucial role in the pathogenesis of the Alzheimer’s disease.
Graphical Abstract
1. Introduction
Dementia is a chronic or progressive illness that is characterised by impaired cognitive capacity beyond what could be considered a consequence of normal aging. The most common form of it is the Alzheimer’s disease (AD) accompanied by the symptoms such as memory loss, difficulty in solving problems, comprehension, calculation and learning, disorientation, impaired learning ability etcCitation1. According to the data from European Prevention of Alzheimer's Dementia, AD affects more than 40 million people worldwide and its prevalence is expected to double over the next 20 years. Moreover, AD is currently the fourth leading cause of death in people over 65 years old in the world, which makes it one of the major health, social, and economic concern of the society worldwideCitation2,Citation3.
Many pathological aspects of AD have been discovered in the course of over 100 years of research and observation of AD patients. One of the most important finding was the identification and functional characterisation of neurotransmitter acetylcholine (ACh)Citation4. Indeed, the investigation of biopsy tissue and post-mortem brain tissues from AD patients showed reduced choline acetyltransferase activity, ACh synthesis, choline uptake and ACh release. Moreover, the impairment of cognitive functions is also associated with degeneration of cholinergic neurons and loss of cholinergic neurotransmission. In line with this discovery, novel therapeutic approaches were developed to “correct” or “compensate” for neurochemical alterations in the cholinergic systemCitation5. Besides, other disrupted neurotransmitter system attracted particular attention for targeting AD pathological pathways, such as N-methyl-d-aspartate (NMDA) receptorsCitation6 dopaminergic systemCitation7 serotoninergic systemCitation8 and others.
Building on the aforementioned, currently approved drugs for AD rivastigmine, galantamine and donepezil, as well as discontinued tacrine (), improve cholinergic neurotransmission by inhibition of acetylcholinesterase (AChE, E.C. 3.1.1.7), enzyme responsible for degradation of AChCitation9. While AChE predominates in the healthy brain, butyrylcholinesterase (BChE, E.C. 3.1.1.8) is considered to play a minor role in the regulation of synaptic ACh levels. This scenario is modified in the context of AD, as the activity of AChE remains unchanged and BChE activity progressively increasesCitation10. The AChE inhibitors (AChEIs) are used for symptomatic treatment of AD shedding light on the importance of AChE which still remain a highly viable classic target for development of new drug candidatesCitation11.
Figure 1. Currently used AChEIs donepezil, galantamine and rivastigmine. Tacrine is no longer approved for AD treatment. Memantine act as NMDA receptor antagonist. Structures of human AChE (PDB ID: 4EY7) and GluN1A-GluN2B NMDA receptor (PDB ID: 4PE5) were downloaded from Protein Data Banka (http://www.rcsb.org) and created with PyMol viewer 1.3.
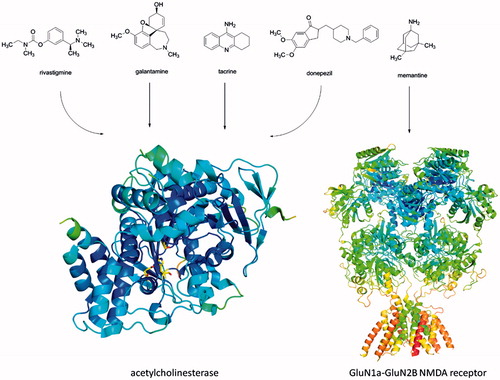
Another drug approved for AD is memantine () which acts as antagonist of NMDA receptorCitation12–14. The connection between the constant stimulation of NMDA receptors and the cognitive deficits seen in AD is based primarily on the principle of excitotoxicity and the death of nerve cells is caused by chronic neuronal activationCitation15.
Another two major players in the pathogenesis of AD postulated that formation of amyloid deposits and neurofibrillary tangles are responsible for the onset of the disease. Accordingly, the amyloid-β (Aβ) hypothesis implies that AD therapy should restore the Aβ homeostasis in the brain by altering the production and clearance of AβCitation16. There are several strategies possible applied for the maintenance/reduction of Aβ levels in the brain: prevention or reduction of Aβ formation, removal of existing amyloid deposits through immunotherapy, prevention or reduction of Aβ aggregation, and enhancement of Aβ clearanceCitation17. Unfortunately, all efforts to develop Aβ-targeted therapies have failed to date. Moreover, recent studies have shown that both prevention of amyloid deposition and removal of amyloid do not, by themselves, lead to improved cognition in ADCitation18. Another important therapeutic target involved in the pathogenesis of AD is the tau protein. Tau is ubiquitous in neurons and plays an important role in microtubule assembly and stabilisation of neuronal microtubule networkCitation19. In AD patients brain, high levels of hyperphosphorylated tau can be found intracellularly which lead to a generation of aberrant aggregates that are toxic to neuronsCitation20. None of the developed anti-tau drug candidates has also advanced to clinical practice to date. It has to be mentioned that current trend in drug design of novel AD drugs switches from anti-Aβ to anti-tau therapyCitation18.
Oxidative stress is considered as one of the key players in the aetiology and progression of various neurodegenerative disorders. Abundant data suggests that oxidative stress may induce not only cellular damage, but also DNA repair system breakdown or mitochondrial malfunction. All of these events largely contribute to aging and neurodegeneration, a phenomena observed in AD or Parkinson’s diseases progressionCitation21. A group of reactive oxygen species (ROS) contain highly reactive and more or less short-lived molecules derived from oxygen. Among these, free radicals such as superoxide, hydroxyl radical, or hydrogen peroxide can be found being responsible for the cytotoxic effectCitation22. Many biological systems have been implicated in ROS production like mitochondria, NADPH oxidases, xanthine oxidase, peroxisomes, or endoplasmatic reticulumCitation23. Increased ROS levels may be down-regulated by several defence systems including antioxidant enzymes or endogenous small-molecule antioxidants (e.g. superoxide dismutase, glutathione peroxidase, catalase, peroxiredoxins, tri-peptide glutathione, vitamins E, and C)Citation24. On the other hand, low levels of ROS have been shown to be involved in physiological processes like cellular signalling, pro-survival pathways or activation of transcription factors regulating cellular response to ROSCitation25.
In general, oxidative stress could be regarded as imbalance between the generation of ROS and malfunction of natural antioxidant system. The depletion of antioxidant system or overproduction of ROS leads to pathological conditions denoted as oxidative stress. Under these conditions, brain is the most vulnerable organ in the body. Brain is characterised by the highest oxygen consumption and contains redox-active metals like iron or copper catalysing ROS formation. Furthermore, high levels of polyunsaturated fatty acids in the brain represent a good substrate for lipid peroxidationCitation26.
The intertwined pathophysiological pathways of AD point to linkage between the Aβ accumulation and increased oxidative stress resulting in mitochondrial malfunction and energy collapse in the early stages of the diseaseCitation27. Vice versa, oxidative stress may also escalate the production and aggregation of Aβ and mediate the phosphorylation of the tau proteinCitation28.
Administration of antioxidants for treating neurodegenerative disorders has brought contradictory results. On one side, the results from animal studies implied potential benefit of this therapy, on the other side clinical trials precluded to meet the expected outcomes and benefitsCitation29. Several explanations could be addressed to low efficacy of antioxidants in the treatment of AD. The reason for such behaviour can be found in insufficient dose of antioxidants, improper therapy management, or insufficient therapy duration. Other reasons could be also highlighted such as: (i) oxidative damage may not be the primary cause but the consequence of vicious cycle accompanying the pathophysiological processes of AD; (ii) single antioxidant may not be capable to sufficiently counteract the complex cascade of oxidative stress; (iii) earlier antioxidant administration would be better since most of the clinical trials focused on patients with advanced AD; (iv) patients should be more cautiously selected for clinical trials (i.e. patients with low levels of endogenous antioxidants are better responders)Citation30,Citation31.
Based on the above-mentioned reasons, multimodal approach combining antioxidant properties with other relevant targets involved in AD pathology is an interesting approach. In recent year, several articles describing the potential of novel compounds including antioxidant properties have been publishedCitation11,Citation32,Citation33. This review article is focused on the drug design and development of novel multi-target directed ligands (MTDLs) amalgamating donepezil as core scaffold extending its biological profile primarily against oxidative stress.
2. Overview of in vitro methods for determination of antioxidant activity
Antioxidant capacity is connected with compounds able to protect a biological system against the damaging influence of ROS and reactive nitrogen species (RNS). The general capacity of the compound to scavenge ROS and/or RNS is called the total antioxidant activity (TAC)Citation34. Nowadays, several chemical assays in combination with highly sensitive detection technologies are massively exploited for measurement of antioxidant activity of compounds through different mechanisms, including scavenging activity against specific radicals or ROS, hydrogen atom transfer (HAT), single electron transfer (ET), metal chelation, and reducing powerCitation35–37. In vitro methods for measuring antioxidant capability described in this section differ from each other in reaction condition and mechanism, substrate that is oxidised, target probe/species, and type of detection. These assays usually utilize a chemical system involving free radicals or other ROS (an oxidant), an oxidizable probe (for some assays is needless) and antioxidants.
HAT-based methods evaluate the capability of antioxidants to scavenge free radicals by hydrogen donation to form a stable compound. Most of these methods control competitive reaction kinetics and the effect is determined from the kinetic curvesCitation38. Among others, oxygen radical absorbance capacity (ORAC) method, total peroxyl radical-trapping antioxidant parameter (TRAP) assay using β-phycoerythrin or fluorescein as the fluorogenic probe and total oxyradical scavenging capacity (TOSC) test belong to HAT-based methodsCitation39,Citation40. ET-based methods measure antioxidant capacity to reduce a probe (transfer one electron to reduce radicals, carbonyls, metals etc.). This process leads to colour change of the probe (an increase or decrease of the probe absorbance at a specific wavelength) after removing of an electron from the antioxidant. The stage of colour change depends on the concentration of antioxidantCitation40. 2,2-Diphenyl-1-picrylhydrazyl (DPPH) radical scavenging, trolox equivalent antioxidant capacity (TEAC), 2,2-azinobis-(3-ethylbenzothiazoline-6-sulfonic) acid (ABTS) radical cation decolourisation, ferric reducing antioxidant power (FRAP) and cupric reducing antioxidant capacity (CUPRAC) assays are the most frequently used techniques of ET-based methodsCitation41–45. Although HAT and ET have different mechanism, both of them are able to reveal the nature of antioxidant profile in the tested compound.
2.1. TEAC assay
TEAC assay is a simple and convenient method for determination of TAC based on the ability of antioxidants to scavenge the stable ABTS radical cation (a blue–green chromophore with an absorption maximum wavelength at 734 nm). Potential antioxidants can neutralize the ABTS radical cation by either radical quenching through hydrogen atom donation or by direct reduction through electron donation. Thereby, the antioxidants decolorize ABTS radical cation and spectrophotometrically can be measured a decrease in absorbance (the loss of its colour). This depends on the intrinsic antioxidant activity, concentration sample and also reaction durationCitation35,Citation44,Citation46. Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxyl acid, water soluble analogue of vitamin E) can be used as standard antioxidant and results of the experiments are usually expressed as Trolox equivalentCitation40. This assay is usually classified as ET-based method.
2.2. ORAC assay
ORAC assay is relatively novel test tube analysis measuring the antioxidant scavenging activity against the peroxyl radical produced by a generator such as 2,2′-azobis(2-amidinopropane) dihydrochloride (AAPH), 2,2′-azobis(2,4-dimethylnaleronitrile) (AMVN) or 2,2-azobis(2-amidinopropane) hydrochloride (ABAP)Citation37,Citation47. The ability of potential antioxidant to slow or stop the radical reaction is observed. The peroxyl radical reacts with a fluorescent probe (fluorescein or β-phycoerythrin) resulting in decrease of fluorescence that can be recorded using spectrofluorimeter. Usually, Trolox is used as a reference and established ORAC values of the tested potential antioxidants are reported as Trolox equivalent. It is well-accepted that the higher ORAC values the better antioxidant ability of the compound. Considering the fact that ORAC assay measures hydrogen atom donating ability of antioxidants, it belongs to HAT-based methodsCitation35,Citation46,Citation48.
2.3. DPPH assay
DPPH radical scavenging assay is probably the most frequently used method. It is characterised as an ET-based method with HAT mechanism. Mechanistically, electron donation of antioxidants neutralizes DPPH radical. DPPH is a cell permeable and stable free radical with a deep violet colour (with an absorption maximum wavelength at around 520 nm). Obviously, reaction between DPPH and appropriate antioxidant is connected with its colour change by decreasing the absorbance of the systemCitation35,Citation36,Citation38.
2.4. FRAP assay
FRAP assay is a typical ET-based non-radical method depending on the reduction of ferric ion (Fe(III))-ligand complex by the antioxidants to navy blue colour ferrous (Fe(II)) complex. Antioxidant capacity is therefore defined as an increase of absorbance at around 593 nm and obtained results can be expressed as Fe2+ equivalents or relative to the antioxidant standards. Normally, 2,4,6-tripyridyl-s-triazine (TPTZ) is used in this assay as the iron-binding ligand and trolox or ascorbic acid as antioxidant standardsCitation34,Citation37. In order to keep solubility of iron, FRAP assay is carried out under low acidic pH (pH = 3.6) which is far from the physiological pH values. Therefore, it is not possible to measure the antioxidants containing thiol groupsCitation49.
2.5. CUPRAC assay
CUPRAC assay was developed as a modification of FRAP assay replacing iron by copper as oxidant. This method determines the ability of antioxidants to reduce cupric ion (Cu(II))-ligand complex to cuprous ion (Cu(I)) complex (chromophore with absorption maximum at 450 nm). Neocuprine (2,9-dimethyl-1,10-phenanthroline) is usually used as ligand. In contrast with FRAP assay, CUPRAC assay is carried out at pH = 7 and therefore enlarge the versatility of this method to other substrates including thiol-containing compoundsCitation49.
3. Donepezil derivatives with antioxidant properties
3.1. Coumarin hybrids
Coumarin derivatives as AChEIs deserved closer attention because these types of naturally occurring as well as chemically developed compounds possess a wide range of pharmacological activities not only favourable for AD treatment but also for other maladies, and have been broadly reviewedCitation50,Citation51. Several studies indicating that coumarins are capable of inhibiting AChE by binding to its PAS have spurred the design and synthesis of novel coumarin derivatives as potent AChEIsCitation52–55.
A large number of coumarins endowed with high AChE inhibitory activities have been recently describedCitation56–59. 6,7-Dimethoxy-3-substituted coumarins linked to a benzylamine moiety placed at an appropriate distance from the heterocyclic ring were designed and reported ()Citation60. Inhibitory potency was chiefly affected by the distance between the amide attached to the coumarin moiety at the 3-position and the benzylamine fragment, with potency enhancement up to a trimethylene bridge. A small drop in inhibitory activity was observed when the amide group was replaced by an inverted amide group or isosteric ester. The same consideration as for donepezil claims that inhibitory activity declines when both methoxy groups attached to coumarin are removed. Compound 1, a cis-3-amino-cyclohexanecarboxylic acid derivative, displayed AChE inhibitory potency comparable to that of the reference donepezil (1: bAChE IC50 = 7.6 nM; hAChE IC50 = 43 nM; eqBChE IC50 = 33 µM; ). The study confirmed a reversible, mixed-type model of inhibition indicating dual binding site character.
Figure 2. Donepezil-related coumarin derivatives.
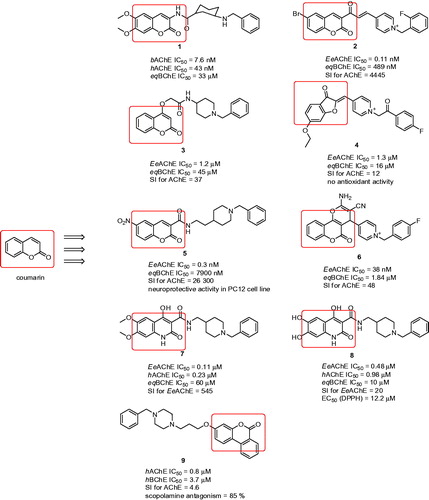
Prof Shafiee’s group reported a variety of synthetic approaches leading to biologically active coumarinsCitation61–65. In a preceding study, it was proved that the N-benzylpyridinium scaffold is required for superior activityCitation66. First, coumarin ring derivatives connected to differently substituted N-benzylpyridinium moieties were investigated ()Citation67. The attachment of these moieties utilised an α,β-unsaturated carbon linker to confer conformational restriction, which made it possible to study substituent modifications regardless of any conformational alterations in the linker. The target compounds demonstrated AChE IC50 values in the range of moderate to outstanding, achieving picomolar concentrations with the most active derivative being 2 (EeAChE IC50 = 0.11 nM; eqBChE IC50 = 489 nM; SI for AChE = 4445; ). SAR disclosed that the activity of these compounds mainly depends on the steric and electronic features of the substituents located on the benzyl moiety. Moreover, the activity was sensitive to the size of substituents at the para position of this moiety, with analogues containing bulky groups being weaker AChEIs. Coumarin ring modification was also performed, considering that insertion of a methoxy group (C-6) or bromine atom (C-8) is necessary for activity enhancement. In addition, two different methods, DPPH (1,1-diphenyl-1-picrylhydrazyl) and FRAP (ferric reducing antioxidant power) proved an absence of antioxidant ability compared to the reference compound (ascorbic acid). In the docking studies with TcAChE, 2 revealed a similar arrangement to that of the reference drug donepezil. Importantly, docking studies confirmed the significance of positive charge within the N-benzylpyridinium moiety, as it can provide cation–π interaction with Tyr334 in the TcAChE active site. This might be the reason for the 2-fold increased activity of 2 compared to donepezil.
A series of 4-hydroxycoumarin derivatives connected via an alkoxy amide spacer to N-phenylpiperazine or N-benzylpiperidine scaffolds was designedCitation68. In the study, compound 3 bearing an N-(1-benzylpiperidin-4-yl)acetamide appendage displayed the highest inhibitory activity against EeAChE (3: EeAChE IC50 = 1.2 µM; eqBChE IC50 = 45 µM; SI for AChE = 38; ). SAR revealed that the anti-AChE activity of these compounds was influenced mainly by the type of the cyclic amine attached to the 2-oxo- or 4-oxoalkoxycoumarin skeleton. Docking studies performed on TcAChE indicated that Phe330 is responsible for ligand recognition and trafficking by forming cation–π interaction with the N-benzylpiperidine moiety.
In another study, coumarin and 3-coumaranone derivatives encompassing the phenacyl pyridinium moiety were preparedCitation69. In particular, compound 4 with a fluoro atom at the para position of the phenacyl moiety emerged as the most potent AChEI, with an IC50 value of 1.3 µM (for EeAChE; eqBChE IC50 = 15.8 µM; SI for AChE = 12; ). Based on kinetic studies, the mode of inhibition indicated mixed type. Compounds in this subset were also tested for their antioxidant properties using FRAP assay. However, none of the compounds displayed significant antioxidant activity in comparison with ascorbic acid as reference, except the analogues containing a methoxy group or catechol group on the phenacyl moiety. Moreover, all compounds in the series exhibited desirable ADME properties with the ability to penetrate BBB, having polar surface area (PSA) less thanCitation70 60–70 Å2.
Two structural motifs combining N-benzylpiperidine with coumarin that are connected either via a carboxamide or N-ethylcarboxamide linker were designed and synthesisedCitation71. Compounds containing the latter linker were more active than their counterparts with carboxamide linker. The most active derivative was found to be 5, bearing a nitro group at position 6 of the coumarin ring, which was 46-fold more potent than donepezil (EeAChE IC50 = 0.3 nM; eqBChE IC50 = 7900 nM; SI for AChE = 26 300; ). Several compounds were determined for cell viability using MTT assay, which indicated no toxicity at concentrations of 1–100 µM. Neuroprotective properties against H2O2 in differentiated PC12 cells were evaluated within the 1–100 µM range, verifying that pre-treatment with these compounds significantly protected neurons against cell death at all the compound’s tested concentrations. The docking studies revealed that the higher flexibility of the N-ethylcarboxamide linker in compound 5 might lead to more facile accommodation of the compound into the active site, with better dual binding site inhibition of AChE. This was confirmed by kinetic analysis, which assigned a mixed type mode of inhibition.
Fused coumarins, namely 5-oxo-4,5-dihydropyrano[3,2-c]chromene derivatives linked to an N-benzylpyridinium scaffold were developed as AChEIsCitation72. The most potent compound was found in the 4-pyridinium series (6: EeAChE IC50 = 38 nM; eqBChE IC50 = 1.84 µM; SI for AChE = 48; ), while the 3-pyridinium series exhibited still good, but rather lower anti-AChE activity in comparison to the 4-substituted series. The SAR including molecular modelling studies disclosed that the presence of electron-withdrawing groups such as fluoro- or chloro- on the appropriate position of the benzylic pendent could lead to reinforcing of π–π interaction with some aromatic residues in the PAS of AChE. Noteworthy, the pyranochromene moiety is oriented within CAS, while the benzyl moiety provides interaction with PAS residues. Such dual binding site character is in agreement with the data obtained from kinetic analysis pointing to mixed type inhibition. Based on the predicted values of BBB penetration, all compounds in the series might be able to permeate into the CNS. Moreover, calculated LC50 values (50% of lethal concentration) indicated that these compounds might show neither acute toxicity nor mutagenic effect, the latter according to AMES test data. Finally, all compounds fulfilled the Lipinski criteria of drug likeness. More recently other authors have extended their study into hybridising the tetrahydroaminoquinoline and coumarin scaffolds. However, this combination does not contain any structural feature related to donepezilCitation73.
Quinolone–benzylpiperidine ChEIs with high radical scavenging activities were established by Pudlo et al.Citation74 Quinolone-containing compounds exhibited a wide variety of biological activities, one of which is ROS scavenging ability and in some broad context can be considered as coumarin derivativesCitation75,Citation76. The described compounds showed mostly moderate selectivity for EeAChE over eqBChE. In vitro experiments singled out to two compounds (7: EeAChE IC50 = 0.11 µM; hAChE IC50 = 0.23 µM eqBChE IC50 = 60 µM; SI for EeAChE = 545; and 8: EeAChE IC50 = 0.48 µM; hAChE IC50 = 0.98 µM eqBChE IC50 = 10 µM; SI for EeAChE = 20; ) as being the best in term of ChE inhibition. The SAR in the series can be enlightened as follows: (i) a methylene linkage between quinolone-carboxamide and N-benzylpiperidine improved anti-AChE potency; (ii) N-substituted quinolone derivatives displayed reduced activity and solubility compared to unsubstituted compounds; (iii) a catechol moiety on the quinolone ring conferred the best radical-scavenging activity. With regard to antioxidant properties, the EC50 (DPPH assay) for compound 8 (EC50 (DPPH) = 12.2 µM) was found to be similar to that of standard quercetin and 2-fold higher than for curcumin. The docking studies demonstrated that a quinolone-attached catechol moiety is involved in interactions with AChE in a similar way to the dimethoxyindanone motif from donepezil with N-benzylpiperidine being stacked against Trp286 (hAChE) in the CAS.
Ellagitannins constitute one of the major classes of polyphenolic natural products. Their chemical structures are basically composed of a central sugar core, typically d-glucopyranose, to which are esterified gallic acid (i.e. 3,4,5-trihydroxybenzoic acid) unitsCitation77. Ellagitannins are macromolecules with no bioavailability; however, they can be fully converted in the human gastrointestinal flora to urolithins (i.e. hydroxylated 6H-benzo[c]chromen-6-one derivatives), which do not provide any AChE/BChE inhibition activityCitation78. In an attempt to develop urolithin-related compounds endowed with anti-ChE properties, rivastigmine-like and donepezil-like analogues were designed employing the 6H-benzo[c]chromen-6-one moiety present in urolithinsCitation79. Since central action is required in the treatment of AD, more saturated 7,8,9,10-tetrahydrobenzo[c]chromen-6-one derivatives related to tetrahydrocannabinol were also designed with regard to their potential to penetrate the CNS. Focused on donepezil-related analogues, three different regions were included into the SAR study: (i) the saturation level of the benzo[c]chromen-6-one moiety; (ii) the carbon chain length bridging the benzo[c]chromen-6-one system to the amine group; (iii) the various amine substitutions. All members in this category displayed selective pattern behaviour for AChE, but were far behind donepezil or galantamine. The most active derivative also displayed comparable activity to that of donepezil and rivastigmine in the scopolamine induced passive avoidance test (9: hAChE IC50 = 0.8 µM; hBChE IC50 = 3.7 µM; SI for AChE = 4.6; scopolamine antagonism = 85%; ).
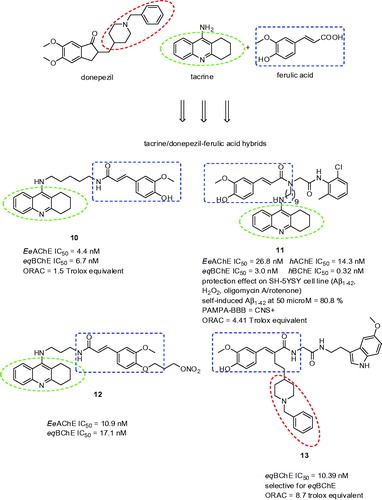
3.2. Ferulic acid and curcumin hybrids
Ferulic acid is a natural phenolic acid possessing potent anti-oxidative and anti-inflammatory activitiesCitation80. With these precedents in mind, ferulic acid has become largely exploited scaffold for design of novel multipotent compounds like tacrine-ferulic acid hybrids with potential implication for AD treatmentCitation81–83. Examples of tacrine-ferulic acid hybrids are shown in (compounds 10, 11, and 12). Inspired by these studies, twelve novel donepezil-ferulic acid hybrids were prepared as potent non-selective EeAChE and eqBChE inhibitors using one-pot Ugi reactionCitation84. In general, all the compounds exerted balanced inhibition profile being less active EeAChE inhibitors and more potent eqBChE inhibitors related to donepezil. In addition, these hybrids displayed stronger antioxidant power than the reference compounds ferulic acid and melatonin. Based on the preliminary results, the study highlighted eqBChE selective derivative 13 (eqBChE IC50 = 10.39 nM; ORAC = 8.7 Trolox equivalent; ).
Figure 3. Ferulic acid hybrids possessing antioxidant and anticholinesteratic properties.
Ferulic acid-O-alkylamines can be classified as MTDLs combining ferulic acid with N-benzylpiperidine fragment into single moleculeCitation85. The pharmacological assessment of this subset consisted of screening inhibition against EeAChE, eqBChE, where some of the compounds were reevaluated on ratAChE, ratBChE, hAChE, and hBChE. Next, the Aβ42 self-aggregation and disaggregation tests, antioxidant properties assessment, cytotoxicity, cell protective effects on H2O2 induced PC12 cell injury, prediction of BBB permeation were performed for the selected compound 14 (EeAChE IC50 = 2.1 µM; eqBChE IC50 = 21 nM; SI for BChE = 101; ratAChE IC50 = 1.8 µM; ratBChE IC50 = 8.6 µM; hAChE IC50 = 3.8 µM; hBChE IC50 = 70 nM; inhibition of Aβ42 at 25 µM = 50%; disaggregation Aβ42 at 50 µM = 38%; ORAC = 0.55 Trolox equivalent, ) that was chosen based upon its anti-cholinergic properties. Moreover, the study was supplemented by in vivo data counting acute toxicity and the step-down passive avoidance tests. The latter was aimed to observe whether 14 can improve the contextual memory in scopolamine-induced mice. Indeed, 14 exerted improvement in cognitive decline when compared with the control group.
Figure 4. Curcumin-based hybrids with antioxidant properties.
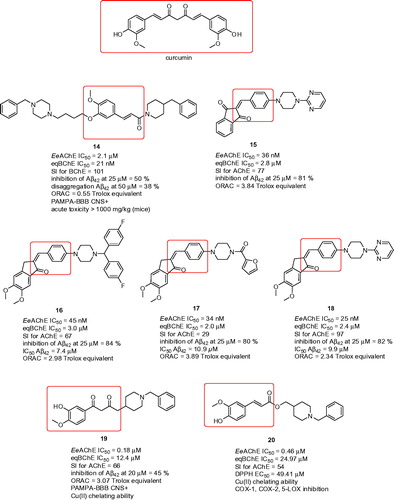
Curcumin () is another compound of natural origin derived from Curcuma longa L. (Zingiberaceae)Citation86. Its chemical structure can be regarded as biosynthetic dimer related to phenylalanine and cinnamic acid, respectivelyCitation87. In traditional medicine, dried turmeric root was a remedy in several pathological conditions, among others skin diseases, wounds, rheumatisms, asthma, allergies, sinusitis, hepatic disorders, intestinal worms, and generic inflammationCitation88. The neuroprotective potential of curcumin or mixture of curcumin derivatives, denoted as curcuminoids, has been confirmed not only in in vitro but also in vivo by attenuating inflammation and microglia activation in AD mouse modelCitation89. Curcumin have attracted researchers attention since it has been found to exert radical scavenging and iron chelation properties in brain tissue homogenateCitation90. Apart from that, it displayed inhibition ability of AChE, protection in PC12 cell line against Aβ-induced damage and also direct inhibition of Aβ42 fibrillogenesisCitation91,Citation92. All these protective characteristic either of curcumin or curcuminoids are amplified via its anti-inflammatory action and downregulation of Aβ production through BACE1 expressionCitation93,Citation94. Furthermore, curcumin can also modulate the phosphorylation of tau protein impeding the formation of neurofibrillary tangles (NFTs)Citation95.
Taken the aforementioned precedents together, curcumin have become an important building block in designing novel compounds with potential implication in neurodegenerative diseases. One such work rationally designed a series of 2-(4-(4-substituted piperazin-1-yl)benzylidene)-1H-indene-1,3(2H)-diones comprising indanone moiety, curcumin fragment and piperazine six-membered ring into one moleculeCitation96. Multipotent profile of these compounds was confirmed in several experiments. Indeed, novel hybrids displayed micromolar to two-digit nanomolar inhibition potency against EeAChE and one-digit micromolar to sub-micromolar eqBChE inhibition ability. Accordingly, the hybrids resulted in preferential AChE inhibition over BChE. With regard to AChE affinity, phenyl ring substitution favoured para substituted electron-withdrawing groups (EWGs) such as trifluoromethyl group over unsubstituted, electron-donating groups (EDGs) or disubstituted ones. Improvement in anti-cholinestrease activity was reached by replacing phenyl moiety with other heterocycles. In this context, remarkable activity was demonstrated by pyrimidine derivative 15 (EeAChE IC50 = 36 nM; eqBChE IC50 = 2.8 µM; SI for AChE = 77; inhibition of Aβ42 at 25 µM = 81%; ORAC = 3.84 Trolox equivalent; ) being slightly more potent than donepezil under experimental conditions. Kinetic experiments indicated mixed-type pattern inhibition of 15. Heterocyclic ring attachment to piperazine moiety also significantly improved inhibition of Aβ42 self-aggregation. The latter was confirmed by Thioflavin–T (ThT) fluorescence assay as well as by transmission electron microscopy either through direct interaction or by blocking the PAS of AChE. Antioxidant capacity, established by ORAC-FL and H2O2-induced SH-SY5Y cell-based stress assays, ranged from 0.55 to 3.52 of Trolox value throughout the series. This is consistent with the design where amalgamating curcumin scaffold into novel hybrids broadens the compounds profile. Neuroprotective profile was also elucidated using the model of H2O2- or Aβ-induced neurotoxicity.
Very close strategy was applied in another family combining piperazine, 5,6-dimethoxy-2,3-dihydro-1H-inden-1-one moiety and curcumin fragmentCitation97. The preparation exploited Knoevenagel condensation to form novel (E)-2-(4-(4-(substitued)piperazin-1-yl)benzylidene)-5,6-dimethoxy-2,3-dihydro-1H-inden-1-one derivatives. Given the structural similarity to previous subset, the inhibition potency also ranged from micromolar to low-nanomolar and micromolar to low-micromolar scales against EeAChE and eqBChE, respectively. The structure–activity relationship (SAR) analysis disclosed that activity is more pronounced for para-substituted phenyl ring even by introduction of electron rich moieties like methyl, 2,4-dimethyl or 4-methoxy groups. The introduction of linker between phenyl ring and adjacent piperazine had detrimental effect on ChE affinity. Again, phenyl replacement by heterocycles like furoyl, pyridine, or pyrimidine yielded in notable activity enhancement. Based on these results, three compounds emerged as the new leads with comparable inhibition ability to donepezil (16: EeAChE IC50 = 45 nM; eqBChE IC50 = 3.0 µM; SI for AChE = 67; inhibition of Aβ42 at 25 µM = 84%; Aβ42 IC50 = 7.4 µM; ORAC = 2.98 Trolox equivalent; 17: EeAChE IC50 = 34 nM; eqBChE IC50 = 2.0 µM; SI for AChE = 29; inhibition of Aβ42 at 25 µM = 80%; Aβ42 IC50 = 10.9 µM; ORAC = 3.89 Trolox equivalent; 18: EeAChE IC50 = 25 nM; eqBChE IC50 = 2.4 µM; SI for AChE = 97; inhibition of Aβ42 at 25 µM = 82%; Aβ42 IC50 = 9.9 µM; ORAC = 2.34 Trolox equivalent ).
Fusing β-diketonate-phenyl substituted analogs with N-benzylpiperidine provided novel class of MTDLs targeting EeAChE, eqBChE, metal imbalance, oxidative stress pathway and Aβ42 self-aggregation processCitation98. N-Benzylpiperidine attachment was conjugated either via the α-carbon between two ketone groups or via terminal carbon of curcumin residue appendage. The length of the tether between basic scaffolds played crucial role, the highest inhibitory potency was coined to two methylene units. 3-Hydroxy-4-methoxy disubstitution of curcumin phenyl ring emerged as the most favourable in terms of prevailing high AChE inhibition and escalated selectivity. Antioxidant activity was associated with free phenolic group in phenyl of curcumin skeleton. The most potent hybrid 19 (EeAChE IC50 = 0.18 µM; eqBChE IC50 = 12.4 µM; SI for AChE = 66; inhibition of Aβ42 at 20 µM = 45%; ORAC = 3.07 Trolox equivalent; ) under the study also revealed the highest inhibition rate of Aβ42 self-aggregation by 45% at 20 µM. In concept with the design of these templates, kinetic characterisation of 19 revealed dual binding site character. This data was consistent with in silico experiments, however proposing that N-benzylpiperidine moiety of donepezil is oriented outwards the active gorge giving interaction with Trp279 from the PAS. To supplement therapeutic profile, the metal-chelating properties of 19 towards selected biometals like Cu(II), Fe (II), and Zn (II), were inspected. The latter might prevent from the slow accumulation of Aβ42 and thus impede its precipitation and crosslinkingCitation99. Indeed, compound 19 displayed 1:1 stoichiometry with Cu(II) thus could provide positive outcome likewise clioquinol or PBT2Citation100,Citation101.
Feruloyl-donepezil hybrids were developed as another example of novel and potent MTDLsCitation102. Variation of the substituent groups in the aromatic region of N-benzylpiperidine derived from donepezil as well as aromatic part of feruloyl were explored in the effort to identify novel drug candidate. Unsubstituted N-benzylpiperidine derivatives showed sub-micromolar EeAChE inhibition activity and significant selectivity over eqBChE (SI >50). In this regard, the study highlighted the hybrid 20 (EeAChE IC50 = 0.46 µM; eqBChE IC50 = 24.97 µM; SI for AChE = 54; DPPH EC50 = 49.41 µM; ) being non-competitive AChE inhibitor with a Ki value of 1.04 µM. Almost all of the derivatives were effective in scavenging free radicals, however, not reaching the activity of ferulic acid itself as parent compound. This results might be sufficiently explained by analogy with the effect of para-hydroxy substituted cinnamic acids, in which the oxygen atom of the hydroxyl group is able to share a positive charge and thereby increase radical stabilisation through conjugation extensionCitation103. To counteract the intracellular ROS formation induced by H2O2 in SH-SY5Y cells was measured for 20 suggesting also its high effectiveness in the cellular model. Moreover, 20 selectively chelated Cu(II) and Fe(II) but not Fe(III) and Zn(II) thus broadening its biological profile. Neuroprotective profile of 20 was also proved when neurotoxicity was elicited by Aβ42 oligomers using SH-SY5Y cells. Given the fact that curcumin mediates its anti-inflammatory effects, among others, via inhibition of cyclooxygenase 1 (COX-1), cyclooxygenase 2 (COX-2), and 5-lypoxygenase (5-LOX) authors were also encouraged to see the effect of 20 towards these enzymes. Indeed, 20 decreased the activity of COX-1, COX-2, and 5-LOX by 40, 27, and 30%, respectively, measured via immunosorbent assay in mice serum. To confirm the latter in vivo, the carrageenan-induced paw oedema assay was conducted pointing out to the same mechanism of action as indomethacin.
3.3. Selenium derivatives with antioxidant properties
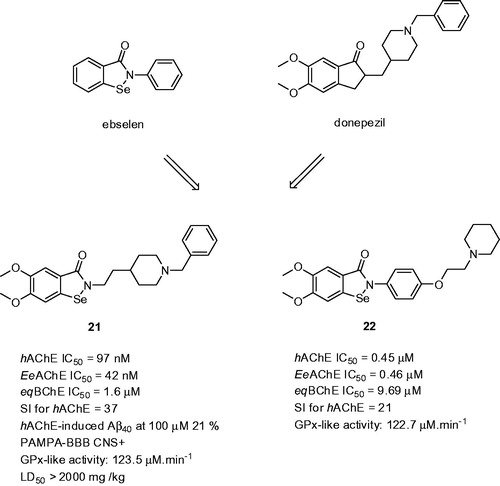
Selenium (Se) is an essential trace mineral nutrient with multiple roles in the growth and function of living animal cells, also providing protection against free radical-induced cell damageCitation104. Several studies have indicated that not only the Se levels decrease with age, which may be in contrast with in the progression of ADCitation105. Ebselen [2-phenyl-1,2-benzisoselenazol-3(2H)-one; is a lipid-soluble derivative mimicking glutathione peroxidase in the way that it is able to protect cells by catalysing the reduction of peroxides by glutathioneCitation106. Moreover, ebselen exerts anti-inflammatory activity and is able to inhibit iron-induced tau phosphorylation, both actions required for AD treatmentCitation107. Fusion strategy of ebselen and donepezil led to the discovery of so-called “selenpezil” compounds ()Citation108. SAR analysis revealed importance of the methoxy groups in the benzoselenazol-3(2H)-one moiety for inhibition activity. The length of the linker between the benzoselenazol-3(2H)-one moiety and piperidine ring is also important for maintaining activity, with a two-carbon spacer exhibiting the highest inhibition potency. Several biological assays highlighted compound 21, which was able to inhibit AChE-induced Aβ40 aggregation (21: hAChE IC50 = 97 nM; EeAChE IC50 = 42 nM; eqBChE IC50 = 1.6 µM; SI for AChE = 37; hAChE-induced Aβ40 at 100 µM 21%; ). Compound 21 exerted glutathione peroxidase-like activity, inferring the compound’s detoxification of hydroperoxides. Antioxidant properties were further confirmed indicating its scavenging ability for H2O2 and peroxynitrite removal. Based on PAMPA-BBB assay, the compound could be considered as centrally active. Further in vivo results with mice showed no acute toxicity or mortality, even after administration of high doses (2000 mg/kg of 21). Other glutathione-peroxidase (GPx) mimics based on ebselen were reported more recentlyCitation109. As shown in for the most active derivative 22, the substitution at C-4 of the ebselen phenyl ring was carried out with attachment of different tertiary amines (pyrrolidine, piperidine, and N,N-dimethyl-1-phenylmethanamine) through an ether-methylene chain or methylene chain of variable length. Compound 22 possesses good inhibitory potency against ChE (hAChE IC50 = 0.45 µM; EeAChE IC50 = 0.46 µM; eqBChE IC50 = 9.7 µM; SI for AChE = 21; ) and antioxidant activity against H2O2 in vitro.
Figure 5. “Selenpezil” derivatives 21 and 22 based on fused ebselen with donepezil.
3.4. Donepezil-like analogues with altered indane motif
New pyridinylmethylene-indanone derivatives endowed with metal-chelating and anti-cholinesterase properties were recently developed by the Meng groupCitation110. Different amines (piperidinyl, pyrrolidinyl, or N,N-diethylamine) were introduced into the C-6 position of the indanone moiety attached by a polymethylene bridge of variable size. The study highlighted the analogues with a piperidinyl substituent and a short spacer (two-carbon linker) as the most active ones. With regard to the aurone scaffold, a double bond was crucial for preservation of high affinity to EeAChE. Pyridinylmethylene-indanone derivatives exerted rather moderate potency against eqBChE, thus being very selective for AChE. Amongst them, compound 23 exhibited the highest AChE inhibitory activity, being 47-fold more potent than tacrine and 14-fold more active than donepezil (EeAChE IC50 = 1.8 nM; eqBChE IC50 = 9.5 µM; SI for AChE = 5250; ). Kinetic studies indicated a mixed type of inhibition. Moreover, 23 showed chelation ability towards biometals such as Cu(II), Fe(III) and Zn(II), suggesting its multifunctionality in AD treatment. In pursuit of promising results with aurone derivatives, the Nadri group prepared and evaluated a novel series of pyridinium analogs maintaining the planar conformation of the aurone ringCitation66. The SAR indicated that introduction of a fluorine atom at position C-2 or C-4 of the benzyl fragment led to increased anti-AChE activity, which is in agreement with data previously reported. Furthermore, it was observed that a methoxy group at C-6 of the benzofuranone moiety delivered higher activity than the ethoxy and propoxy analogues. In this subset, compound 24 was highlighted as the most potent against EeAChE (EeAChE IC50 = 10 nM; ). Subsequently this pyridinium series was enlarged by using the more polar 5,6-dimethoxybenzofuranone scaffold, producing results very similar to those of 24Citation111. As anticipated, compound 25 bearing a fluorine atom at position C-2 of the benzyl moiety showed the highest anti-AChE potency, being less active than its 6-methoxy counterpart 24 (25: EeAChE IC50 = 52 nM; eqBChE IC50 = 1620 nM; SI for AChE = 31; ). Inspired by these findings, Shafiee and co-workers designed and synthesised indoline-based AChEIs, also bearing the benzylpyridinium moietyCitation112. Screening assay revealed very potent inhibitory activities (EeAChE IC50 = 0.44–12.8 nM) exceeding the standard donepezil. Of these, the 2-chorobenzyl derivative (26: EeAChE IC50 = 0.44 nM; eqBChE IC50 = 1370 nM; SI for AChE = 3113; ) became the most potent compound against AChE. The SAR disclosed that substitution of the C-2 or C-3 position of the N-benzyl pendent by a halogen, methyl or methoxy group significantly improved the anti-AChE activity, while substitution at C-4 of the N-benzyl group diminished the activity. The observed IC50 values for eqBChE displayed that almost all compounds in the series exceeded the activity of donepezil towards this enzyme. Likewise, substitution of the N-benzyl moiety proved to have a positive effect on anti-BChE activity. The docking studies indicated three types of interactions involved in the attachment of ligand to TcAChE, such as hydrophobic interaction, π–π interaction and cation–π interaction. The latter is responsible for interaction between the charged nitrogen of 26 and the mid-gorge site comprising Phe330, thus facilitating ligand recognition. Kinetic analysis performed on 26 was in agreement with the proposed binding mode of AChE displaying mixed-type inhibition (Ki = 1.14 nM). The prediction of ADMET analysis using web-based application suggested a centrally active character for these compounds, low acute toxicity according to the calculated LC50 values, and no mutagenic effect according to the AMES testCitation113. Another survey focused on benzofuran-derived N-benzylpyridinium bromides highlighted derivative 27 as being 7-fold more potent than donepezil (27: EeAChE IC50 = 4.1 nM; )Citation114. Docking studies with 27 revealed that the N-benzylpyridinium part of 27 was situated around Trp84, in the vicinity of the catalytic site. The positively charged nitrogen contributes in formation of a π–cation interaction with aromatic residues (Phe330 and Tyr334) at the mid-gorge recognition site.
Figure 6. Indane-based ChEIs 23–32.
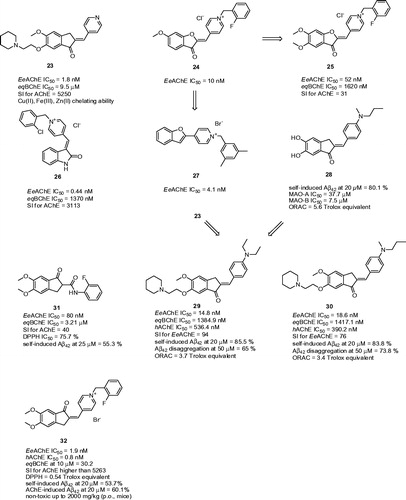
Benzylideneindanone derivatives showed an interesting and broad biological profile potentially applicable for AD treatmentCitation115. In particular, compound 28 () gave the greatest inhibitory potency toward self-induced Aβ42 aggregation (80.1% at 20 µM). Moreover, 28 was an excellent antioxidant (ORAC = 5.60 Trolox equivalent) and monoamine oxidase (MAO)-A/B inhibitor (MAO-A IC50 = 37.7 µM; MAO-B IC50 = 7.5 µM). 28 was also found to be a good biometal chelator; it inhibited Cu(II)-induced Aβ aggregation and it could also disassemble well-structured Aβ fibrils. Another series of indanone derivatives combining the excellent AChE inhibitory profile of 23 with the anti-Aβ aggregation properties of 28 were further developedCitation116. Indeed, the novel hybrids were potent inhibitors of EeAChE with IC50 values in the nanomolar range. It is also important to note that the compounds showed weaker inhibition activities against hAChE and they also demonstrated slightly higher inhibitory potency against eqBChE than donepezil. The two most active compounds 29 (EeAChE IC50 = 14.8 nM; eqBChE IC50 = 1384.9 nM; hAChE IC50 = 536.4 nM; SI for EeAChE = 94; ) and 30 (EeAChE IC50 = 18.6 nM; eqBChE IC50 = 1417.1 nM; hAChE IC50 = 390.2 nM; SI for EeAChE = 76; ) were used for other biological investigation. Kinetic studies with 29 indicated a mixed type inhibition, and hence indanone members could simultaneously target PAS and CAS of AChE. All the derivatives demonstrated excellent antioxidant activity with ORAC values of 1.67–3.88 Trolox equivalents. Compared to curcumin, both compounds exerted markedly higher inhibitory activity in self-mediated Aβ42 aggregation assay (at 20 µM 52.1%, 85.5%, and 83.8% for curcumin, 29 and 30, respectively). The number of Aβ fibrils was significantly decreased after their incubation with 29 and 30 (at 50 µM 65.0% and 73.8% disaggregation rates for 29 and 30, respectively). Based on PAMPA-BBB assay, it can be predicted that 29 and 30 can permeate BBB.
Modified donepezil analogues bearing a secondary aromatic amide moiety displayed interesting pharmacological profile including inhibitory effects on ChEs and Aβ42 aggregation with antioxidant and metal chelating abilitiesCitation117. 5,6-Dimethoxy-indanone ring from donepezil was maintained attaching ortho-, meta-, or para-substituted secondary aromatic amines via carbonyl linker. All the compounds revealed selective AChE inhibition potency ranging from 0.08 to 0.92 µM concentrations observing that higher activity is bound to EWG-containing substituents on aromatic moiety. The latter is consistent with previous studiesCitation118,Citation119. The highest inhibition activity reached p-fluoro substituted benzamide 31 being mixed-type AChE inhibitor (31: EeAChE IC50 = 80 nM; eqBChE IC50 = 3.21 µM; SI for AChE = 40; ). All the meta- and para-substituted compounds conferred more positive contribution toward BChE inhibition than ortho-substituted compounds. When incubated with Aβ42, 31 displayed 55.3% propensity to inhibit Aβ self-aggregation at 25 µM. In general, all the hybrids in the subset demonstrated both moderate antioxidant properties and ability to complex Zn(II). All these features are presumably coined to the presence of beta-keto-amide moiety in the structure.
N-Benzylpyridinium with 5,6-dimethoxy-indanone were constructed primarily targeting ChEs, oxidative stress and Aβ pathological pathwaysCitation120. The similar pattern for AChE inhibition was observed for 31 as well as for 32 bearing 2-fluoro substituent. The inhibitory activity against AChE was 21-fold higher than for donepezil with the negligible BChE inhibitory activity and significant AChE selectivity (32: EeAChE IC50 = 1.9 nM; hAChE IC50 = 0.8 nM; eqBChE at 10 µM = 30.2%; SI for EeAChE >5263, ). In the kinetic study, 32 was found to be mixed-type AChE inhibitor. 32 is potentially able to counteract Aβ pathological pathway via two distinct mechanisms: (i) by direct interaction with Aβ42 (53.7% inhibition at 20 µM) and (ii) by interaction with PAS of AChE to decrease the AChE-accelerated Aβ aggregation (60.1% inhibition at 20 µM). In general, all the pyridinium salts were moderate antioxidants. With this respect, 32 yielded as the most potent free radical scavenger possessing DPPH value 0.54 of Trolox equivalent. Moreover, 32 increased cell viability of PC12 cell line in the presence of both Aβ and H2O2 insults indicating its neuroprotective profile. Finally, 32 can be classified as centrally active based on the observations from the PAMPA-BBB assay. No acute toxicity was observed when orally administered to mice at doses up to 2000 mg/kg.
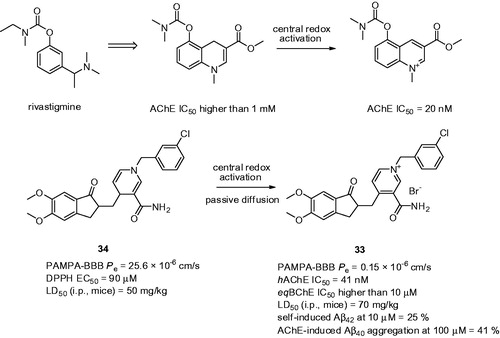
Bio-oxidizable prodrugs represents interesting concept to relieve the potential side effects. The first-generation of bio-oxidizable AChEI masking the positive charge in rivastigmine was developed intending to transport the pro-drug to the brain avoiding the compounds peripheral side effects. Indeed, the proof-of-concept further revealed the central redox-activation presumably mediated via NADH dehydrogenase resulting into exclusive central cholinergic activation (AChEperipheral IC50 > 1 mM; AChEcentral IC50 = 20 nM; )Citation121. These results encouraged researchers to apply this approach on donepezil templateCitation122. Accordingly, piperidine moiety was replaced by a 1,4-dihydropyridine ring containing EWG at C-3 position that constitutes a key element/stabilizer in the design of the pro-drug. Importantly, redox-activation step mediated by oxidoreductases does not only convert the pro-drug to pyridinium drug but also entails a “locked-in” effect in the brain. By that it prevents peripherals cholinergic side effects while enabling prolonged duration of AChEI in brain tissue. The majority of the pyridinium salts exhibited potent, hAChE nanomolar scale inhibition potency in the same range as donepezil and low inhibitory activity against eqBChE. Compound 33 stands out the most interesting from the subset (33: hAChE IC50 = 41 nM; eqBChE IC50 > 10 µM; ) showing AChE dual-binding site character. Multipotent profile of 33 was corroborated by inhibition of AChE-induced Aβ40 aggregation (41% at 100 µM) and inhibition of self-induced Aβ42 aggregation (25% at 10 µM). High permeability value (Pe) of pro-drug 34 over 33 underlined the lipophilic character of 34 to be eligible to permeate through BBB (Pe for 34 = 25.6 × 10−6 cm/s; Pe for 33 = 0.15 × 10−6 cm/s; high BBB permeation is predicted when Pe value >4.0 × 10−6 cm s−1, low BBB permeation equals Pe < 2.0 × 10−6 cm s−1 and uncertain BBB permeation lies in between Pe value ranging from 2.0 × 10−6 cm s−1 to 4.0 × 10−6 cm s−1)Citation123,Citation124. The incubation of 34 in 10% fresh mice brain homogenate led to oxidation product 33 in 35% yield after 180 min. Moreover, 34 exhibited a noticeable radical scavenging activity (DPPH EC50 = 90 µM). Under these conditions, 34 is converted into 33 as the main oxidation product which forms robust antioxidant system. Neither 34 nor 33 displayed significant genotoxicity. In vivo toxicity evaluation determined LD50 values in mice for 34, 33, and donepezil as follows – 50, 70, and 22.5 mg/kg, respectively. The repeated administration of 34 at 10 mg/kg did not cause any macroscopic and microscopic tissue alterations thus presuming its relative safety.
Figure 7. Bio-oxidizable pro-drugs 34 forming charged entities after oxidative activation 33. The proof-of-concept was firstly validated on rivastigmine-like analogue – the upper part of the Figure.
3.5. Melatonin hybrids
Melatonin (N-acetyl-5-methoxytryptamine, ) is a ubiquitous compound responsible not only for the regulation of circadian rhythm but also for a wide range of other biological activities. This endogenous molecule is able to directly scavenge reactive oxygen and nitrogen species and furthermore, it stimulates the activity of antioxidant enzymes and their expressionCitation125. Moreover, some studies indicated that melatonin can attenuate tau hyperphosphorylationCitation126, reduce the burden of Aβ loadCitation127, and preclude kainic acid-induced microglial and astroglial responses thus having anti-inflammatory potentialCitation128. Decreased melatonin in serum and cerebrospinal fluid (CSF) and the loss of melatonin diurnal rhythm are frequently observed in AD patientsCitation129. Due to the aforementioned benefits, low toxicity and no significant side effects, melatonin is largely used as a scaffold for design of various multifunctional compounds, a number of them being melatonin-donepezil hybridsCitation130.
Figure 8. The MTDLs combining melatonin and donepezil templates.
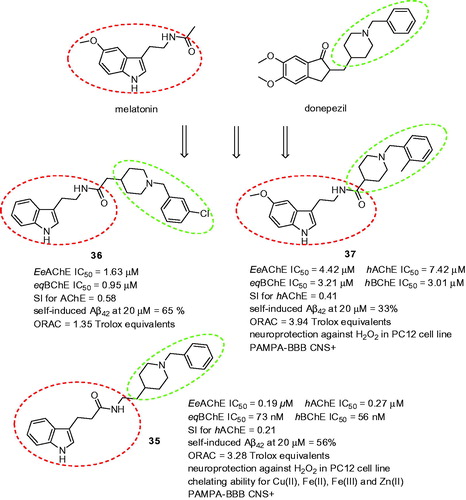
The design of three series of multifunction compounds combining donepezil and melatonin has been reportedCitation131. Although the design approach was rather simple it produced potent hybrids with a plethora of biological activities. The lead compounds consisted of substituted or unsubstituted N-benzyl piperidine and indole fragments joined via a carboxamide linker. All of the synthesised derivatives showed good inhibitory activities against EeAChE and eqBChE and their human equivalents, the most potent being 35 (EeAChE IC50 = 193 nM; eqBChE IC50 = 73 nM; hAChE IC50 = 273 nM; hBChE IC50 = 56 nM, SI for hAChE = 0.21; ). Kinetic study performed with this ligand indicated a mixed-type inhibition mode. Almost all the compounds exhibited higher activity for BChE than for AChE, thus being more selective BChEIs. Most of the hybrids also displayed moderate to good (33–65%) inhibition potencies for Aβ42 self-induced aggregation at 20 µM. Within this context, the most potent inhibitor throughout the subset was 36 (). Not surprisingly, good antioxidant properties were achieved with ORAC values ranging between 0.82 and 3.94-fold of Trolox equivalents. The most active with this respect was 37 (3.94-fold of Trolox value; ) bearing 5-methoxy group at indole ring and 2-methyl group at benzene ring. The capacity to protect the rat pheochromocytoma cell line PC12 cells against oxidative stress-associated death induced by H2O2 was tested on selected derivatives. All of the compounds exhibited neuroprotective effects at concentrations ranging from 1.25 to 10 µM. Derivative 35 revealed almost the same capacity as Trolox at 10 µM and much better than melatonin. Moreover, 35 demonstrated good biometal-chelating ability towards Cu(II), Fe(II), Fe(III), and Zn(II). Amide bond was designated as potential metal chelating unit in the molecule.
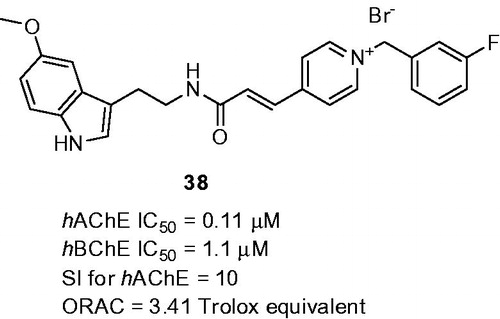
The rational combination of melatonin and N-benzylpyridinium bromides as multipotent compounds with enhanced solubility profile was reportedCitation132. The basic indole core was either substituted with 5-mehoxy group or remained unsubstituted. Interestingly, in both cases, these structural templates preserved remarkable antioxidant properties ranging from 1.14 to 3.66 Trolox equivalents. All the compounds also showed ability to inhibit both human ChEs with preferential inhibition of hAChE over hBChE. However, the most active analogue in this subset, melatonin-N-benzylpyridinium bromide hybrid 38 (hAChE IC50 = 0.11 µM; hBChE IC50 = 1.1 nM; SI for AChE = 10; ORAC = 3.41 Trolox equivalent; ) exerted even 10-fold decreased lower potency to inhibit hAChE than donepezil. Further assessments of 38 confirmed its dual binding site character of hAChE (mixed-type pattern inhibition, docking studies) and its ability to protect SH-SY5Y cells against oxidative stress induced by H2O2 in vitro. Interestingly, 38 revealed opposite arrangement in the enzyme cavity compared to donepezil which might be responsible for its lower activity.
Figure 9. Melatonin-N-benzylpyridinium bromide hybrid 38.
3.6. 8-Hydroxyquinoline derivatives
The combination of donepezil moieties, i.e. N-benzylpiperidine or its isoster N-benzylpiperazine, with metal chelating core from clioquinol/PBT2 (), i.e. 8-hydroxyquinoline (8HQ), resulted in novel multipotent compoundsCitation133. PBT2 and cliquinol are well established neuroprotective agents with potential therapeutic implication in AD, Parkinson’s disease or Huntigton’s diseaseCitation134,Citation135. Authors of the study investigated the effect on cholinesterase activity of EWGs as well as EDGs in different position of benzylic ring of donepezil. Interestingly, initial screening assay revealed that most of these 8-hydroxyquinoline derivatives were hBChE selective showing from 49.2% to 89.1% inhibitory potency at 40 µM compounds concentration. Among these hybrids, compounds bearing any substituent at 2-position of benzylic fragment revealed the highest hBChE inhibitory activity. Replacing the piperazine nucleus by 4-aminopiperidine one showed drastic effect in term of the hAChE affinity enhancement for this enzyme. The authors of the study speculated that this effect might be explained by higher degree of flexibility. The introduction of 8HQ core, mimicking the dimethoxyindanone moiety from donepezil, embedded compounds with neuroprotective properties. In this regard, the authors evaluated Aβ anti-aggregating properties of these compounds reaching moderate to high inhibition potencies (Aβ42 = 19.1–65.0% at 50 µM), the highest value was quite close to well-known anti-aggregating compound curcumin (73.7%)Citation136. The ability of complexing Cu(II) and Zn(II) was also investigated by UV–Vis difference spectroscopy. The results of this assay for selected compounds showed that these could effectively complex metal ions in physiological conditions, i.e. phosphate buffer, pH = 7.4. Additionally, all the compounds displayed antioxidant ability, some of them even increasing the capability of Trolox in scavenging free radicals. Finally, PAMPA-BBB predicted their potential to reach the therapeutic targets in CNS. In the subset, 8HQ analogue 39 showed balanced multipotent profile (hBChE IC50 = 5.71 µM; PAMPA-BBB CNS+; ).
Figure 10. The most potent analogues 39–42 related to donepezil-8HQ hybrids.
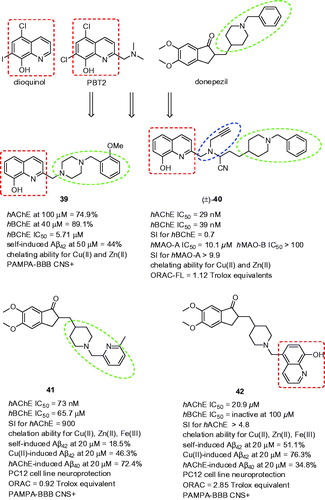
Another series of 8HQ hybrids bearing N-benzylpiperidine was enriched with propargylamine fragment to extend the range of biological activities of MAOs-inhibitionCitation137. This combination led to the development of derivatives with inhibition potency against ChEs and MAOs, metal-chelating capacity and antioxidant properties. Compound with most interesting profile racemic analog (±)-40 was selected as the drug-lead candidate being hAChE/hBChE nanomolar inhibitor (hAChE IC50 = 29 nM, hBChE IC50 = 39 nM; ). Moreover, it also showed to be a selective MAO-A inhibitor in comparison to MAO-B (hMAO-A IC50 = 10.1 µM, hMAO-B IC50 > 100 µM). The latter was expected based upon the N-propargyl moiety attachmentCitation138. The kinetic studies showed mix-type inhibition for both ChEs and irreversible MAO-A inhibition. Derivative (±)-40 was also found as metal-chelating agent for Cu(II) and Zn(II). The antioxidant properties of (±)-40 was carried out by different in vitro assays, the most of them showing positive result.
Hybridisation of phenyl ring of N-benzylpiperidine moiety by pyridine, quinolone, or 8HQ yielded into novel multipotent donepezil analogsCitation139. Mostly, the hybrids exerted prevailing AChE inhibitory selective profile not making the differences, whether small or bulkier substituents were introduced. The most active compound in term of the highest inhibition potency bore 2-fluoro-benzyl moiety (EeAChE IC50 = 43 nM, hAChE IC50 = 32 nM; ) being more active than template donepezil. Phenyl replacement by pyridylmethyl group in compound 41 (EeAChE IC50 = 85 nM, hAChE IC50 = 73 nM; EqBChE IC50 = 20.5 µM; hBChE IC50 = 65.7 µM; SI for hAChE = 900, ) retained high anti-AChE activity, however, not overwhelming the potency of donepezil. Attaching 8HQ to piperidine moiety had detrimental effect for both AChE and BChE inhibition. Obviously, 41 acted as mixed-type inhibitor of hAChE which was also in agreement with the docking protocol. Indanone moiety preferably occupied PAS of hAChE via plausible π–π interaction with Trp286 whereas pyridylmethyl-substituted piperidine was anchored to CAS mainly via cation–π interaction to Trp84. The fluorescence emission spectra indicated that 41 is able to complex with Cu(II), Zn(II), and Fe(III). For Cu(II)-41, 1:1 stoichiometry was found. When tested at 20 µM, 41 displayed 18.5, 46.3, and 72.4% inhibition of Aβ42 self-induced aggregation, Cu(II)-induced Aβ42 aggregation and hAChE-induced Aβ40 aggregation, respectively. Note that the remarkable anti-amyloid profile also achieved the hybrid bearing 8HQ fragment 42 (51.1%, 76.3%, and 34.8% inhibition of Aβ42 self-induced aggregation, Cu(II)-induced Aβ42 aggregation, and hAChE-induced Aβ40 aggregation, respectively, ). The crosstalk between Aβ and 41 and 42 was also corroborated by the data from cell viability and neuroprotection against Aβ42-induced and Cu(II)-Aβ42-induced toxicity assays indicating that these hybrids possess neuroprotection by improving cell viability in PC12 cell line. Based on the structure aspects, 42 displayed more potent antioxidant scavenging properties than 41 (ORAC-FL = 0.92 and 2.85 Trolox equivalent for 41 and 42, respectively). Before in vivo testing, compounds were tested for their potential permeation through BBB using PAMPA-BBB assay. As expected, 41 and 42 showed satisfied potential for BBB penetration. Mice treated with 41 (10 mg/kg) demonstrated comparable results to that of donepezil administered at 5 mg/kg to reverse scopolamine-induced memory deficit in the step-through passive avoidance test. No alterations were observed in the levels of AST and ALT suggesting 41 as a safe compound.
3.7. Donepezil-based derivatives of glutamic acid
An interesting approach to novel multifunctional donepezil-based derivatives for the treatment of AD was applied by using glutamic acid as a suitable biocompatible linkerCitation140,Citation141. This α-amino acid was chosen because of the appropriate distance between α-NH and γ-COOH groups that allows simultaneous interaction for the pharmacophoric groups within the CAS and PAS of AChE. The three fragments were coined with l-glutamic acid in the first family of these derivatives exploiting: (i) N-benzylpiperidine moiety from donepezil chosen to inhibit CAS of AChE; (ii) N-protecting group able to interact with PAS of AChE along conferring neuroprotection against oxidative stress; and (iii) a lipophilic alkyl ester that would facilitate penetration into the CNS through BBB ()Citation140. All the prepared compounds showed good inhibition of the hAChE with IC50 values in the sub-micromolar range (0.10–0.53 µM). They also inhibited hBChE with IC50 in micromolar scale. The majority of the measured analogues showed significant ability to displace the propidium cation from the PAS thus suggesting its ability to inhibit Aβ aggregation promoted by AChE. The in vitro PAMPA-BBB assay showed Pe values of measured glutamic acid derivatives over the CNS + limit presuming their passive permeation. The authors also studied cell viability and neuroprotective effects of synthetised derivatives against death induced in human neuroblastoma cell line SH-SY5Y by various toxic insults related to oxidative stress triggered by H2O2 and the mixture of rotenone and oligomycin A (R/O). According to the results all tested compounds protected cells from damage induced by H2O2 and were able to protect neuroblastoma cells against both exogenous and mitochondrial ROSCitation140. Compound 43 () was selected for further pharmacological evaluation. It displayed voltage-gated calcium channel blockadeCitation142 infarct volume reduction in a photothrombotic stroke-model in mice and neuroprotection against sodium and calcium overload in motor neuron-like NSC-34 cells, as a model of amyotrophic lateral sclerosisCitation143,Citation144.
Figure 11. Donepezil-based derivatives with glutamic acid.
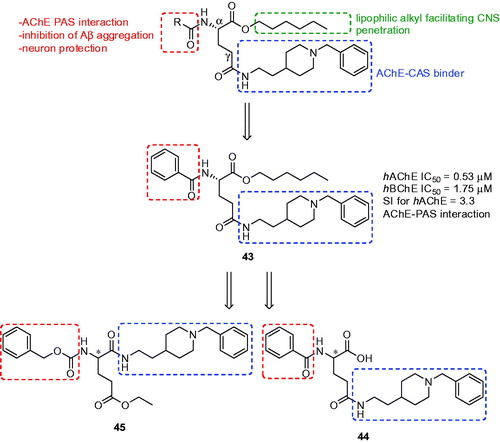
Hybrid 43 was chosen as a lead compound for the design of second series of l- and d-Glu N-benzylpiperidine derivatives (. All new compounds showed good inhibition ability of both human ChE with IC50 values in the low- and submicromolar range. The amino acid chirality seemed to have little influence on the hAChE inhibition, with the exception of l-44 () that was two orders more active than d-44. All tested compounds displayed preference for hAChE with the exception of enantiomers derived from 45 () with N-benzylpiperidine fragment in the α-carboxyl group where d-isomer proved 10-fold better hBChE inhibition profile over l-45. The majority of the compounds displayed neuroprotection behaviour ranging from 13% to 46% using R/O as toxic insult, and from 32% to 52% in OGD/R protocol in human neuroblastoma cell line SH-SY5Y. In both models, l-isomers displayed better protection than their d-counterparts, the best neuroprotection values showed compound l-44. Tested compounds were inactive at 10 µM as antioxidants, with the exception of derivatives bearing indole-3-carbonyl moiety that showed radical clearance around 70–73% of the Trolox value. However, the good neuroprotective properties of these compounds towards counteracting free radicals could be presumably associated to the activation of endogenous antioxidant pathways rather than their ability to capture these radicals. Compound l-45 was, like in the case of l-43, chosen for additional pharmacological assays based on its balanced biological profile and high predicted CNS-permeability. Derivative l-45 displayed voltage-gated calcium channel blockade being 6-fold less potent than l-43. On the other hand, in the neuroprotection study in a tissue model of cerebral ischemia, compound l-45 was more efficient than the reference compound l-43.
3.8. Lipoic acid hybrids
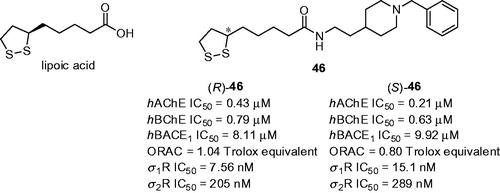
Lipoic acid (LA, ) is a natural antioxidant with ability to scavenge free radicals in both membrane and aqueous domainsCitation145. These features are mostly attributed to chelating redox-active transition metals, increasing the levels of reduced glutathione and down-regulating inflammatory processes. Besides having direct antioxidant potential, the relevance for using LA in drug design is underlined by the fact that it can also regenerate other biogenic antioxidantsCitation146. Therefore, LA is considered as privileged scaffold in designing novel biologically active compounds. One such series composes of LA with attached N-benzylpiperidine hybrids () to target three AD therapeutic objectives: (i) AChE; (ii) β-secretase-1 (BACE1) and (iii) σ-1 receptor (σ1R)Citation147. Initial hits derived from LA demonstrated ability to reduce oxidative stress and provide neuroprotectionCitation148. Novel family displayed moderate inhibition of hChE with IC50 values in the micro- to sub-micromolar rangeCitation147. The best inhibition was demonstrated by derivatives 46 with two carbon linker between lipoic acid and N-benzylpiperidine moiety. The inhibition activity of racemic mixture (R,S)-46 (hAChE IC50 = 0.39 µM, hBChE IC50 = 1.23 µM) and enantiomers (R)-46 (hAChE IC50 = 0.43, hBChE IC50 = 0.79 µM, ) and (S)-46 (hAChE IC50 = 0.21 µM, hBChE IC50 = 0.63 µM, ) did not show any noticeable activity differences. Each enantiomer of 46 was found to be good hBACE1 inhibitors with IC50 values in the low micromolar range, the best being however the racemic mixture with hBACE1 IC50 = 5.65 µM. According to PAMPA-BBB assay both enantiomers were predicted to reach CNS. The affinities of LA-based analogues 46 for σ1R and σ2R were determined in competition experiment with radio-ligands. Tested compounds showed good affinities for σRs, with Kis between the low-micromolar and the low-nanomolar scale. The racemic mixture and both enantiomers exhibited preference for the σ1R subtype with the selectivity ratio against σ2R > 19. The neurogenic studies on enantiomer (R)-46 to assess its potential ability to promote differentiation of rat brain stem cells into a neuronal phenotype demonstrated that (R)-46 was able not only to promote early neurogenesis but also stimulated neuronal maturation.
Figure 12. Structures of lipoic acid and the most active lipoic-N-benzylpiperidine hybrids (R)-46 and (S)-46.
3.9. Other donepezil hybrids with antioxidant properties
The design of N-benzylpiperidine and diarylthiazole-fused hybrids was based on the previous work where authors have designed and developed vicinally substituted diaryltriazines 47 ()Citation149. The design of the novel hybrids replaced the morpholino/piperazinoethyl side chain by N-benzylpiperidine fragment that was bound to diaryltriazine moietyCitation150. It was presumed that diaryltriazine would mimic dimethoxyindanone of donepezil being able to reside in the PAS of AChE. In general, the study disclosed 45 novel compounds combining two basic moieties either via aminomethylene or carboxamide linker. The most promising one was 48 that showed inhibitory activity against both ChEs in sub- and low-micromolar range (48: hAChE IC50 = 0.30 µM, hBChE IC50 = 1.84 µM, ), respectively, and exhibited potent inhibition of AChE-induced Aβ42 aggregation at 10 µM concentration (27.65%). The enzyme kinetic study of 48 indicated a mixed type of AChE inhibition being in line with the design of these hybrids. The PAMPA assay predicted its ability to cross the BBB by passive diffusion. The effect of 48 on cell viability and neuroprotective potential against oxidative stress were evaluated using the human neuroblastoma SH-SY5Y cell line. Compound 48 imposed negligible cytotoxicity even at 80 µM concentration to the human neuroblastoma SH-SY5Y cells and showed 39.6% neuroprotection (SH-SY5Y cells, tested at 10 µM of 48; H2O2 insult). In the DPPH assay 48 was found to exhibit free radical scavenging activity (55 and 70% at 10 µM and 20 µM concentrations, respectively). These in vitro results prompted following in vivo studies. Derivative 48 showed ROS scavenging and antiapoptotic properties against Aβ42-insult in the primary rat hippocampal neuron cultures. Intracerebroventricular injection of Aβ42 to rats worsened hippocampal-dependent memory working which was improved when treated with 48. The treatment with this drug attenuated the antiamnestic effect cause by scopolamine. Moreover, it also reduced the brain levels of malondialdehyde suggesting its antioxidant properties with simultaneous catalase level elevation. 48 also significantly attenuated the levels of Aβ42 and p-tau and demonstrated significant antiapoptotic potential by lowering the levels of cleaved caspase-3 and cleaved-poly(ADP-ribose) polymerase-1 (PARP) as assessed by Western blot analysis. According to the pharmacokinetic analysis, 48 has good oral absorption and elimination at moderate rate compared to the absorption phase.
Figure 13. The most active AChEIs 48 and 49 from N-benzylpiperidine-diarylthiazole, 5,6-dimethoxy-indanone benzenamides, and N′-(4-benzylpiperidin-/piperazin-/benzhydrylpiperazin-1-yl)alkylamine families, respectively.
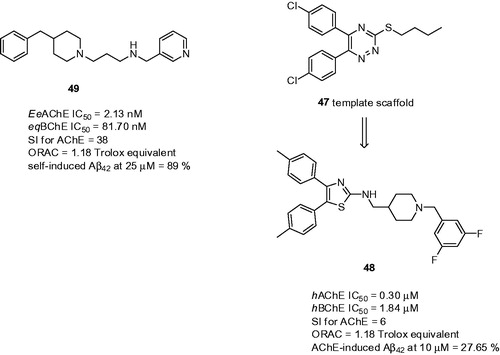
N′-(4-Benzylpiperidin-/piperazin-/benzhydrylpiperazin-1-yl)alkylamine derivatives were developed to target the multifaceted nature of ADCitation151. The 4-benzylamine motif was combined with an appropriate alkyl spacer of varying length with electron rich substituents such as hydroxy, di-, and tri-methoxy groups on the aromatic ring and/or a nitrogen-containing heterocyclic system. These were previously shown to play an important role in inhibiting Aβ aggregation and scavenging of a variety of reactive oxygen speciesCitation152. Regarding ChE inhibition, the SAR studies revealed that 4-benzylpiperidine derivatives bearing a terminal pyridine ring and a vanillyl group on the benzyl ring were the most effective in inhibiting both ChEs, with compound 49 (EeAChE IC50 = 2.13 nM; eqBChE IC50 = 81.7 nM; SI for AChE = 38; ) being the most potent AChEI in the series. Kinetic analysis with 49 indicated mixed-type inhibition. The ability to inhibit self-induced Aβ42 aggregation at 25 µM ranged between 54 and 89%; the most active compound was 49, displaying anti-amyloid properties at the upper limit. Docking studies performed with 49 on EeAChE revealed significant interactions in the CAS and PAS binding sites, as the pyridine ring was firmly bound to the catalytic site of AChE in the bottom of the gorge, through a favourable π–π interaction with active site residue Trp84. At the cavity entrance, 4-benzylpiperidine was stacked against the indole ring of Trp279 through π–π stacking interactions, which is inconsistent with donepezil superimposition. Generally, all compounds exerted strong antioxidant properties ranging between 0.68–4.45 Trolox values. It was shown that the presence of a vanillyl ring at the terminal end is crucial for providing strong radical scavenging ability. The predicted ADME properties revealed that all compounds fulfil drug-like criteria, exhibit low in silico possible toxicity risk, and could be considered as novel nootropic agents based on prediction of activity spectra for substances (PASS) predictionCitation153.
4. Conclusion
AD constitute the major health problem in the world. The estimated number of patients needing treatment is 40 million worldwide with expectation to nearly double by 2040Citation3. Owing to the alarming number of AD patients and population ageing, it is clear that effective cure is urgently required. Currently, marketed drugs provide only symptomatic and short-term relief and their effectiveness do not treat the cause of the dementiaCitation9.
Compelling evidence imposed AD as multi-factorial disease. Its pathogenesis is composed of many different mechanisms, interaction among them generates a highly complex web of different pathwaysCitation154. Based upon multifactorial aetiology of AD, it is clear that it can be hardly treated by administration of drug targeting solely one pathological condition. For almost three decades, the attention was paid to AChEIs. The cholinergic hypothesis was during this period slightly modified, the paradigm shifted from solely AChEIs towards BChEIs or non-selective ChE inhibitors. This is caused by the fact that cortical levels of BChE show a significant increase in AD progressionCitation155. Moreover, high BChE levels are associated with the formation of neuritic plaques and neurofibrillary tanglesCitation156. From this point of view, the marketed drug rivastigmine possesses favourable profile offering dual and sustained AChE/BChE inhibitionCitation157. On the other hand, when other hypothesis dealing with the onset and progression of AD have been postulated like Aβ and tau hypothesis, it is evident that pharmacodynamics and pharmacokinetic properties of ChEIs are not sufficient to tackle AD. For instance, further studies with galantamine demonstrated its ability to allosterically modulate α7 homomeric and α4β2 heteromeric nicotinic ACh receptors to alleviate some of the cognitive deficits associated with ADCitation158. Intriguing features also favour using donepezil for the treatment of AD. Since its approval, the secondary non-cholinergic role to down-regulate formation of toxic Aβ oligomers and to decrease brain tissue Aβ plaques has been formulatedCitation159.
Based on data from plethora epidemiological studies several compounds with antioxidant activity have already entered to clinical evaluation to uncover their potential in prevention of cognitive decline and treatment of mild cognitive impairment in AD. Among them, curcumin, LA and other antioxidants not mentioned in this review like vitamin E, resveratrol, ubiquinone (coenzyme Q10), and pramipexole have been clinically investigated. The most of the compounds reached Phase I and II, with the exception of resveratrol. Although available data do not warrant the doubtless use of antioxidants in AD, they are hampered by extremely poor comparability. Moreover, the major profit of radical-free scavengers may lie in their administration in prodromal stage of AD. Thus, the absence of substantial clinical benefit of antioxidants in AD is not disproved to date.
MTDLs have emerged as a promising approach for management of ADCitation160,Citation161. Several ways how to build these novel and multi-potent ligands exist. From this perspective, framework combination is mostly used either by linking, fusing, or merging two entities in order to implement two or more abilities into single moleculeCitation162. Several issues are commonly associated with MTDLs designing/development when created by linking or fusing based upon their poor drug-likeness. Being concerned by this fact, we are aware that the true MTDLs will have to possess balanced physico-chemical propertiesCitation163–165. Another challenging task is to obtain MTDLs with balanced activity/affinity profile towards different targets. This can be nicely described as a pair of scales where one activity improvement mostly results into loss/decrease of other. Thus, those who are involved in drug development process like medicinal chemists, pharmacologists, or biochemist have to bear in mind that compounds needs to be optimised by addressing activities in the same concentration ranges. From this point of view, only a very few MTDLs with well-balanced activities/affinities aimed at neurodegenerative disorders are reported today like triazinones (dual BACE-1 and glycogen synthase kinase-3β inhibitors) aminobenzimidazoles (dual BChE and human cannabinoid subtype 2 receptor agonists) or the most advanced drug ladostigil (clinical trial phase 2, AChE/BChE, monoamine oxidase B inhibitors)Citation138,Citation166,Citation167. This is, however, not the case of almost all the ligands reported within this review and thus the true hybrid combining balanced physico-chemical characteristics, antioxidant, and anti-cholinesterase properties is eagerly awaited.
In this review, we have turned our attention to profiling donepezil scaffold into novel chemical entities by conferring the antioxidant properties. The unique features of donepezil chemical scaffold allowed the chemist to mostly retain drug-like characteristics while exacerbate others. Antioxidant profile was mostly achieved by introducing known scaffolds responsible for radical scavenging effect like coumarin, ferulic acid, 8HQ, lipoic acid, and others. It remains speculative whether these agents are able to slow-down the progression of AD acting as symptomatic drugs or they may treat the cause of the disease. In our opinion, the most interesting compounds disclosed herein are bio-oxidizable pro-drug 34 with its activated adduct 33 and “selenpezil” derivatives 21 and 22, both exerting in vivo efficacy. It has to be also noted that oxidative stress closely correlates with inflammationCitation168. However, only large clinical studies may provide clear answer as inflammation encompasses dozens of highly interactive molecular mediators and mechanisms, some of them potentially helpful and some of them potentially harmful. Be that as it may, we believe that current knowledge from AD pathogenesis open new route for MTDLs combining ChEIs with antioxidant properties, as the oxidative stress is inherent part of neurodegenerative diseases.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
Related Research Data
References
- Sanabria-Castro A, Alvarado-Echeverría I, Monge-Bonilla C. Molecular pathogenesis of Alzheimer's disease: an update. Ann Neurosci 2017;24:46–54.
- Ritchie CW, Molinuevo JL, Truyen L, et al. Development of interventions for the secondary prevention of Alzheimer’s dementia: the European Prevention of Alzheimer’s Dementia (EPAD) project. Lancet Psychiatry 2016;3:179–86.
- Marešová P, Mohelská H, Dolejš J, Kuča K. Socio-economic aspects of Alzheimer's disease. Curr Alzheimer Res 2015;12:903–11.
- Bartus RT, Dean RL, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982;217:408–14.
- Francis P, Palmer A, Snape M, Wilcock G. The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry 1999;66:137–47.
- Francis PT. The interplay of neurotransmitters in Alzheimer's disease. CNS Spectr 2005;10:6–9.
- Mann DM, Yates PO, Marcyniuk B. Dopaminergic neurotransmitter systems in Alzheimer’s disease and in Down’s syndrome at middle age. J Neurol Neurosurg Psychiatry 1987;50:341–4.
- Rodríguez JJ, Noristani HN, Verkhratsky A. The serotonergic system in ageing and Alzheimer's disease. Prog Neurobiol 2012;99:15–41.
- Zemek F, Drtinova L, Nepovimova E, et al. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin Drug Saf 2014;13:759–74.
- Nordberg A, Ballard C, Bullock R, et al. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim Care Companion CNS Disord 2013;15. doi:10.4088/PCC.12r01412
- Spilovska K, Korabecny J, Nepovimova E, et al. Multitarget tacrine hybrids with neuroprotective properties to confront Alzheimer’s disease. Curr Top Med Chem 2017;17:1006–26.
- Spilovska K, Zemek F, Korabecny J, et al. Adamantane - a lead structure for drugs in clinical practice. Curr Med Chem 2016;23:3245–66.
- Farlow MR, Graham SM, Alva G. Memantine for the treatment of Alzheimer's disease: tolerability and safety data from clinical trials. Drug Saf 2008;31:577–85.
- Horak M, Holubova K, Nepovimova E, et al. The pharmacology of tacrine at N-methyl-d-aspartate receptors. Prog Neuropsychopharmacol Biol Psychiatry 2017;75:54–62.
- Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem Int 2004;45:583–95.
- Hamley IW. The amyloid beta peptide: a chemist's perspective. Role in Alzheimer's and fibrillization. Chem Rev 2012;112:5147–92.
- Barage SH, Sonawane KD. Amyloid cascade hypothesis: pathogenesis and therapeutic strategies in Alzheimer's disease. Neuropeptides 2015;52:1–18.
- Giacobini E, Gold G. Alzheimer disease therapy–moving from amyloid-β to tau. Nat Rev Neurol 2013;9:677–86.
- Avila J, Lucas JJ, Perez M, Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiol Rev 2004;84:361–84.
- Forman MS, Trojanowski JQ, Lee VM-Y. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med 2004;10:1055–63.
- Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev 2012;2012:428010.
- Bolisetty S, Jaimes EA. Mitochondria and reactive oxygen species: physiology and pathophysiology. Int J Mol Sci 2013;14:6306–44.
- Kim GH, Kim JE, Rhie SJ, Yoon S. The role of oxidative stress in neurodegenerative diseases. Exp Neurobiol 2015;24:325–40.
- Dasuri K, Zhang L, Keller JN. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med 2013;62:170–85.
- Patten DA, Germain M, Kelly MA, Slack RS. Reactive oxygen species: stuck in the middle of neurodegeneration. J Alzheimers Dis 2010;20 Suppl 2:S357–S67.
- Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci 2010;2:12.
- Radi E, Formichi P, Battisti C, Federico A. Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimers Dis 2014;42 Suppl 3:S125–S52.
- Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev 2013;2013:316523.
- Praticò D. Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends Pharmacol Sci 2008;29:609–15.
- Firuzi O, Miri R, Tavakkoli M, Saso L. Antioxidant therapy: current status and future prospects. Curr Med Chem 2011;18:3871–88.
- Murphy MP. Antioxidants as therapies: can we improve on nature?. Free Radic Biol Med 2014;66:20–3.
- Nepovimova E, Korabecny J, Dolezal R, et al. Tacrine-trolox hybrids: a novel class of centrally active, nonhepatotoxic multi-target-directed ligands exerting anticholinesterase and antioxidant activities with low in vivo toxicity. J Med Chem 2015;58:8985–9003.
- Rosini M, Andrisano V, Bartolini M, et al. Rational approach to discover multipotent anti-Alzheimer drugs. J Med Chem 2005;48:360–3.
- Pinchuk I, Shoval H, Dotan Y, Lichtenberg D. Evaluation of antioxidants: scope, limitations and relevance of assays. Chem Phys Lipids 2012;165:638–47.
- Alam MN, Bristi NJ, Rafiquzzaman M. Review on in vivo and in vitro methods evaluation of antioxidant activity. Saudi Pharm J 2013;21:143–52.
- Balasaheb Nimse S, Pal D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv 2015;5:27986–8006.
- Pisoschi AM, Negulescu GP. Methods for total antioxidant activity determination: a review. Biochem Anal Biochem 2011;1:106.
- Karadag A, Ozcelik B, Saner S. Review of Methods to Determine Antioxidant Capacities. Food Anal Methods 2009;2:41–60.
- Apak R, Gorinstein S, Böhm V, et al. Methods of measurement and evaluation of natural antioxidant capacity/activity (IUPAC Technical Report). Pure Appl Chem 2013;85:957–98.
- Huang D, Ou B, Prior RL. The chemistry behind antioxidant capacity assays. J Agric Food Chem 2005;53:1841–56.
- Benzie IF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem 1996;239:70–6.
- Özyürek M, Güçlü K, Tütem E, et al. A comprehensive review of CUPRAC methodology. Anal Methods 2011;3:2439–53.
- Re R, Pellegrini N, Proteggente A, et al. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol Med 1999;26:1231–7.
- van den Berg R, Haenen GRMM, van den Berg H, Bast A. Applicability of an improved Trolox equivalent antioxidant capacity (TEAC) assay for evaluation of antioxidant capacity measurements of mixtures. Food Chem 1999;66:511–7.
- Sharma OP, Bhat TK. DPPH antioxidant assay revisited. Food Chem 2009;113:1202–5.
- Shahidi F, Zhong Y. Measurement of antioxidant activity. J Funct Foods 2015;18:757–81.
- Niki E. Free radical initiators as source of water- or lipid-soluble peroxyl radicals. Methods Enzymol 1990;186:100–8.
- Cao G, Prior RL. Measurement of oxygen radical absorbance capacity in biological samples. Methods Enzymol 1999;299:50–62.
- López-Alarcón C, Denicola A. Evaluating the antioxidant capacity of natural products: a review on chemical and cellular-based assays. Anal Chim Acta 2013;763:1–10.
- Peng X-M, Damu GLV, Zhou C-H. Current developments of coumarin compounds in medicinal chemistry. Curr Pharm Des 2013;19:3884–930.
- Venugopala KN, Rashmi V, Odhav B. Review on natural coumarin lead compounds for their pharmacological activity. BioMed Res Int 2013;2013:963248.
- Anand P, Singh B, Singh N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer's disease. Bioorg Med Chem 2012;20:1175–80.
- Castro A, Martinez A. Peripheral and dual binding site acetylcholinesterase inhibitors: implications in treatment of Alzheimer's disease. Mini Rev Med Chem 2001;1:267–72.
- Radić Z, Reiner E, Taylor P. Role of the peripheral anionic site on acetylcholinesterase: inhibition by substrates and coumarin derivatives. Mol Pharmacol 1991;39:98–104.
- Radić Z, Reiner E, Simeon V. Binding sites on acetylcholinesterase for reversible ligands and phosphorylating agents. A theoretical model tested on haloxon and phosphostigmine. Biochem Pharmacol 1984;33:671–7.
- Catto M, Nicolotti O, Leonetti F, et al. Structural insights into monoamine oxidase inhibitory potency and selectivity of 7-substituted coumarins from ligand- and target-based approaches. J Med Chem 2006;49:4912–25.
- Gnerre C, Catto M, Leonetti F, et al. Inhibition of monoamine oxidases by functionalized coumarin derivatives: biological activities, QSARs, and 3D-QSARs. J Med Chem 2000;43:4747–58.
- Pisani L, Catto M, Giangreco I, et al. Design, synthesis, and biological evaluation of coumarin derivatives tethered to an edrophonium-like fragment as highly potent and selective dual binding site acetylcholinesterase inhibitors. ChemMedChem 2010;5:1616–30.
- Pisani L, Muncipinto G, Miscioscia TF, et al. Discovery of a novel class of potent coumarin monoamine oxidase B inhibitors: development and biopharmacological profiling of 7-[(3-chlorobenzyl)oxy]-4-[(methylamino)methyl]-2H-chromen-2-one methanesulfonate (NW-1772) as a highly potent, selective, reversible, and orally active monoamine oxidase B inhibitor. J Med Chem 2009;52:6685–706.
- Catto M, Pisani L, Leonetti F, et al. Design, synthesis and biological evaluation of coumarin alkylamines as potent and selective dual binding site inhibitors of acetylcholinesterase. Bioorg Med Chem 2013;21:146–52.
- Khoobi M, Emami S, Dehghan G, et al. Synthesis and free radical scavenging activity of coumarin derivatives containing a 2-methylbenzothiazoline motif. Arch Pharm (Weinheim) 2011;344:588–94.
- Khoobi M, Foroumadi A, Emami S, et al. Coumarin-based bioactive compounds: facile synthesis and biological evaluation of coumarin-fused 1,4-thiazepines. Chem Biol Drug Des 2011;78:580–6.
- Khoobi M, Alipour M, Zarei S, et al. A facile route to flavone and neoflavone backbones via a regioselective palladium catalyzed oxidative Heck reaction. Chem Commun Camb Engl 2012;48:2985–7.
- Khoobi M, Ramazani A, Foroumadi A, et al. Highly cis-diastereoselective synthesis of coumarin-based 2,3-disubstituted dihydrobenzothiazines by organocatalysis. Helv Chim Acta 2012;95:660–71.
- Khoobi M, Ramazani A, Foroumadi AR, et al. Efficient microwave-assisted synthesis of 3-benzothiazolo and 3-benzothiazolino coumarin derivatives catalyzed by heteropoly acids. J Iran Chem Soc 2012;8:1036–42.
- Nadri H, Pirali-Hamedani M, Shekarchi M, et al. Design, synthesis and anticholinesterase activity of a novel series of 1-benzyl-4-((6-alkoxy-3-oxobenzofuran-2(3H)-ylidene) methyl) pyridinium derivatives. Bioorg Med Chem 2010;18:6360–6.
- Alipour M, Khoobi M, Foroumadi A, et al. Novel coumarin derivatives bearing N-benzyl pyridinium moiety: potent and dual binding site acetylcholinesterase inhibitors. Bioorg Med Chem 2012;20:7214–22.
- Razavi SF, Khoobi M, Nadri H, et al. Synthesis and evaluation of 4-substituted coumarins as novel acetylcholinesterase inhibitors. Eur J Med Chem 2013;64:252–9.
- Alipour M, Khoobi M, Nadri H, et al. Synthesis of some new 3-coumaranone and coumarin derivatives as dual inhibitors of acetyl- and butyrylcholinesterase. Arch Pharm (Weinheim) 2013;346:577–87.
- Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx J Am Soc Exp Neurother 2005;2:541–53.
- Asadipour A, Alipour M, Jafari M, et al. Novel coumarin-3-carboxamides bearing N-benzylpiperidine moiety as potent acetylcholinesterase inhibitors. Eur J Med Chem 2013;70:623–30.
- Khoobi M, Alipour M, Sakhteman A, et al. Design, synthesis, biological evaluation and docking study of 5-oxo-4,5-dihydropyrano[3,2-c]chromene derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors. Eur J Med Chem 2013;68:260–9.
- Khoobi M, Alipour M, Moradi A, et al. Design, synthesis, docking study and biological evaluation of some novel tetrahydrochromeno [3’,4’:5,6]pyrano[2,3-b]quinolin-6(7H)-one derivatives against acetyl- and butyrylcholinesterase. Eur J Med Chem 2013;68:291–300.
- Pudlo M, Luzet V, Ismaïli L, et al. Quinolone-benzylpiperidine derivatives as novel acetylcholinesterase inhibitor and antioxidant hybrids for Alzheimer disease. Bioorg Med Chem 2014;22:2496–507.
- Detsi A, Bouloumbasi D, Prousis KC, et al. Design and synthesis of novel quinolinone-3-aminoamides and their alpha-lipoic acid adducts as antioxidant and anti-inflammatory agents. J Med Chem 2007;50:2450–8.
- Naito Y, Yoshikawa T, Tanigawa T, et al. Hydroxyl radical scavenging by rebamipide and related compounds: electron paramagnetic resonance study. Free Radic Biol Med 1995;18:117–23.
- Pouységu L, Deffieux D, Malik G, et al. Synthesis of ellagitannin natural products. Nat Prod Rep 2011;28:853–74.
- Seeram NP, Henning SM, Zhang Y, et al. Pomegranate juice ellagitannin metabolites are present in human plasma and some persist in urine for up to 48 hours. J Nutr 2006;136:2481–5.
- Gulcan HO, Unlu S, Esiringu I, et al. Design, synthesis and biological evaluation of novel 6H-benzo[c]chromen-6-one, and 7,8,9,10-tetrahydro-benzo[c]chromen-6-one derivatives as potential cholinesterase inhibitors. Bioorg Med Chem 2014;22:5141–54.
- Sgarbossa A, Giacomazza D, di Carlo M. Ferulic acid: a hope for alzheimer's disease therapy from plants. Nutrients 2015;7:5764–82.
- Benchekroun M, Bartolini M, Egea J, et al. Novel tacrine-grafted Ugi adducts as multipotent anti-Alzheimer drugs: a synthetic renewal in tacrine-ferulic acid hybrids. ChemMedChem 2015;10:523–39.
- Chen Y, Sun J, Fang L, et al. Tacrine–ferulic acid–nitric oxide (NO) donor trihybrids as potent, multifunctional acetyl- and butyrylcholinesterase inhibitors. J Med Chem 2012;55:4309–21.
- Fang L, Kraus B, Lehmann J, et al. Design and synthesis of tacrine–ferulic acid hybrids as multi-potent anti-Alzheimer drug candidates. Bioorg Med Chem Lett 2008;18:2905–9.
- Benchekroun M, Ismaili L, Pudlo M, et al. Donepezil-ferulic acid hybrids as anti-Alzheimer drugs. Future Med Chem 2015;7:15–21.
- Sang Z, Pan W, Wang K, et al. Design, synthesis and evaluation of novel ferulic acid-O-alkylamine derivatives as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur J Med Chem 2017;130:379–92.
- Verghese J. Isolation of curcumin from Curcuma longa L. rhizome. Flavour Fragr J 1993;8:315–9.
- Katsuyama Y, Kita T, Funa N, Horinouchi S. Curcuminoid biosynthesis by two type III polyketide synthases in the herb Curcuma longa. J Biol Chem 2009;284:11160–70.
- Ammon HP, Wahl MA. Pharmacology of Curcuma longa. Planta Med 1991;57:1–7.
- Wang Y-J, Thomas P, Zhong J-H, et al. Consumption of grape seed extract prevents amyloid-beta deposition and attenuates inflammation in brain of an Alzheimer’s disease mouse. Neurotox Res 2009;15:3–14.
- Dairam A, Fogel R, Daya S, Limson JL. Antioxidant and iron-binding properties of curcumin, capsaicin, and S-allylcysteine reduce oxidative stress in rat brain homogenate. J Agric Food Chem 2008;56:3350–6.
- Park S-Y, Kim DSHL. Discovery of natural products from Curcuma longa that protect cells from beta-amyloid insult: a drug discovery effort against Alzheimer’s disease. J Nat Prod 2002;65:1227–31.
- Kim H, Park B-S, Lee K-G, et al. Effects of naturally occurring compounds on fibril formation and oxidative stress of beta-amyloid. J Agric Food Chem 2005;53:8537–41.
- Shimmyo Y, Kihara T, Akaike A, et al. Epigallocatechin-3-gallate and curcumin suppress amyloid beta-induced beta-site APP cleaving enzyme-1 upregulation. Neuroreport 2008;19:1329–33.
- Derosa G, Maffioli P, Simental-Mendía LE, et al. Effect of curcumin on circulating interleukin-6 concentrations: a systematic review and meta-analysis of randomized controlled trials. Pharmacol Res 2016;111:394–404.
- Tang M, Taghibiglou C. The mechanisms of action of curcumin in Alzheimer's disease. J Alzheimers Dis 2017;58:1003–16.
- Mishra CB, Manral A, Kumari S, et al. Design, synthesis and evaluation of novel indandione derivatives as multifunctional agents with cholinesterase inhibition, anti-β-amyloid aggregation, antioxidant and neuroprotection properties against Alzheimer’s disease. Bioorg Med Chem 2016;24:3829–41.
- Mishra CB, Kumari S, Manral A, et al. Design, synthesis, in-silico and biological evaluation of novel donepezil derivatives as multi-target-directed ligands for the treatment of Alzheimer’s disease. Eur J Med Chem 2017;125:736–50.
- Yan J, Hu J, Liu A, et al. Design, synthesis, and evaluation of multitarget-directed ligands against Alzheimer’s disease based on the fusion of donepezil and curcumin. Bioorg Med Chem 2017;25:2946–55.
- Szutowicz A, Bielarczyk H, Zyśk M, et al. Early and late pathomechanisms in Alzheimer's disease: from zinc to amyloid-β neurotoxicity. Neurochem Res 2017;42:891–904.
- Ritchie CW, Bush AI, Mackinnon A, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol 2003;60:1685–91.
- Sampson EL, Jenagaratnam L, McShane R. Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. Cochrane Database Syst Rev 2012;2012:CD005380.
- Dias KST, de Paula CT, Dos Santos T, et al. Design, synthesis and evaluation of novel feruloyl-donepezil hybrids as potential multitarget drugs for the treatment of Alzheimer’s disease. Eur J Med Chem 2017;130:440–57.
- Cai Y-Z, Mei S, null, Jie X, null, et al. Structure-radical scavenging activity relationships of phenolic compounds from traditional Chinese medicinal plants. Life Sci 2006;78:2872–88.
- Allmang C, Wurth L, Krol A. The selenium to selenoprotein pathway in eukaryotes: more molecular partners than anticipated. Biochim Biophys Acta 2009;1790:1415–23.
- Berr C, Nicole A, Godin J, et al. Selenium and oxygen-metabolizing enzymes in elderly community residents: a pilot epidemiological study. J Am Geriatr Soc 1993;41:143–8.
- Wilson SR, Zucker PA, Huang RRC, Spector A. Development of synthetic compounds with glutathione peroxidase activity. J Am Chem Soc 1989;111:5936–9.
- Xie L, Zheng W, Xin N, et al. Ebselen inhibits iron-induced tau phosphorylation by attenuating DMT1 up-regulation and cellular iron uptake. Neurochem Int 2012;61:334–40.
- Luo Z, Sheng J, Sun Y, et al. Synthesis and evaluation of multi-target-directed ligands against Alzheimer’s disease based on the fusion of donepezil and ebselen. J Med Chem 2013;56:9089–99.
- Luo Z, Liang L, Sheng J, et al. Synthesis and biological evaluation of a new series of ebselen derivatives as glutathione peroxidase (GPx) mimics and cholinesterase inhibitors against Alzheimer’s disease. Bioorg Med Chem 2014;22:1355–61.
- Meng F-C, Mao F, Shan W-J, et al. Design, synthesis, and evaluation of indanone derivatives as acetylcholinesterase inhibitors and metal-chelating agents. Bioorg Med Chem Lett 2012;22:4462–6.
- Nadri H, Pirali-Hamedani M, Moradi A, et al. 5,6-Dimethoxybenzofuran-3-one derivatives: a novel series of dual Acetylcholinesterase/Butyrylcholinesterase inhibitors bearing benzyl pyridinium moiety. Daru J Fac Pharm Tehran Univ Med Sci 2013;21:15.
- Akrami H, Mirjalili BF, Khoobi M, et al. Indolinone-based acetylcholinesterase inhibitors: synthesis, biological activity and molecular modeling. Eur J Med Chem 2014; 84:375–81.
- Cheng F, Li W, Zhou Y, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 2012;52:3099–105.
- Baharloo F, Moslemin MH, Nadri H, et al. Benzofuran-derived benzylpyridinium bromides as potent acetylcholinesterase inhibitors. Eur J Med Chem 2015;93:196–201.
- Huang L, Lu C, Sun Y, et al. Multitarget-directed benzylideneindanone derivatives: anti-β-amyloid (Aβ) aggregation, antioxidant, metal chelation, and monoamine oxidase B (MAO-B) inhibition properties against Alzheimer’s disease. J Med Chem 2012;55:8483–92.
- Huang L, Miao H, Sun Y, et al. Discovery of indanone derivatives as multi-target-directed ligands against Alzheimer’s disease. Eur J Med Chem 2014;87:429–39.
- Yerdelen KO, Koca M, Anil B, et al. Synthesis of donepezil-based multifunctional agents for the treatment of Alzheimer's disease. Bioorg Med Chem Lett 2015;25:5576–82.
- Koca M, Yerdelen KO, Anil B, et al. Design, synthesis and biological activity of 1H-indene-2-carboxamides as multi-targeted anti-Alzheimer agents. J Enzyme Inhib Med Chem 2016;31:13–23.
- Yerdelen KO, Tosun E. Synthesis, docking and biological evaluation of oxamide and fumaramide analogs as potential AChE and BuChE inhibitors. Med Chem Res 2015;24:588–602.
- Lan J-S, Zhang T, Liu Y, et al. Design, synthesis and biological activity of novel donepezil derivatives bearing N-benzyl pyridinium moiety as potent and dual binding site acetylcholinesterase inhibitors. Eur J Med Chem 2017;133:184–96.
- Bohn P, Gourand F, Papamicaël C, et al. Dihydroquinoline carbamate derivatives as “bio-oxidizable” prodrugs for brain delivery of acetylcholinesterase inhibitors: [11C] radiosynthesis and biological evaluation. ACS Chem Neurosci 2015;6:737–44.
- Peauger L, Azzouz R, Gembus V, et al. Donepezil-based central acetylcholinesterase inhibitors by means of a ‘bio-oxidizable’ prodrug strategy: design, synthesis, and in vitro biological evaluation. J Med Chem 2017;60:5909–26.
- Di L, Kerns EH, Fan K, et al. High throughput artificial membrane permeability assay for blood-brain barrier. Eur J Med Chem 2003;38:223–32.
- Lemes LFN, de Andrade Ramos G, de Oliveira AS, et al. Cardanol-derived AChE inhibitors: towards the development of dual binding derivatives for Alzheimer’s disease. Eur J Med Chem 2016;108:687–700.
- Miller E, Morel A, Saso L, Saluk J. Melatonin redox activity. Its potential clinical applications in neurodegenerative disorders. Curr Top Med Chem 2015;15:163–9.
- Li X-C, Wang Z-F, Zhang J-X, et al. Effect of melatonin on calyculin A-induced tau hyperphosphorylation. Eur J Pharmacol 2005;510:25–30.
- Lahiri DK. Melatonin affects the metabolism of the beta-amyloid precursor protein in different cell types. J Pineal Res 1999;26:137–46.
- Rosales-Corral S, Tan D-X, Reiter RJ, et al. Orally administered melatonin reduces oxidative stress and proinflammatory cytokines induced by amyloid-beta peptide in rat brain: a comparative, in vivo study versus vitamin C and E. J Pineal Res 2003;35:80–4.
- Wu Y-H, Swaab DF. The human pineal gland and melatonin in aging and Alzheimer's disease. J Pineal Res 2005;38:145–52.
- Ramos E, Egea J, de Los Ríos C, et al. Melatonin as a versatile molecule to design novel multitarget hybrids against neurodegeneration. Future Med Chem 2017;9:765–80.
- Wang J, Wang Z-M, Li X-M, et al. Synthesis and evaluation of multi-target-directed ligands for the treatment of Alzheimer's disease based on the fusion of donepezil and melatonin. Bioorg Med Chem 2016;24:4324–38.
- Luo X-T, Wang C-M, Liu Y, Huang Z-G. New multifunctional melatonin-derived benzylpyridinium bromides with potent cholinergic, antioxidant, and neuroprotective properties as innovative drugs for Alzheimer’s disease. Eur J Med Chem 2015;103:302–11.
- Prati F, Bergamini C, Fato R, et al. Novel 8-hydroxyquinoline derivatives as multitarget compounds for the treatment of Alzheimer’s disease. ChemMedChem 2016;11:1284–95.
- Adlard PA, Cherny RA, Finkelstein DI, et al. Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 2008;59:43–55.
- Nguyen T, Hamby A, Massa SM. Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington’s disease mouse model. Proc Natl Acad Sci USA 2005;102:11840–5.
- Simoni E, Caporaso R, Bergamini C, et al. Polyamine conjugation as a promising strategy to target amyloid aggregation in the framework of Alzheimer’s disease. ACS Med Chem Lett 2016;7:1145–50.
- Wu M-Y, Esteban G, Brogi S, et al. Donepezil-like multifunctional agents: design, synthesis, molecular modeling and biological evaluation. Eur J Med Chem 2016;121:864–79.
- Korábečný J, Nepovimová E, Cikánková T, et al. Newly developed drugs for Alzheimer’s disease in relation to energy metabolism, cholinergic and monoaminergic neurotransmission. Neuroscience 2018;370:191–206.
- Wang Z-M, Cai P, Liu Q-H, et al. Rational modification of donepezil as multifunctional acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease. Eur J Med Chem 2016;123:282–97.
- Arce MP, Rodríguez-Franco MI, González-Muñoz GC, et al. Neuroprotective and cholinergic properties of multifunctional glutamic acid derivatives for the treatment of Alzheimer’s disease. J Med Chem 2009;52:7249–57.
- Monjas L, Arce MP, León R, et al. Enzymatic and solid-phase synthesis of new donepezil-based L- and d-glutamic acid derivatives and their pharmacological evaluation in models related to Alzheimer’s disease and cerebral ischemia. Eur J Med Chem 2017;130:60–72.
- Maroto M, de Diego AMG, Albiñana E, et al. Multi-target novel neuroprotective compound ITH33/IQM9.21 inhibits calcium entry, calcium signals and exocytosis. Cell Calcium 2011;50:359–69.
- Lorrio S, Gómez-Rangel V, Negredo P, et al. Novel multitarget ligand ITH33/IQM9.21 provides neuroprotection in in vitro and in vivo models related to brain ischemia. Neuropharmacology 2013;67:403–11.
- Moreno-Ortega AJ, Al-achbili LM, Alonso E, et al. Neuroprotective effect of the novel compound ITH33/IQM9.21 against oxidative stress and Na+ and Ca2+ overload in motor neuron-like NSC-34 cells. Neurotox Res 2016;30:380–91.
- Di Domenico F, Barone E, Perluigi M, Butterfield DA. Strategy to reduce free radical species in Alzheimer’s disease: an update of selected antioxidants. Expert Rev Neurother 2015;15:19–40.
- Castañeda-Arriaga R, Alvarez-Idaboy JR. Lipoic acid and dihydrolipoic acid. a comprehensive theoretical study of their antioxidant activity supported by available experimental kinetic data. J Chem Inf Model 2014;54:1642–52.
- Estrada M, Pérez C, Soriano E, et al. New neurogenic lipoic-based hybrids as innovative Alzheimer’s drugs with σ-1 agonism and β-secretase inhibition. Future Med Chem 2016;8:1191–207.
- Rosini M, Simoni E, Bartolini M, et al. Exploiting the lipoic acid structure in the search for novel multitarget ligands against Alzheimer’s disease. Eur J Med Chem 2011;46:5435–42.
- Irannejad H, Amini M, Khodagholi F, et al. Synthesis and in vitro evaluation of novel 1,2,4-triazine derivatives as neuroprotective agents. Bioorg Med Chem 2010;18:4224–30.
- Shidore M, Machhi J, Shingala K, et al. Benzylpiperidine-linked diarylthiazoles as potential anti-Alzheimer’s agents: synthesis and biological evaluation. J Med Chem 2016;59:5823–46.
- Meena P, Nemaysh V, Khatri M, et al. Synthesis, biological evaluation and molecular docking study of novel piperidine and piperazine derivatives as multi-targeted agents to treat Alzheimer’s disease. Bioorg Med Chem 2015;23:1135–48.
- Ono K, Hasegawa K, Naiki H, Yamada M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J Neurosci Res 2004;75:742–50.
- Poroikov VV, Filimonov DA, Ihlenfeldt W-D, et al. PASS biological activity spectrum predictions in the enhanced open NCI database browser. J Chem Inf Comput Sci 2003;43:228–36.
- De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol Rev 2010;90:465–94.
- Perry EK, Perry RH, Blessed G, Tomlinson BE. Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol Appl Neurobiol 1978;4:273–7.
- Guillozet AL, Smiley JF, Mash DC, Mesulam MM. Butyrylcholinesterase in the life cycle of amyloid plaques. Ann Neurol 1997;42:909–18.
- Grossberg GT. Cholinesterase Inhibitors for the Treatment of Alzheimer's disease: getting on and staying on. Curr Ther Res Clin Exp 2003;64:216–35.
- Pepeu G, Giovannini MG. Cholinesterase inhibitors and memory. Chem Biol Interact 2010;187:403–8.
- Dong H, Yuede CM, Coughlan CA, et al. Effects of donepezil on amyloid-beta and synapse density in the Tg2576 mouse model of Alzheimer's disease. Brain Res 2009;1303:169–78.
- Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem 2005;48:6523–43.
- Cavalli A, Bolognesi ML, Minarini A, et al. Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem 2008;51:347–72.
- Prati F, Cavalli A, Bolognesi ML. Navigating the chemical space of multitarget-directed ligands: from hybrids to fragments in Alzheimer's disease. Mol Basel Switz 2016;21:466.
- Panek D, Więckowska A, Wichur T, et al. Design, synthesis and biological evaluation of new phthalimide and saccharin derivatives with alicyclic amines targeting cholinesterases, beta-secretase and amyloid beta aggregation. Eur J Med Chem 2017;125:676–95.
- Wager TT, Hou X, Verhoest PR, Villalobos A. Central nervous system multiparameter optimization desirability: application in drug discovery. ACS Chem Neurosci 2016;7:767–75.
- Mezeiova E, Korabecny J, Sepsova V, et al. Development of 2-methoxyhuprine as novel lead for Alzheimer’s disease therapy. Mol Basel Switz 2017;22:E1265.
- Dolles D, Nimczick M, Scheiner M, et al. Aminobenzimida-zoles and structural isomers as templates for dual-acting butyrylcholinesterase inhibitors and hCB2 R ligands to combat neurodegenerative disorders. ChemMedChem 2016;11:1270–83.
- Prati F, De Simone A, Bisignano P, et al. Multitarget drug discovery for Alzheimer's disease: triazinones as BACE-1 and GSK-3β inhibitors. Angew Chem Int Ed Engl 2015;54:1578–82.
- Wyss-Coray T, Rogers J. Inflammation in Alzheimer Disease: a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2012;2:a006346.