
Abstract
A practical and transition metal-free one-pot domino synthesis of diversified (1,3,4-oxadiazol-2-yl)anilines has been developed employing isatins and hydrazides as the starting materials, in the presence of molecular iodine. The prominent feature of this domino process involves consecutive condensation, hydrolytic ring cleavage, and an intramolecular decarboxylation, in a one-pot process that leads to the oxidative formation of a C–O bond. Fluorescence properties of some of the representative molecules obtained in this way were studied. The synthesised 2-(1,3,4-oxadiazolo-2-yl)aniline-benzene sulphonamides (8a–o) were screened for their carbonic anhydrase (CA, EC 4.2.1.1) inhibitory activity. Most of the compounds exhibited low micromolar to nanomolar activity against human (h) isoforms hCA I, hCA II, hCA IV, and XII, with some compounds displaying selective CA inhibitory activity towards hCA II with KIs of 6.4–17.6 nM.
Introduction
Construction of O–heterocyclic ring systems via intramolecular C–O bond formation has become an emerging tool in drug discovery. Accordingly, many efforts have been devoted to this activity, and remarkable results have been achieved to date. Among these, the traditional intramolecular Pd-catalysed Hartwig–BuchwaldCitation1 and copper-catalysedCitation2 Ullmann-type C–O coupling of aryl halides with hydroxyl moieties, and in an alternative approach, the direct dehydrogenative coupling occurs between C–H and O–H bondsCitation3, leading to various functionalised compounds. In most cases, these elaborative designs implied complex catalytic systems (based on Pd(II), Cu(II), Rh(III), and Ru(III) derivatives) and multi-step processes for the preparation of diversely functionalised derivatives, such as, furan, pyrrole, pyrazole, isoquinoline, indole, benzoxazole, and carbazole ring systemsCitation4. However, oxidative decarboxylation leading to construction of C–heteroatom bonds, particularly the C–O and the C–N bonds, has received significantly less attention. In recent years, in the perspective of green chemistry, most of the organic chemists have switched to metal-free reactions to reduce the burden of toxicity. In this context, iodine and hypervalent iodine reagents have emerged as inexpensive, versatile, and environmentally more friendly reagentsCitation5. Structural features and the reactivity pattern of these iodine compounds in many aspects are similar to those of the transition metal compounds applied for such purposes. Up until now, many efforts have been made to directly functionalise C–H bonds for the construction of C–C and C–heteroatoms bonds by employing iodine or hypervalent iodine reagentsCitation6,Citation7. Wang et al. demonstrated a facile access to various heterocycles (quinazoline, oxazole, and pyridine) through the tandem oxidative coupling reactions using iodine as catalyst and tert-butyl-hydroperoxide (TBHP) as the oxidantCitation8. Furthermore, Ma et al. proposed the synthesis of imidazo[1,2-a]pyridines via oxidative coupling of 2-aminopyridine with 1,3-diketones in the presence of tetra-butylammonium iodide (TBAI), TBHP, and BF3·etherateCitation9. Very recently Tang et al. reported iodine-catalysed radical oxidative annulation for the synthesis of dihydrofurans and indolisinesCitation10. Interestingly, I2 (or hypervalent iodine derivatives) also promoted the oxidative decarboxylation of amino acids and β,γ-unsaturated carboxylic acidsCitation11. Intrigued by these advances, herein we envisioned a metal-free, iodine-mediated domino strategy involving intramolecular decarboxylative coupling of isatins, and hydrazides for the synthesis of 2-(1,3,4-oxadiazol-2-yl)aniline derivatives (Scheme 1).
Scheme 1. Transition metal-free domino oxidative decarboxylation for the formation of 1,3,4-oxadiazole.
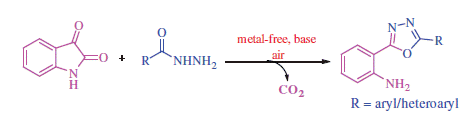
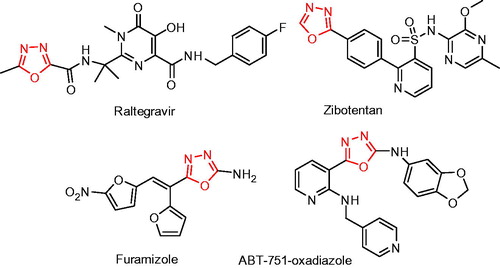
1,3,4-Oxadiazole motif is an important five-membered aromatic heterocyclic ring present in many bioactive moleculesCitation12,Citation13, including anticancer, antibacterial, anti-inflammatory, anti-diabetic, antiviral, anticonvulsant, analgesic, and antifungal agentsCitation12,Citation13. Some of the drugs and drug candidates, such as raltegravir, zibotentan, furamizole, and ABT-751-oxadiazole possessing 1,3,4-oxadiazole moieties are depicted in . Apart from biology, their applications have also been extended to material chemistry due to their unique optoelectronic propertiesCitation14.
Figure 1. Some of the bioactive compounds containing 1,3,4-oxadiazole moiety.
To date, a number of synthetic protocols have been described in the literature to access 1,3,4-oxadiazolesCitation15–17. They include: (i) oxidative cyclisation of N-acylhydrazones with FeCl3, CAN, PbO2, hypervalent iodines, chloramine T, KMnO4, Br2, HgO/I2; (ii) From 1,2-diacylhydrazones via cyclodehydration by employing PPA, POCl3, SOCl2, and H2SO4; (iii) Arylation of preformed 2-substituted 1,3,4-oxadiazoles through C–H activationCitation18. Additionally, Guin et al. successfully accomplished 2,5-disubstituted 1,3,4-oxadiazoles from N-arylidenearoyl hydrazides using Cu(OTf)2Citation19. Recently, Xu et al. demonstrated an easy access to synthesise 2-(1,3,4-oxadiazol-2-yl)anilines by employing CuI as the catalystCitation20. Nevertheless, the problems associated with these protocols, including the use of expensive, hazardous materials, or inefficient multi-step processes endowed them with a limited applicability. Therefore, more general and eco-friendly strategies for the synthesis of functionalised 1,3,4-oxadiazoles from easily available starting materials are still highly desirable. This prompted us to explore a simpler and more efficient protocol which is reported in this article.
Materials and methods
Chemistry
All solvents were purified and dried using standard methods prior to use. Commercially available reagents were used without further purification. All reactions involving air- or moisture-sensitive compounds were performed under a nitrogen atmosphere using dried glassware and syringe techniques to transfer solutions. Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel 60 F-254 aluminium plates. Melting points were determined on Stuart digital melting-point apparatus/SMP 30 in open capillary tubes and uncorrected. Nuclear magnetic resonance (1H-NMR, 13 C-NMR) spectra were recorded using an Avance Bruker 500 MHz, 125 MHz spectrometer in DMSO-d6. Chemical shifts reported in parts per million (ppm) with TMS as an internal reference, and the coupling constants (J) expressed in hertz (Hz). Splitting patterns are denoted as follows: s, singlet; d, doublet; t, triplet; m, multiplet; dd, doublet of doublet. HRMS were determined with Agilent QTOF mass spectrometer 6540 series instrument and were performed in the ESI techniques at 70 eV.
General procedure for the preparation of 2–(1,3,4-oxadiazol-2-yl) aniline derivatives (3a–u), (6a–g), and (8a–o)
A glass tube charged with a mixture of the desired isatin (0.5 mmol), aryl or heteroaryl hydrazide (0.5 mmol), I2 (100 mol%), K2CO3 (1.5 equiv.), and then 3 ml of DMSO at room temperature, was sealed and the resulting mixture was stirred under microwave irradiation at 160 °C until the disappearance of the reactants (monitored by TLC in 20% EtOAc and hexane). Iodine was then quenched by the addition of 10% aqueous Na2S2O3 and the product was extracted with EtOAc (3 × 25 ml). The combined extract was washed with brine, dried over anhydrous Na2SO4 and was concentrated under reduced pressure. The residue was purified by column chromatography on 60–120 silica gel using a mixture of EtOAc (bp 77 °C) and petroleum ether (bp 42–60 °C) as eluent to afford the desired product (correspondingly, 3a–3u/6a–6g/8a–o) as a yellow solid (yield, 69–92%).
2-(5-(p-Tolyl)-1,3,4-oxadiazol-2-yl)aniline (3a)
Yellow solid, Yield: 114 mg (91%), m.p 174–175 °C. 1H NMR (500 MHz, CDCl3) δ 8.02 (d, J = 7.7 Hz, 2H), 7.85 (d, J = 7.6 Hz, 1H), 7.31 (dd, J = 23.7, 7.5 Hz, 3H), 6.79 (dd, J = 14.7, 7.7 Hz, 2H), 5.89 (s, 2H), and 2.44 (s, 3H). 13 C NMR (126 MHz, DMSO) δ 164.5, 162.6, 148.2, 142.6, 132.9, 130.4, 128.2, 127.0, 120.9, 116.3, 116.1, 104.5, and 21.6. HRMS (ESI) m/z [M + H]+ calculated for C15H14N3O 252.1131, found 252.1136.
2-(5-(4-Methoxy-2-methylphenyl)-1,3,4-oxadiazol-2-yl)aniline (3b)
Yellow solid, Yield: 126 mg (90%), m.p 148.5–150 °C. 1H NMR (500 MHz, DMSO) δ 8.02 (d, J = 8.7 Hz, 1H), 7.80 (dd, J = 8.0, 1.4 Hz, 1H), 7.31–7.25 (m, 1H), 7.03 (d, J = 2.4 Hz, 1H), 6.99 (dd, J = 8.7, 2.6 Hz, 1H), 6.92 (d, J = 8.3 Hz, 1H), 6.78 (s, 2H), 6.73–6.68 (m, 1H), 3.84 (s, 3H), and 2.68 (s, 3H). 13 C NMR (126 MHz, DMSO) δ 163.8, 162.7, 161.8, 148.2, 140.30, 132.80, 131.13, 128.1, 117.3, 116.3, 116.0, 115.3, 112.6, 104.6, 55.8, and 22.3. HRMS (ESI) m/z: [M + H]+ calculated for C16H16N3O2 282.1237, found 282.1240.
2-(5-(3,4,5-Trimethoxyphenyl)-1,3,4-oxadiazol-2-yl)aniline (3c)
Yellow solid, Yield: 149 mg (91%), m.p 169–171 °C. 1H NMR (500 MHz, DMSO) δ 7.94 (dd, J = 8.0, 1.4 Hz, 1H), 7.39 (s, 2H), 7.30 (ddd, J = 8.5, 7.2, 1.5 Hz, 1H), 6.93 (dd, J = 8.3, 0.6 Hz, 1H), 6.79 (s, 2H), 6.75–6.69 (ddd,.1H), 3.92 (s, 6H), 3.77 (s, 3H). 13 C NMR (126 MHz, DMSO) δ 164.7, 162.4, 153.9, 148.3, 141.0, 133.0, 128.4, 118.9, 116.3, 115.9, 104.5, 104.4, 60.7, and 56.6. HRMS (ESI) m/z: [M + H]+ calculated for C17H18F3N3O4 328.1292, found 328.1291.
2-(5-(4-(Trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)aniline (3d)
Yellow solid, Yield: 129 mg (84%), m.p 195–197 °C. 1H NMR (500 MHz, DMSO) δ 8.34 (d, J = 8.0 Hz, 2H), 7.98 (d, J = 8.0 Hz, 2H), 7.88 (d, J = 7.8 Hz, 1H), 7.30 (t, J = 7.5 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 6.80 (s, 2H), and 6.72 (t, J = 7.4 Hz, 1H). 13 C NMR (126 MHz, DMSO) δ 165.3, 161.4, 148.5, 133.2, 128.3, 127.9, 126.7 (d, J = 3.7 Hz), 125.3, 123.1, 116.4, 116.0, and 104.1. HRMS (ESI) m/z: [M + H]+ calculated for C15H11F3N3O 306.0849, found 306.0846.
2-(5-(4-Fluorophenyl)-1,3,4-oxadiazol-2-yl)aniline (3e)
Yellow solid, Yield: 106 mg (83%), m.p 170–172 °C. 1H NMR (500 MHz, CDCl3) δ 8.21–8.10 (m, 2H), 7.84 (dd, J = 7.9, 1.3 Hz, 1H), 7.32–7.27 (m, 1H), 7.26–7.19 (m, 2H), 6.80 (dd, J = 15.8, 7.8 Hz, 2H), and 5.83 (s, 2H). 13 C NMR (126 MHz, CDCl3) δ 165.7, 163.77, 162.00, 147.1, 132.6, 129.20 (d, J = 8.9 Hz), 127.7, 120.24 (d, J = 3.3 Hz), 116.8, 116.5, 116.3, and 105.61. HRMS (ESI) m/z: [M + H]+ calculated for C14H11FN3O 256.0881, found 256.0878.
2-(5-(4-Bbromophenyl)-1,3,4-oxadiazol-2-yl)aniline (3f)
Yellow solid, Yield: 134 mg (85%), m.p 186–187 °C. 1H NMR (500 MHz, DMSO) δ 8.10–8.06 (m, 2H), 7.89–7.82 (m, 3H), 7.30 (ddd, J = 8.5, 7.2, 1.5 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 6.79 (d, J = 7.3 Hz, 2H), and 6.74–6.69 (m, 1H). 13 C NMR (126 MHz, DMSO) δ 164.9, 161.8, 148.4, 133.1, 132.9, 129.0, 128.3, 125.9, 123.0, 116.4, 116.0, and 104.3. HRMS (ESI) m/z: [M + H]+ calculated for C14H11BrN3O 316.0080, found 316.0078.
2-(5-(3,5-Dichlorophenyl)-1,3,4-oxadiazol-2-yl)aniline (3g)
Yellow solid, Yield: 126 mg (83%), m.p 210–212 °C. 1H NMR (500 MHz, DMSO) δ 8.16 (d, J = 1.9 Hz, 2H), 7.98 (dd, J = 8.0, 1.5 Hz, 1H), 7.92 (t, J = 1.9 Hz, 1H), 7.30 (ddd, J = 8.5, 7.1, 1.6 Hz, 1H), 6.92 (dd, J = 8.4, 0.7 Hz, 1H), 6.79 (s, 2H), and 6.73–6.68 (m, 1H). 13 C NMR (126 MHz, DMSO) δ 165.4, 160.5, 148.5, 135.6, 133.3, 131.64, 128.7, 127.0, 125.5, 116.3, 116.0, and 104.0. HRMS (ESI) m/z [M + H]+ calculated for C14H10Cl2N3O 306.0915, found 306.0918.
2-(5-(2-Methyl-5-nitrophenyl)-1,3,4-oxadiazol-2-yl)aniline (3i)
Yellow solid, Yield: 120 mg (81%), m.p 186.5–188 °C. 1H NMR (500 MHz, DMSO) δ 8.12 (d, J = 8.3 Hz, 1H), 8.00–7.96 (m, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.65 (dd, J = 8.0, 1.3 Hz, 1H), 7.33–7.27 (m, 1H), 6.94 (d, J = 8.3 Hz, 1H), 6.80 (s, 2H), 6.73–6.66 (m, 1H), 2.52 (s, 3H). 13 C NMR (126 MHz, DMSO) δ 165.4, 159.3, 148.4, 146.2, 145.3, 133.9, 133.4, 132.0, 128.0, 125.3, 117.6, 116.5, 116.1, 103.8, and 21.2. HRMS (ESI) m/z: [M + H]+ calculated for C15H13N4O3 297.0982, found 297.0976.
4-Chloro-2-(5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)aniline (3j)
Yellow solid, Yield: 137 mg (81%), m.p 196–198 °C. 1H NMR (500 MHz, DMSO) δ 8.34 (t, J = 44.9 Hz, 2H), 8.17–7.76 (m, 3H), 7.32 (d, J = 7.2 Hz, 1H), 6.95 (s, 3H). 13 C NMR (126 MHz, DMSO) δ 164.2, 161.8, 147.3, 132.9, 128.16, 127.45, 127.19, 126.7 (d, J = 3.7 Hz), 125.33, 119.26, 118.3, and 105.1. HRMS (ESI) m/z: [M + H]+calculated for C15H10ClF3N3O 340.0459, found 340.0462.
4-Chloro-2-(5-(4-fluorophenyl)-1,3,4-oxadiazol-2-yl)aniline (3k)
Yellow solid, Yield: 117 mg (81%), m.p 176–177.5 °C. 1H NMR (500 MHz, DMSO) δ 8.29–8.23 (m, 2H), 7.91 (d, J = 2.5 Hz, 1H), 7.50–7.44 (m, 2H), 7.31 (dd, J = 8.9, 2.5 Hz, 1H), 6.96 (d, J = 8.9 Hz, 1H), 6.92 (s, 2H). 13 C NMR (126 MHz, DMSO) δ 165.67, 163.78, 162.0, 147.1, 132.7, 130.0 (d, J = 9.1 Hz), 127.1, 120.0 (d, J = 3.1 Hz), 119.2, 118.2, 117.1, 116.9, and 105.4. HRMS (ESI) m/z: [M + H]+ calculated for C14H10ClFN3O 290.0941, found 290.0939.
2-(5-(4-Bromophenyl)-1,3,4-oxadiazol-2-yl)-4-chloroaniline (3l)
Yellow solid, Yield: 145 mg (83%), m.p 196–197 °C. 1H NMR (500 MHz, DMSO) δ 8.17–8.13 (m, 2H), 7.93 (d, J = 2.5 Hz, 1H), 7.87–7.82 (m, 2H), 7.33 (dd, J = 8.9, 2.5 Hz, 1H), 6.97–6.91 (m, 3H). 13 C NMR (126 MHz, DMSO) δ 163.9, 162.2, 147.2, 132.9, 132.8, 129.2, 127.1, 126.1, 122.9, 119.2, 118.2, and 105.3. HRMS (ESI) m/z: [M + H]+ calculated for C14H10BrClN3O 349.9690, found 349.9698.
4-Chloro-2-(5-(3,5-dichlorophenyl)-1,3,4-oxadiazol-2-yl)aniline (3m)
Yellow solid, Yield: 137 mg (81%), m.p 229–231 °C. 1H NMR (500 MHz, DMSO) δ 8.24 (d, J = 1.8 Hz, 2H), 8.06 (d, J = 2.5 Hz, 1H), 7.92 (t, J = 1.8 Hz, 1H), 7.32 (dd, J = 8.9, 2.5 Hz, 1H), 6.97–6.92 (m, 3H). 13 C NMR (126 MHz, DMSO) δ 164.4, 160.8, 147.3, 135.6, 133.06, 131.7, 127.4, 126.8, 125.75, 119.35, 118.2, and 105.06. HRMS (ESI) m/z: [M + H]+ calculated for C14H9Cl3N3O 339.9806, found 339.9811.
4-Chloro-2-(5-(5-methoxy-2-methylphenyl)-1,3,4-oxadiazol-2-yl)aniline (3n)
Yellow solid, Yield: 140 mg (89%), m.p 158–15.5 °C. 1H NMR (500 MHz, DMSO) δ 8.08 (d, J = 8.7 Hz, 1H), 7.79 (d, J = 2.4 Hz, 1H), 7.30 (dd, J = 8.9, 2.5 Hz, 1H), 7.02 (d, J = 2.1 Hz, 1H), 6.99–6.90 (m, 4H), 3.84 (s, 3H), and 2.67 (s, 3H). 13 C NMR (126 MHz, DMSO) δ 162.9, 162.74, 161.8, 147.0, 140.4, 132.4, 131.3, 126.9, 119.1, 118.1, 117.3, 115.1, 112.6, 105.6, 55.87, and 22.3. HRMS (ESI) m/z: [M + H]+ calculated for C16H15ClN3O2 316.0847, found 316.0844.
4-Bromo-2-(5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)aniline (3o)
Yellow solid, Yield: 157 mg (82%), m.p 167–168 °C. 1H NMR (500 MHz, DMSO) δ 8.40 (d, J = 7.4 Hz, 2H), 8.09–7.93 (m, 3H), 7.42 (d, J = 8.4 Hz, 1H), 6.97 (s, 2H), 6.91 (d, J = 8.8 Hz, 1H). 13 C NMR (126 MHz, DMSO) δ 164.2, 161.8, 147.6, 135.6, 130.0, 128.1, 127.4, 126.74 (d, J = 3.7 Hz), 125.3, 118.6, 106.2, and 105.85. HRMS (ESI) m/z: [M + H]+ calculated for C15H10BrF3N3O 383.9984, found 383.9990.
2-(5-(3,5-Dimethoxyphenyl)-1,3,4-oxadiazol-2-yl)-4-fluoroaniline (3p)
Yellow solid, Yield: 139 mg (88%), m.p 152–154 °C. 1H NMR (500 MHz, DMSO) δ 7.78 (dd, J = 9.6, 3.0 Hz, 1H), 7.28 (t, J = 8.4 Hz, 2H), 7.21 (td, J = 8.7, 3.0 Hz, 1H), 6.94 (dd, J = 9.1, 4.8 Hz, 1H), 6.76 (t, J = 2.2 Hz, 1H), 6.70 (s, 2H), 3.87 (s, 6H). 13 C NMR (126 MHz, DMSO) δ 164.1 (d, J = 2.6 Hz), 162.6, 161.4, 154.5, 152.7, 145.2, 125.2, 120.8 (d, J = 22.5 Hz), 117.8 (d, J = 7.4 Hz), 113.4 (d, J = 24.4 Hz), 105.4, 104.4, 104.01 (d, J = 8.2 Hz), and 56.11 (s). HRMS (ESI) m/z: [M + H]+ calculated for C16H15FN3O3 316.1092, found 316.1094.
2-(5-(4-Fluorophenyl)-1,3,4-oxadiazol-2-yl)-4-(trifluoromethoxy)aniline (3q)
Yellow solid, Yield: 129 mg (80%), m.p 183–184 °C. 1H NMR (500 MHz, DMSO) δ 8.30–8.23 (m, 18H), 7.87 (d, J = 2.6 Hz, 8H), 7.48 (t, J = 8.8 Hz, 18H), 7.32 (dd, J = 9.0, 2.0 Hz, 9H), 7.02 (s, 5H), 7.00 (s, 17H). 13 C NMR (126 MHz, DMSO) δ 165.6, 163.7 (d, J = 9.4 Hz), 162.1, 147.5, 138.1 (d, J = 2.0 Hz), 130.1 (d, J = 9.2 Hz), 126.5, 121.8, 120.9, 120.3 (d, J = 3.0 Hz), 119.8, 117.6, 117.1 (d, J = 22.5 Hz), and 104.2. HRMS (ESI) m/z: [M + H]+ calculated for C15H10F4N3O2 340.0755, found 340.0759.
4-(Trifluoromethoxy)-2-(5-(3,4,5-trimethoxyphenyl)-1,3,4-oxadiazol-2-yl)aniline (3r)
Yellow solid, Yield: 172 mg (87%), m.p 203.5–205 °C. 1H NMR (500 MHz, DMSO) δ 7.93 (s, 1H), 7.44 (s, 2H), 7.32 (s, 1H), 7.01 (s, 3H), 3.93 (s, 6H), 3.77 (s, 3H). 13 C NMR (126 MHz, DMSO) δ 163.7, 162.8, 153.9, 147.4, 141.2, 138.1, 126.4, 121.0, 118.7, 117.6, 104.9, 104.2, 60.7, and 56.8. HRMS (ESI) m/z: [M + H]+ calculated for C18H17F3N3O5 411.1166, found 411.1169.
2-(5-(3,5-Dimethylphenyl)-1,3,4-oxadiazol-2-yl)-4-(trifluoromethoxy)aniline (3s)
Yellow solid, Yield: 144 mg (84%), m.p 199–201 °C. 1H NMR (500 MHz, DMSO) δ 7.78 (dd, J = 9.6, 3.0 Hz, 1H), 7.28 (t, J = 8.4 Hz, 2H), 7.21 (td, J = 8.7, 3.0 Hz, 1H), 6.94 (dd, J = 9.1, 4.8 Hz, 1H), 6.76 (t, J = 2.2 Hz, 1H), 6.70 (s, 2H), 3.87 (s, 6H). 13 C NMR (126 MHz, DMSO) δ 163.6, 163, 147.4, 139.2, 138, 133, 126.4, 124.8, 123.4, 120.9, 117.6, 104.3, and 21.1. HRMS (ESI) m/z: [M + H]+ calculated for C17H15F3N3O 350.1162, found 350.1158.
2-(5-(5-Methoxy-2-methylphenyl)-1,3,4-oxadiazol-2-yl)-4-nitroaniline (3t)
Yellow solid, Yield: 139 mg (85%), m.p 240–241 °C. 1H NMR (500 MHz, DMSO) δ 8.63 (t, J = 9.8 Hz, 1H), 8.21–7.92 (m, 4H), 7.10–6.99 (m, 3H), 3.84 (d, J = 20.0 Hz, 3H), 2.67 (d, J = 25.2 Hz, 3H). 13 C NMR (126 MHz, DMSO) δ 163.3, 162.2, 162.0, 153.0, 140.5, 136.3, 131.4, 128.0, 125.4, 117.3, 116.2, 114.9, 112.7, 103.9, 55.9, and 22.3. HRMS (ESI) m/z: [M + H]+ calculated for C16H15N4O4 327.1088, found 327.1089.
2-(5-(3,5-Dimethoxyphenyl)-1,3,4-oxadiazol-2-yl)-4-methoxyaniline (3u)
Yellow solid, Yield: 150 mg (92%), m.p 143.5–145 °C. 1H NMR (500 MHz, DMSO) δ 8.30–8.23 (m, 18H), 7.87 (d, J = 2.6 Hz, 8H), 7.48 (t, J = 8.8 Hz, 18H), 7.32 (dd, J = 9.0, 2.0 Hz, 9H), 7.02 (s, 5H), and 7.00 (s, 17H). 13 C NMR (126 MHz, DMSO) δ 164.80, 162.41, 161.47, 150.35, 143.13, 125.36, 121.72, 118.03, 111.04, 105.08, 104.13, 104.12, 56.19, and 56.08. HRMS (ESI) m/z: [M + H]+ calculated for C17H18N3O4 328.1292, found 328.1290.
2-(5-(Pyridin-4-yl)-1,3,4-oxadiazol-2-yl)aniline (6a)
Yellow solid, Yield: 108 mg (90%), m.p 174–175 °C. 1H NMR (500 MHz, DMSO) δ 8.85 (dd, J = 4.5, 1.6 Hz, 2H), 8.07 (dd, J = 4.5, 1.5 Hz, 2H), 7.89 (dd, J = 8.0, 1.4 Hz, 1H), 7.31 (ddd, J = 8.5, 7.2, 1.5 Hz, 1H), 6.94 (d, J = 7.9 Hz, 1H), 6.81 (s, 2H), 6.76–6.68 (m, 1H). 13 C NMR (126 MHz, DMSO) δ 165.5, 161.0, 151.3, 148.6, 133.4, 130.9, 128.4, 120.7, 116.5, 116.0, and 104.0. HRMS (ESI) m/z: [M + H]+ calculated for C13H11N4O 239.0927, found 239.0928.
2-(5-(Isoquinolin-3-yl)-1,3,4-oxadiazol-2-yl)aniline (6b)
Yellow solid, Yield: 126 mg (87%), m.p 256–257 °C. 1H NMR (500 MHz, CDCl3) δ 9.43 (s, 1H), 8.69 (s, 1H), 8.13–7.96 (m, 3H), 7.83 (t, J = 7.4 Hz, 1H), 7.76 (t, J = 7.3 Hz, 1H), 7.35–7.27 (m, 1H), 6.86–6.79 (m, 2H). 13 C NMR (126 MHz, DMSO) δ 165.20, 162.55, 153.88, 148.50, 136.96, 135.62, 133.17, 132.25, 130.01, 129.37, 128.43, 128.2, 128.1, 121.16, 116.50, 116.11, and 104.40. HRMS (ESI) m/z: [M + H]+ calculated for C17H13N4O 289.1084, found 289.1088.
2-(5-(1H-indazol-3-yl)-1,3,4-oxadiazol-2-yl)aniline (6c)
Yellow solid, Yield: 95 mg (69%) m.p 281–283 °C. 1H NMR (500 MHz, DMSO) δ 14.05 (s, 1H), 8.27 (d, J = 8.2 Hz, 1H), 7.83 (dd, J = 8.0, 1.3 Hz, 1H), 7.73 (t, J = 8.5 Hz, 1H), 7.54 (dd, J = 8.2, 7.1 Hz, 1H), 7.40 (dd, J = 7.9, 7.2 Hz, 1H), 7.35–7.29 (m, 1H), 6.96 (d, J = 8.0 Hz, 1H), 6.85 (s, 2H), 6.75 (t, J = 7.5 Hz, 1H). 13 C NMR (126 MHz, DMSO) δ 164.03, 158.68, 148.42, 141.46, 133.08, 130.12, 128.06, 127.81, 123.35, 121.28, 121.09, 116.48, 116.20, 111.64, and 104.36. HRMS (ESI) m/z: [M + H]+ calculated for C15H12N5O 278.1036, found 278.1038.
4-Fluoro-2-(5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl)aniline (6d)
Yellow solid, Yield: 99 mg (77%), m.p 226–227.5 °C. 1H NMR (500 MHz, DMSO) δ 8.86 (dd, J = 4.4, 1.6 Hz, 2H), 8.12 (dd, J = 4.4, 1.6 Hz, 2H), 7.75 (dd, J = 9.6, 3.0 Hz, 1H), 7.24 (ddd, J = 8.9, 8.4, 3.0 Hz, 1H), 6.96 (dd, J = 9.1, 4.8 Hz, 1H), 6.73 (s, 2H). 13 C NMR (126 MHz, DMSO) δ 164.8, 161.2, 154.5, 152.6, 151.3, 145.5, 130.7, 121.2 (d, J = 23.7 Hz), 120.8, 118.07 (d, J = 7.5 Hz), 113.3 (d, J = 22.5 Hz), and 103.6 (d, J = 8.3 Hz). HRMS (ESI) m/z: [M + H]+ calculated for C13H10FN4O 257.0833, found 257.0837.
4-Chloro-2-(5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl)aniline (6e)
Yellow solid, Yield: 115 mg (84%), m.p 208–209 °C. 1H NMR (500 MHz, DMSO) δ 8.86 (dd, J = 7.1, 2.7 Hz, 2H), 8.19–8.07 (m, 2H), 7.94 (dd, J = 4.9, 2.5 Hz, 1H), 7.33 (ddd, J = 7.2, 4.0, 2.1 Hz, 1H), 7.02–6.89 (m, 3H). 13 C NMR (126 MHz, DMSO) δ 164.53, 161.34, 151.34, 147.41, 133.11, 130.77, 127.28, 120.88, 119.29, 118.35, and 105.05. HRMS (ESI) m/z: [M + H]+ calculated for C13H10ClN4O 273.0538, found 273.0537.
4-Chloro-2-(5-(isoquinolin-3-yl)-1,3,4-oxadiazol-2-yl)aniline (6f)
Yellow solid, Yield: 119 mg (74%), m.p 213–214 °C. 1H NMR (500 MHz, DMSO) δ 9.52 (s, 1H), 8.88 (s, 1H), 8.27 (s, 2H), 7.90 (d, J = 37.9 Hz, 3H), 7.34 (s, 1H), 6.99 (s, 3H). 13 C NMR (126 MHz, DMSO) δ 164.0, 162.77, 153.9, 147.3, 136.7, 135.6, 132.8, 132.2, 130.0, 129.4, 128.4, 128.1, 126.9, 121.4, 119.2, 118.3, and 105.3. HRMS (ESI) m/z: [M + H]+ calculated for C17H12ClN4O 323.0694, found 323.0688.
2-(5-(Isoquinolin-3-yl)-1,3,4-oxadiazol-2-yl)-4-methoxyaniline (6g)
Yellow solid, Yield: 118 mg (74%), m.p 190–191 °C. 1H NMR (500 MHz, DMSO) δ 9.54 (s, 1H), 8.86 (s, 1H), 8.27 (dd, J = 17.0, 8.1 Hz, 2H), 7.94 (t, J = 7.5 Hz, 1H), 7.86 (t, J = 7.5 Hz, 1H), 7.36 (d, J = 2.7 Hz, 1H), 7.04 (dd, J = 9.0, 2.8 Hz, 1H), 6.93 (d, J = 9.0 Hz, 1H), 6.49 (s, 2H), 3.80 (s, 3H).13 C NMR (126 MHz, DMSO) δ 165.1, 162.5, 153.8, 150.3, 143.2, 136.9, 135.6, 132.2, 130.1, 129.3, 128.4, 128.1, 122.0, 121.2, 118.2, 110.5, 104.1, and 56.2. HRMS (ESI) m/z: [M + H]+ calculated for C18H15N4O2 319.1190, found 319.1191.
4-(5-(2-Aminophenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8a)
Yellow solid, Yield: 118 mg (75%), m.p 282–283 °C. 1H NMR (500 MHz, DMSO) δ 8.38–8.31 (m, 2H), 8.06 (d, J = 8.5 Hz, 2H), 7.89 (dt, J = 12.5, 6.3 Hz, 1H), 7.61 (s, 2H), 7.34–7.27 (m, 1H), 6.93 (t, J = 7.0 Hz, 1H), 6.79 (d, J = 13.9 Hz, 2H), 6.76–6.70 (m, 1H). 13 C NMR (126 MHz, DMSO) δ 165.2, 161.6, 148.5, 147.0, 133.2, 128.4, 127.7, 127.1, 116.4, 116.0, and 104.2. HRMS (ESI) m/z: [M + H]+ calculated for C14H13N4O3S 317.0703, found 317.0710.
4-(5-(2-Amino-5-methylphenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8b)
Yellow solid, Yield: 117 mg (71%), m.p 278–279 °C.1H NMR (500 MHz, DMSO) δ 8.38–8.31 (m, 2H), 8.09–8.02 (m, 2H), 7.71 (d, J = 0.9 Hz, 1H), 7.59 (s, 2H), 7.14 (dt, J = 14.5, 7.3 Hz, 1H), 6.85 (t, J = 8.8 Hz, 1H), 6.59 (d, J = 21.4 Hz, 2H), 2.26 (s, 3H).13 C NMR (126 MHz, DMSO) δ 165.3, 161.6, 147.1, 146.4, 134.4, 127.7, 127.1, 126.6, 124.6, 116.7, 104.0, and 20.3. HRMS (ESI) m/z: [M + H]+ calculated for C14H13N4O3S 331.0859, found 331.0866.
4-(5-(2-Amino-5-methoxyphenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8c)
Yellow solid, Yield: 130 mg (75%), m.p 253–254 °C.1H NMR (500 MHz, DMSO) δ 8.38 (t, J = 10.3 Hz, 2H), 8.09–8.01 (m, 2H), 7.59 (d, J = 7.1 Hz, 2H), 7.40 (d, J = 2.9 Hz, 1H), 7.03 (dd, J = 9.0, 2.9 Hz, 1H), 6.90 (dd, J = 13.1, 8.5 Hz, 1H), 6.44 (s, 2H), 3.78 (s, 3H). 13 C NMR (126 MHz, DMSO) δ 165.1, 161.7, 150.4, 147.1, 143.3, 127.8, 127.1, 126.6, 122.10, 118.1, 110.8, 103.9, and 56.2. HRMS (ESI) m/z: [M + H]+ calculated for C15H15N4O4S 347.0809, found 347.0812.
4-(5-(2-Amino-5-fluorophenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8d)
Yellow solid, Yield: 117 mg (70%), m.p 290–291 °C.1H NMR (500 MHz, DMSO) δ 8.42–8.36 (m, 2H), 8.08–8.02 (m, 2H), 7.79–7.71 (m, 1H), 7.59 (s, 2H), 7.21 (tdd, J = 11.9, 8.7, 3.0 Hz, 1H), 6.95 (td, J = 9.4, 4.8 Hz, 1H), 6.71 (s, 2H).13 C NMR (126 MHz, DMSO) δ 13 C NMR (126 MHz, DMSO) δ 164.49 (s), 161.92 (s), 154.55 (s), 152.71 (s), 147.22 (s), 145.39 (s), 127.94 (s), 127.08 (s), 126.50 (s), 121.08 (d, J = 23.2 Hz), 118.02 (d, J = 7.5 Hz), 113.33 (d, J = 24.4 Hz), and 103.85 (d, J = 8.2 Hz). HRMS (ESI) m/z: [M + H]+ calculated for C14H12FN4O3S 335.0609, found 335.0621.
4-(5-(2-Amino-5-chlorophenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8e)
Yellow solid, Yield: 117 mg (67%), m.p 306–307 °C.1H NMR (500 MHz, DMSO) δ 8.40 (d, J = 8.4 Hz, 2H), 8.08–8.03 (m, 2H), 7.93 (t, J = 4.8 Hz, 1H), 7.60 (s, 2H), 7.33 (dd, J = 8.9, 2.4 Hz, 1H), 6.95 (dd, J = 17.6, 11.7 Hz, 3H).13 C NMR (126 MHz, DMSO) δ 164.2, 161.9, 147.2 (d, J = 11.0 Hz), 132.9, 127.9, 127.1 (d, J = 13.3 Hz), 126.4, 119.2, 118.3, and 105.2. HRMS (ESI) m/z: [M + H]+ calculated for C14H12ClN4O3S 353.0313, found 353.0305.
4-(5-(2-Amino-5-(trifluoromethoxy)phenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8f)
Yellow solid, Yield: 138 mg (65%), m.p 242–243 °C.1H NMR (500 MHz, DMSO) δ 8.42–8.37 (m, 2H), 8.08–8.03 (m, 2H), 7.90 (d, J = 2.8 Hz, 1H), 7.60 (s, 2H), 7.34 (dt, J = 11.2, 5.6 Hz, 1H), 7.04–7.00 (m, 3H).13 C NMR (126 MHz, DMSO) δ 164.1, 162.0, 147.6, 147.4 (d, J = 51.9 Hz), 147.2, 138.1, 128.0, 127.0, 126.8,126.4, 121.8, 121.1, 119.8, 117.7, and 104.0. HRMS (ESI) m/z: [M + H]+ calculated for C15H12F3N4O3S 401.0525, found 401.0536.
4-(5-(2-Amino-3-fluorophenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8g)
Yellow solid, Yield: 102 mg (61%), m.p 260–261 °C.1H NMR (500 MHz, DMSO) δ 8.38–8.33 (m, 2H), 8.08–8.04 (m, 2H), 7.78 (t, J = 6.3 Hz, 1H), 7.60 (s, 2H), 7.32 (ddd, J = 11.7, 7.9, 1.3 Hz, 1H), 6.79–6.72 (m, 3H).13 C NMR (126 MHz, DMSO) δ 164.5 (d, J = 4.1 Hz), 161.9, 152.2, 150.3, 147.2, 137.1 (d, J = 15.6 Hz), 137.1(d, J = 15.6 Hz), 127.9, 127.1, 126.4, 124.0 (d, J = 3.4 Hz), 118.0 (d, J = 17.9 Hz), 115.7 (d, J = 7.3 Hz), and 106.8 (d, J = 5.6 Hz). HRMS (ESI) m/z: [M + H]+ calculated for C14H12FN4O3S 335.0609, found 335.0611.
4-(5-(2-Amino-3,5-dimethylphenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8h)
Yellow solid, Yield: 112 mg (65%), m.p 295–296 °C.1H NMR (500 MHz, DMSO) δ 8.34 (d, J = 7.6 Hz, 2H), 8.05 (d, J = 7.7 Hz, 2H), 7.60 (s, 3H), 7.07 (s, 1H), 6.44 (s, 2H), 2.22 (d, J = 35.1 Hz, 6H).13 C NMR (126 MHz, DMSO) δ 165.6, 161.6, 147.1, 144.6, 135.2, 127.7, 127.1, 126.6, 125.7, 124.6, 123.7, 104.0, 20.3, and 18.1. HRMS (ESI) m/z: [M + H]+ calculated for C16H17N4O3S 345.1016, found 345.1012.
4-(5-(2-Amino-3,5-dichlorophenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8i)
Yellow solid, Yield: 114 mg (60%), m.p 284–285 °C.1H NMR (500 MHz, DMSO) δ 8.29–8.24 (m, 2H), 8.08–8.02 (m, 2H), 7.60 (s, 2H), 7.52 (t, J = 9.3 Hz, 1H), 6.85 (d, J = 8.5 Hz, 1H), 6.43 (s, 2H).13 C NMR (126 MHz, DMSO) δ 164.1, 161.2, 147.3, 146.5, 133.22 (d, J = 41.0 Hz), 127.9, 127.2, 126.7, 117.5 (d, J = 33.0 Hz), and 106.9. HRMS (ESI) m/z: [M + H]+ calculated for C14H11Cl2N4O3S 384.9923, found 384.9927.
3-(5-(2-Aminophenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8j)
Yellow solid, Yield: 99 mg (63%), m.p 257–258 °C. 1H NMR (500 MHz, DMSO) δ 8.56 (s, 1H), 8.36 (d, J = 7.9 Hz, 1H), 8.07 (d, J = 7.9 Hz, 1H), 7.90–7.82 (m, 2H), 7.62 (s, 2H), 7.34–7.29 (m, 1H), 6.94 (d, J = 8.3 Hz, 1H), 6.81 (s, 2H), 6.73 (dd, J = 11.1, 4.0 Hz, 1H).13 C NMR (126 MHz, DMSO) δ 13 C NMR (126 MHz, DMSO) δ 165.1, 161.6, 148.4, 145.7, 133.2, 130.9, 130.1, 129.0, 128.3, 124.6, 124.0, 116.4, 116.0, and 104.2. HRMS (ESI) m/z: [M + H]+ calculated for C14H13N4O3S 317.0703, found 317.0711.
3-(5-(2-Amino-5-methylphenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8k)
Yellow solid, Yield: 107 mg (65%), m.p 264–265 °C. 1H NMR (500 MHz, DMSO) δ 8.55 (s, 1H), 8.37 (d, J = 7.7 Hz, 1H), 8.07 (d, J = 7.7 Hz, 1H), 7.85 (t, J = 7.8 Hz, 1H), 7.65 (d, J = 22.4 Hz, 3H), 7.15 (d, J = 8.1 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 6.62 (s, 2H), 2.27 (s, 3H).13 C NMR (126 MHz, DMSO) δ 165.2, 161.5, 146.4, 145.7, 134.4, 130.8, 130.2, 129.0, 127.6, 124.6 (d, J = 2.3 Hz),124.0, 116.7, 104.0, and 20.3. HRMS (ESI) m/z: [M + H]+ calculated for C15H15N4O3S 331.0859, found 331.0855.
3-(5-(2-Amino-5-methoxyphenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8l)
Yellow solid, Yield: 119 mg (69%), m.p 240–242 °C. 1H NMR (500 MHz, DMSO) δ 8.57 (t, J = 1.5 Hz, 1H), 8.42–8.37 (m, 1H), 8.09–8.04 (m, 1H), 7.85 (t, J = 7.8 Hz, 1H), 7.64 (s, 2H), 7.38 (d, J = 2.9 Hz, 1H), 7.04 (dd, J = 9.0, 2.9 Hz, 1H), 6.92 (d, J = 9.0 Hz, 1H), 6.45 (s, 2H), 3.78 (s, 3H).13 C NMR (126 MHz, DMSO) δ 165.0, 161.6, 150.36, 145.7, 143.2, 130.8, 130.3, 129.0, 124.5, 124.0, 121.9, 118.1, 110.9, 103.9, and 56.2. HRMS (ESI) m/z: [M + H]+ calculated for C15H15N4O4S347.0809, found 347.0818.
3-(5-(2-Amino-5-fluorophenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8m)
Yellow solid, Yield: 100 mg (60%), m.p225–226 °C. 1H NMR (500 MHz, DMSO) δ 8.59 (t, J = 1.6 Hz, 1H), 8.41 (d, J = 7.8 Hz, 1H), 8.06 (ddd, J = 11.1, 6.1, 4.8 Hz, 1H), 7.85 (t, J = 7.8 Hz, 1H), 7.74–7.67 (m, 1H), 7.63 (s, 2H), 7.23 (td, J = 8.6, 3.0 Hz, 1H), 6.96 (dd, J = 9.1, 4.8 Hz, 1H), 6.71 (s, 2H).13 C NMR (126 MHz, DMSO) δ 164.38, 161.8, 154.5, 152.7, 145.7, 145.3, 130.8, 130.3, 129.1, 124.4, 124.1, 121.1, 121.0 (d, J = 23.1 Hz), 120.9, 118.0 (d, J = 7.4 Hz), 113.3, 113.2 (d, J = 24.3 Hz), 113.1, and 103.8 (d, J = 8.2 Hz). HRMS (ESI) m/z: [M + H]+ calculated for C14H12FN4O3S335.0609, found 335.0617.
3-(5-(2-Amino-5-(trifluoromethoxy)phenyl)-1,3,4-oxadiazol-2-yl)benzenesulfonamide (8n)
Yellow solid, Yield: 120 mg (60%), m.p 253–255 °C. 1H NMR (500 MHz, DMSO) δ 8.56 (s, 1H), 8.37 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 8.3 Hz, 1H), 7.85 (t, J = 7.8 Hz, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.62 (s, 2H), 7.32 (dd, J = 10.7, 8.0 Hz, 1H), 6.80–6.72 (m, 3H).13 C NMR (126 MHz, DMSO) δ 164.4, 161.9, 152.2, 150.3, 145.8, 137.1 (d, J = 15.6 Hz), 130.9, 130.3, 129.2, 124.4, 124.1, 123.9 (d, J = 3.2 Hz), 118.0 (d, J = 17.9 Hz), 115.7 (d, J = 7.2 Hz), and 106.8 (d, J = 5.4 Hz). HRMS (ESI) m/z: [M + H]+ calculated for C15H12F3N4O4S401.0526, found 401.0533.
3-(5-(2-Amino-3-fluorophenyl)-1,3,4-ovxadiazol-2-yl)benzenesulfonamide (8o)
Yellow solid, Yield: 101 mg (61%), m.p 231–233 °C. 1H NMR (500 MHz, DMSO) δ 8.58 (t, J = 1.6 Hz, 1H), 8.45–8.40 (m, 1H), 8.10–8.06 (m, 1H), 7.85 (dd, J = 9.0, 6.6 Hz, 2H), 7.63 (s, 2H), 7.35 (dd, J = 9.1, 2.0 Hz, 1H), 7.05–7.00 (m, 3H).13 C NMR (126 MHz, DMSO) δ 164.0, 161.9, 147.6, 145.7, 138.0 (d, J = 1.9 Hz), 130.8, 130.5, 129.2, 126.7, 124.4, 124.2 (d, J = 33.0 Hz), 124.1, 120.9, 117.7, and 104.09. HRMS (ESI) m/z: [M + H]+ calculated for C14H12FN4O3S335.0609, found 335.0615.
Carbonic anhydrase inhibition assay
An SX.18 MV-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the catalytic/inhibition of various CA isozymesCitation24. Phenol Red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.4) as buffer, 0.1 M Na2SO4 or NaClO4 (for maintaining constant the ionic strength; these anions are not inhibitory in the used concentration), following the CA-catalysed CO2 hydration reaction for a period of 5–10 s. Saturated CO2 solutions in water at 25 °C were used as substrate. Stock solutions of inhibitors were prepared at a concentration of 10 µM (in DMSO-water 1:1, v/v) and dilutions up to 0.01 nM done with the assay buffer mentioned above. At least seven different inhibitor concentrations have been used for measuring the inhibition constant. Inhibitor and enzyme solutions were preincubated together for 10 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. Triplicate experiments were done for each inhibitor concentration, and the values reported throughout the paper are the mean of such results. The inhibition constants were obtained by non-linear least-squares methods using the Cheng–Prusoff equation, as reported earlier, and represent the mean from at least three different determinations. All CA isozymes used here were recombinant proteins obtained as reported earlier by our groupCitation25,Citation26.
Results and discussion
Chemistry
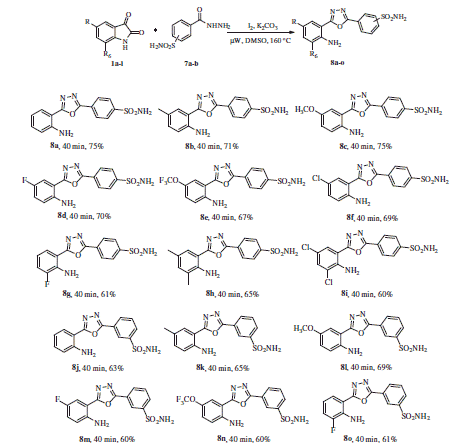
We commenced our investigation with a reaction using an equimolar ratio of isatin and 4-methyl benzohydrazide as model substrates using molecular iodine (100 mol%) and Cs2CO3 (1.0 equiv.) in DMSO at 100 °C (). The desired product was obtained in 71% yield (, entry 1). No product was obtained in the absence of either catalyst or base which suggests that an iodine/base combination is required for the reaction to occur (, entries 2–4). Exploring the possibility for improving the reaction efficiency, the effect of other alkali metal carbonates/other bases on the reaction efficiency was then examined. The transformation underwent smoothly in the presence of K2CO3 to afford the desired product 3a in 80% yield after 12 h (, entry 5),whereas other bases, such as Na2CO3, K3PO4, and NaHCO3 were found to be less effective (, entries 6–8). With an attempt to further optimise the yield of the product, we investigated the influence of various iodine reagents. TBAI, N-iodosuccinimide (NIS) and KI gave poor to moderate yields, i.e. of 18, 45, and 35%, respectively (, entries 9–11). However, phenyliodine(III) diacetate (PIDA), and hydroxy(tosyloxy)iodobenzene (HITB) did not at all lead to the formation of the desired product 3a (, entries 12–13). Furthermore, a series of experiments were also carried out in various other solvents, such as, DMF, MeCN, THF, 1,4-dioxane, EtOH, MeOH, and H2O. From the obtained results, it can be seen that the use of DMSO and DMF at 120 °C gave an almost identical result, albeit with a lower yield in the latter case (, entries 14–15), whereas, MeCN, THF, 1,4-dioxane, EtOH, MeOH, and H2O at reflux temperatures proved to be less effective (, entries 16–21). Furthermore, the iodine loading was also investigated in this reaction, and the yields were dropped to 59 and 52 at 0.75 and 0.5 equiv., respectively (, entries 22–23) of I2, and to a significantly lower value of 33% at 0.2 mol equiv. of I2 (, entry 24). We also conducted a control experiment under nitrogen atmosphere, but the yield under these conditions was diminished to 25%. This indicated that atmospheric O2 played an important role in the above transformation. Surprisingly, when the same reaction was performed under microwave irradiation gave better yield of 3a (91%) within a short span of time (40 min). Indeed, the use of microwave technology has never been mentioned in the literature for the synthesis of 2–(1,3,4-oxadiazol-2-yl)aniline derivatives up until now. Thus the foregoing experiments led to the conclusion that the conditions used under entry 25 of are the optimal ones for the reaction and, therefore, the microwave conditions were employed subsequently for all further reactions to generate compounds 3a–3u/6a–6g/8a–o.
Table 1. Optimisation of the reaction conditions for the synthesis of compound 3aTable Footnotea.
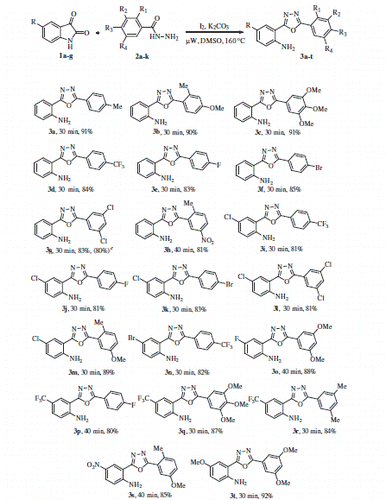
With the optimised conditions in hand, we started our exploration towards finding the potential applicability of this intramolecular decarboxylating domino reaction by attempting to prepare a variety of 1,3,4-oxadiazoles, using a varied set number of substituted isatins and benzohydrazides. The results are summarised in Scheme 2. In this way, a diversified set of 1,3,4-oxadiazoles 3a–3u were obtained in moderate to excellent yields. It was found that the reactions were equally successful with both electron withdrawing (4-CF3, 4-F, 4-Br, and 3,5-dichloro) as well as electron donating substituents [4-Me, 2-Me-4-OMe, and 3,4,5-(OMe)3] on the hydrazide component. However, it may be emphasised that in contrast to other electron-withdrawing substituents, the 4-nitro group-bearing substrates required longer time to complete the reaction satisfactorily (3h).
Scheme 2. One pot synthesis of the 2-(1,3,4-oxadiazo-2-yl)aniline derivatives. (a) Reaction conditions: 1 (1 equiv.), 2 (1.05 equiv.), I2 (1.0 equiv.), K2CO3 (1.5 equiv.) in DMSO (3 ml) under µW irradiation at 160 °C for 30–40 min, (b) isolated yields, and (c) The reaction was performed on gram scale.
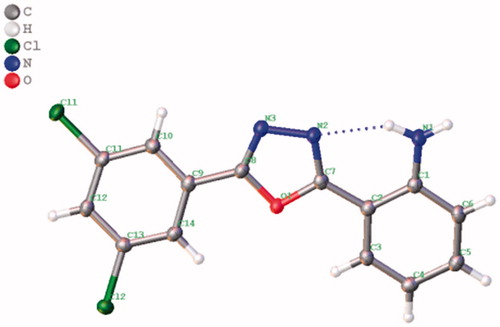
We also studied the effect of electron-donating (–OMe) and electron-withdrawing groups (5-Cl, 5-Br, 5-F, 5-OCF3, and 5-NO2) on the isatin component on the reaction efficiency, in terms of both the yield and the reaction time. Notably, these reactions also underwent smoothly to render the corresponding 1,3,4-oxadiazoles in good to excellent yields of 80–92% (3j–3u). Considering the significance of the heterocyclic scaffolds in organic synthesis and medicinal chemistry, we further investigated as substrate of this protocol a variety of heteroaryl hydrazides, such as isonicotinoylhydrazide, isoquinoline-3-carbohydrazide, and indazole-3-carbohydrazide (Scheme 3). Under the optimal conditions mentioned above, these heteroaryl derivatives smoothly reacted with isatin and provided the corresponding 1,3,4-oxadiazoles in moderate to good yields, i.e. 69–90% (6a–c), whereas, the reactions with 5-chloro, 5-fluoro, and 5-methoxy-isatin required longer reaction times (40 min) to furnish the desired product in satisfying yields (6d–g, 74–84%). The structure of 3g was confirmed by X-ray crystallographic analysis, as depicted in .
Scheme 3. One-pot synthesis of 2-(1,3,4-oxadiazo-2-yl)aniline derivatives from various isatins and heteroaryl hydrazides. Reaction conditions: 4 (1.0 equiv.), 5 (1.05 equiv.), I2 (1.0 equiv.), K2CO3 (1.5 equiv.) in DMSO (3 ml) under µW irradiation at 160 °C for 30–40 min, isolated yields.
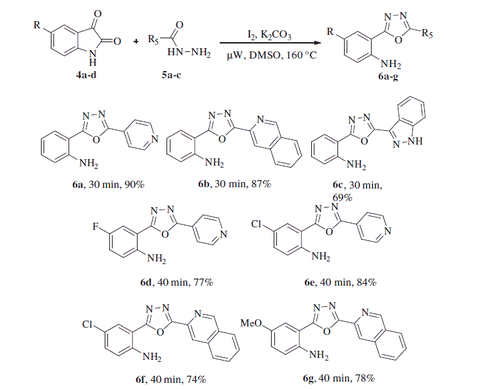
Figure 2. ORTEP diagram of the single crystal structure of compound 3g as determined by X-ray crystallography.
In addition to various substituted aryl and heteroaryl hydrazides, we applied this protocol on hydrazides incorporating a sulfonamide moiety. Under the optimised conditions mentioned earlier, 3 or 4-sulfamoyl benzhydrazides 7a,b reacted smoothly with a variety of substituted isatins to afford different 2-(1,3,4-oxadiazolo-2-yl)aniline-benzene sulfonamides 8a–o (Scheme 4).
Scheme 4. One pot synthesis of 2-(1,3,4-oxadiazolo-2-yl)aniline-benzene sulfonamide derivatives. Reaction conditions: 1 (1.0 equiv.), 7 (1.05 equiv.), I2 (1.0 equiv.), K2CO3 (1.5 equiv.) in DMSO (3 ml) under µW irradiation at 160 °C for 40 min, isolated yields.
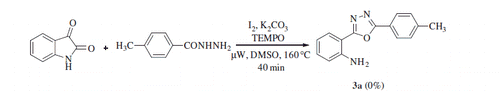
In order to obtain some mechanistic insights into the nature of the reaction, radical trapping experiments were performed by employing TEMPO (0.5 equiv.) under the set of optimised conditions mentioned above. Indeed, no desired product was obtained when the reaction was performed in the presence of TEMPO, suggesting clearly that the reaction took place through a radical pathway (Scheme 5).
Scheme 5. Control experiment using TEMPO.
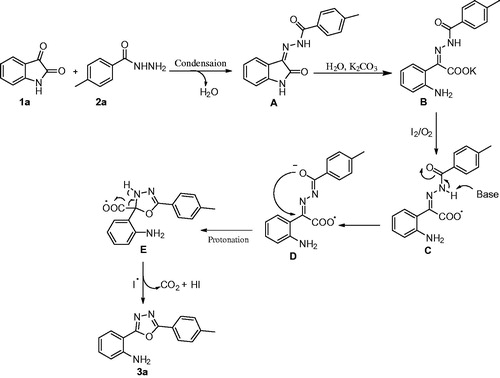
In the light of the obtained results and the work reported in the literatureCitation8,Citation17, a plausible reaction mechanism is proposed, which is shown in , using 1a and 2a as the starting materials for the iodine-mediated domino reaction. Isatin (1a) condenses with the hydrazide (2a) to give the intermediate hydrazone A, which subsequently undergoes a hydrolytic ring cleavage to form the carboxylate B. Iodine in the presence of oxygen, oxidises B to form the radical intermediate C. Subsequently, abstraction of a proton by the base, followed by a cyclisation gave the intermediate E, which by elimination of one molecule of CO2 and HI, led to the formation of the final product 3a.
Figure 3. Possible reaction mechanism for the domino reaction investigated here.
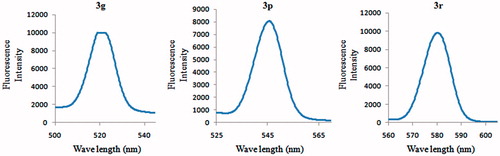
1,3,4-Oxadiazoles are well known for exhibiting a specific fluorescenceCitation21. In this regard, we investigated the excitation and emission spectra of some representative molecules described here in diluted DMSO as solvent. The excitation and emission spectra of compounds 3e and 3o showed prominent shifts to longer wavelengths, in contrast to the other cases in which the shifts were typically only marginal. shows the fluorescence emission spectra of compounds 3g, 3q, and 3s. Fluorescence properties of these compounds suggest that they may hold a potential for applications as chemical probes.
Figure 4. Fluorescence emission spectra of compounds 3g, 3q, and 3s in DMSO.
Carbonic anhydrase inhibition
Carbonic anhydrases (CAs, EC 4.2.1.1) are a superfamily of metalloenzymes, present in most living organisms, in which they catalyses a simple physiological reaction, i.e. the reversible hydration of CO2 to bicarbonate and protons via a ping–pong mechanism. These enzymes are involved in many physiological and pathological processes, such as pH and CO2 homeostasis, respiration and transport of carbon dioxide and bicarbonate between metabolising tissues and lungs, electrolyte secretion in various tissues and organs, biosynthetic reactions (gluconeogenesis, lipogenesis, and ureagenesis); calcification, bone resorption, and tumourigenicity (in mammals)Citation22,Citation23. Dysregulated activities of these carbonic anhydrases were proven to be connected with different human diseases, and inhibition of these enzymes by small molecules represents an efficient strategy in chemotherapeutic intervention. Sulphonamides and their bioisosteres (sulfamates and sulfamides), represents the main class of pharmacologically relevant CA inhibitors. Hence, it was of interest to evaluate the CA inhibitory activity of 2-(1,3,4-oxadiazolo-2-yl)aniline-benzene sulfonamides (8a–o) reported here. Thus, compounds 8a–o were investigated as inhibitors of four catalytically active human (h) CA isoforms, i. e. widespread, cytosolic, hCA I, and hCA II, the membrane-anchored hCA IV, as well as the transmembrane hCA IX, using the clinically used compound acetazolamide as a standard inhibitor.
The inhibition data are shown in . The following structure-activity relationship (SAR) can be delineated from the data of :
Table 2. Inhibition of hCA isoforms I, II, IV, and IX with sulphonamides 8a–o by a stopped-flow CO2 hydrase assayCitation24.
The slow cytosolic isoform hCA I was inhibited by all the examined sulphonamide derivatives 8a–o with inhibition constants (KIs) spanning between 81.4 and 5828.3 nM. Sulphonamides incorporating fluoro (8g) and dimethylaniline moieties (8h) showed medium nanomolar activity (KI of 81.4 and 89.1 nM, respectively). The other analogues were less potent and exhibited high nanomolar to low micromolar inhibitory potency against this isoform (KIs ranging between 222.2 and 5828.3, respectively)Citation25,Citation26.
hCA II, the dominant physiological isozyme, which is an anti-glaucoma drug target, was inhibited by all the tested compounds, with efficacy spanning from the low to the high nanomolar range (KIs of 6.4–515.7 nM, ). Among these, compound 8g displayed the highest inhibitory activity with a KI of 6.4 nM. Compounds 8a–d, 8f, 8h–k, and 8o also showed nanomolar inhibitory activity against this isoform, with KIs in the range of 16.4–86.3 nM, whereas remaining analogues exhibited high nanomolar inhibitory action. Among all these compounds, the 4-sulfamoyl derivatives showed a better CA II inhibitory activity compared to the 3-sulfamoyl derivatives. For example compounds, 8c and 8g were 31 and 13 times more potent than 8l and 8o, respectively. On the other hand, substitution on the aniline fragment also had a significant role on activity, i.e. o-fluoro substituted aniline bearing analogues 8g and 8o were more active in comparison to the p-substituted analogues 8d and 8m. From these observations, it can be clearly demonstrated that the substitution pattern on both phenyl rings had significant effect on the inhibitory activity against hCA II.
hCA IV, which is a membrane-associated isoform majorly expressed in the eye, lungs, and kidneys, being involved among others in glaucoma and retinitis pigmentosa diseases, was not particularly prone to inhibition by the sulphonamides investigated here In fact, all screened molecules (8a–o) displayed micromolar inhibitory activity, except 8j–m and 8o, which showed high nanomolar CA inhibitory activity, with KIs of 437.3–548.3 nM ().
hCA IX, the tumour-associated isoform, was moderately inhibited by all tested compounds with KIs in the range of 140.3–2964.0 nM. Substitution on both phenyl rings did not significantly influence the inhibition profile of these compounds for this isoform. Of the screened compounds, 8c, 8n, and 8o exhibited better CA IX inhibitory profile against this isozyme, with KIs of 140.3–282.1 nM ().
Conclusions
We have developed a more efficient and environmentally friendly protocol for the construction of 2-(1,3,4-oxadiazol-2-yl)aniline derivatives through one-pot domino decarboxylation by employing molecular iodine under microwave irradiation. This strategy works well with various substituted isatins and hydrazides belonging to both aryl and heteroaryl series, also showing good functional group tolerance. Furthermore, this protocol provided various oxadiazoles, which can be used for further functionalisation protocols. The reaction mechanism of this domino reaction was also delineated and presented in this article. Many of the synthesised molecules exhibited fluorescence properties that indicate their potential usefulness in the field of material chemistry. The synthesised 2-(1,3,4-oxadiazolo-2-yl)aniline-benzene sulfonamides (8a–o) were tested for their CA inhibitory activity and it was noticed that compounds 8c, 8g, and 8h displayed promising and selective activity against isoform hCA II with KIs of 16.4, 6.4, and 17.6 nM, respectively. Such compounds may be useful for various applications in which the CA activity must be inhibited, such as for the design of anti-glaucoma, antiobesity, or antitumor agentsCitation27,Citation28.
Acknowledgements
Authors are thankful to DoP, Ministry of Chemicals and Fertilisers, Govt. of India, New Delhi, for the award of a Research Fellowship and to Prof. K. P. R. Kartha, Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER), Sector 67, S. A. S. Nagar, Punjab 160062, India, for his valuable suggestions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- (a) Palucki M, Wolfe JP, Buchwald SL. Synthesis of oxygen heterocycles via a palladium-catalyzed C–O bond-forming reaction. J Am Chem Soc 1996;118:10333–4. (b) Torraca KE, Kuwabe SI, Buchwald SLA. High-yield general method for the catalytic formation of oxygen heterocycles. J Am Chem Soc 2000;122:12907–8. (c) Kuwabe SI, Torraca KE, Buchwald SL. Palladium-catalyzed intramolecular C–O bond formation. J Am Chem Soc 2001;123:12202–6. (d) Ylijoki KEO, Kundig EP. The preparation of 2H-1,4-benzoxazin-3-(4H)-ones via palladium-catalyzed intramolecular C–O bond formation. Chem Commun 2011;47:10608–10.
- (a) Evindar G, Batey RA. Parallel synthesis of a library of benzoxazoles and benzothiazoles using ligand-accelerated copper-catalyzed cyclizations of ortho-halobenzanilides. J Org Chem 2006;71:1802–8. (b) Fang Y, Li C. O-Arylation versus C-Arylation: copper-catalyzed intramolecular coupling of Aryl bromides with 1,3-Dicarbonyls. J Org Chem 2006;71:6427–31. (c) Mestichelli P, Scott MJ, Galloway WRJD, et al. Concise copper-catalyzed synthesis of tricyclic biaryl ether-linked aza-heterocyclic ring systems. Org Lett 2013;15:5448–51. (d) Reddy MN, Swamy KCK. Dual catalysis by Cu(I): facile single step click and intramolecular C–O bond formation leading to triazole tethered dihydrobenzodioxines/benzoxazines/benzoxathiines/benzodioxepines. Org Biomol Chem 2013;11:7350–60. (e) Sudheendran K, Malakar CC, Conrad J, Beifuss U. Copper(I)-catalyzed intramolecular O-arylation for the synthesis of 2,3,4,9-tetrahydro-1H-xanthen-1-ones with low loads of CuCl. J Org Chem 2012;77:10194–210.
- (a) Xiao B, Gong TJ, Liu ZJ, et al. Synthesis of dibenzofurans via Palladium-catalyzed phenol-directed C–H activation/C–O cyclization. J Am Chem Soc 2011;133:9250–3. (b) Wang X, Liu Y, Dai HX, Yu JQ. Pd (II)-catalyzed hydroxyl-directed C–H activation/C–O cyclization: expedient construction of dihydrobenzofurans. J Am Chem Soc 2010;132:12203–5. (c) Ueda S, Nagasawa H. Synthesis of 2-arylbenzoxazoles by copper-catalyzed intramolecular oxidative C–O coupling of benzanilides. Angew Chem Int Ed 2008;47:6411–3. (d) Modak A, Dutta U, Kancherla R, et al. Predictably selective (sp3)C–O bond formation through copper catalyzed dehydrogenative coupling: facile synthesis of dihydro-oxazinone derivatives. Org Lett 2014;16:2602–5. (e) Wei Y, Yoshikai N. Oxidative cyclization of 2-arylphenols to dibenzofurans under Pd (II)/peroxybenzoate catalysis. Org Lett 2011;13:5504–7. (f) Tang L, Pang Y, Yan Q, et al. Synthesis of coumestan derivatives via FeCl3-mediated oxidative ring closure of 4-hydroxy coumarins. J Org Chem 2011;76:2744–52. (g) Cheung CW, Buchwald SL. Room temperature copper(II)-catalyzed oxidative cyclization of enamides to 2,5-disubstituted oxazoles via vinylic C–H functionalization. J Org Chem 2012;77:7526–37.
- (a) He C, Guo S, Ke J, et al. Silver-mediated oxidative C–H/C–H functionalization: a strategy to construct polysubstituted furans. J Am Chem Soc 2012;134:5766–9. (b) Daw P, Chakraborty S, Garg JA, et al. Direct synthesis of pyrroles by dehydrogenative coupling of diols and amines catalyzed by cobalt pincer complexes. Angew Chem Int Ed 2016;55:14373–7. (c) Srimani D, Ben-David Y, Milstein D. Direct synthesis of pyrroles by dehydrogenative coupling of β-Aminoalcohols with secondary alcohols catalyzed by ruthenium pincer complexes. Angew Chem Int Ed 2013;125:4104–7. (d) Li X, He L, Chen H, et al. Copper-catalyzed aerobic C(sp2)–H functionalization for C–N bond formation: synthesis of pyrazoles and indazoles. J Org Chem 2013;78:3636–46. (e) Guimond N, Fagnou K. Isoquinoline synthesis via rhodium-catalyzed oxidative cross-coupling/cyclization of aryl aldimines and alkynes. J Am Chem Soc 2009;131:12050–1. (f) Stuart DR, Bertrand-Laperle M, Burgess KMN, Fagnou K. Indole synthesis via rhodium catalyzed oxidative coupling of acetanilides and internal alkynes. J Am Chem Soc 2008;130:16474–5. (g) Ueda S, Nagasawa H. Synthesis of 2-arylbenzoxazoles by copper-catalyzed intramolecular oxidative C–O coupling of benzanilides. Angew Chem Int Ed 2008;47:6411–3. (h) Tsang WCP, Zheng N, Buchwald SL. Synthesis of 2-arylbenzoxazoles by copper-catalyzed intramolecular oxidative C–O coupling of benzanilides. J Am Chem Soc 2005;127:14560–1.
- Zhdankin VV, Hypervalent iodine chemistry: preparation, structure, and synthetic applications of polyvalent iodine compounds. Chichester, UK: Wiley; 2013.
- (a) Nageswar Rao D, Rasheed SK, Vishwakarma RA, Das P. Hypervalent iodine (III) catalyzed oxidative C–N bond formation in water: synthesis of benzimidazole-fused heterocycles. RSC Adv 2014;4:25600–4. (b) Bagdi AK, Mitra S, Ghosh M, Hajra A. Iodine-catalyzed regioselective thiolation of imidazo[1,2-a]pyridines using sulfonyl hydrazides as a thiol surrogate. Org Biomol Chem 2015;13:3314–20. (c) Chu J, Hsu WT, Wu YH, et al. Substituent electronic effects govern direct intramolecular C–N cyclization of N-(Biphenyl)pyridin-2-amines induced by hypervalent iodine(III) reagents. J Org Chem 2014;79:11395–408. (d) Ma L, Wang X, Yu W, Han B. TBAI-catalyzed oxidative coupling of aminopyridines with β-ketoesters and 1,3-diones—synthesis of imidazo[1,2-a]pyridines. Chem Commun 2011;47:11333–5. (e) Li E, Hu Z, Song L, et al. Synthesis of 1,2,4-triazolo[4,3-a]pyridines and related heterocycles by sequential condensation and iodine-mediated oxidative cyclization. Chem Eur J 2016;22:11022–7. (f) Wu X, Gao Q, Geng X, et al. Iodine-promoted oxidative cross-coupling of unprotected anilines with methyl ketones: a site-selective direct C–H bond functionalization to C4-dicarbonylation of anilines. Org Lett 2016;18:2507–10. (g) Rajeshkumar V, Chandrasekar S, Sekar G. An efficient route to synthesize isatins by metal-free, iodine-catalyzed sequential C(sp3)–H oxidation and intramolecular C–N bond formation of 2′-aminoacetophenones. Org Biomol Chem 2014;12:8512–8. (h) Xu H, Wang F-J, Xin M, Zhang Z. I2-promoted condensation/cyclization of aryl methyl ketones with anilines for facile synthesis of 1,2,4-triarylpyrroles. Eur J Org Chem 2016;2016:925–9. (i) Lamani M, Prabhu KR. Iodine-catalyzed amination of benzoxazoles: a metal-free route to 2-aminobenzoxazoles under mild conditions. J Org Chem 2011;76:7938–44.
- a) Fabry DC, Stodulski M, Hoerner S, Gulder T. Metal-free synthesis of 3,3-disubstituted oxoindoles by iodine(III)-catalyzed bromocarbocyclizations. Chem Eur J 2012;18:10834–8. b) Ji KG, Zhu HT, Yang F, et al. A novel iodine-promoted tandem cyclization: an efficient synthesis of substituted 3,4-diiodoheterocyclic compounds. Chem Eur J 2010;16:6151–4. c) Kim I, Won HK, Choi J, Lee GH. A novel and efficient approach to highly substituted indolizines via 5-endo-trig iodocyclization. Tetrahedron 2007;63:12954–60. d) Yu QF, Zhang YH, Yin Q, et al. Electrophilic ipso-iodocyclization of N-(4-methylphenyl)propiolamides: selective synthesis of 8-methyleneazaspiro[4,5]trienes. J Org Chem 2008;73:3658–61. e) Masdeu C, Gómez E, Williams NAO, Lavilla R. Double insertion of isocyanides into dihydropyridines: direct access to substituted benzimidazolium salts. Angew Chem 2007;119:3103–6.
- a) Zhang J, Zhu D, Yu C, et al. A simple and efficient approach to the synthesis of 2-phenylquinazolines via sp3C–H functionalization. Org Lett 2010;12:2841–3. b) Yan Y, Wang Z. Chem Commun 2011;47:9513–5. c) Yan Y, Zhang Y, Feng C, et al. Selective iodine-catalyzed intermolecular oxidative amination of C(sp3)—H bonds with ortho-carbonyl-substituted anilines to give quinazolines. Angew Chem Int Ed 2012;51:8077–81. d) Wan C, Gao L, Wang Q, et al. Simple and efficient preparation of 2,5-disubstituted oxazoles via a metal-free-catalyzed cascade cyclization. Org Lett 2010;12:3902–5.
- Ma L, Wang X, Yu W, Han B. TBAI-catalyzed oxidative coupling of aminopyridines with β-keto esters and 1, 3-diones—synthesis of imidazo [1,2-a] pyridines. Chem Commun 2011;47:11333–5.
- Tang S, Liu K, Long Y, et al. Iodine-catalyzed radical oxidative annulation for the construction of dihydrofurans and indolizines. Org Lett 2015;17:2404–7.
- a) Boto A, Hernández R, Suárez E. Tandem radical decarboxylation − oxidation of amino acids: a mild and efficient method for the generation of N-acyliminium ions and their nucleophilic trapping. J Org Chem 2000;64:4930–7. b) Boto A, Hernández R, Suárez E. Tandem oxidative radical decarboxylation-β-iodination of amino acids. Application to the synthesis of chiral 2,3-disubstituted pyrrolidines. Tetrahedron Lett 2000;41:2495–8. c) Boto A, Hernández R, Suárez E. Oxidative decarboxylation of α-amino acids: a mild and efficient method for the generation of N-acyliminium ions. Tetrahedron Lett 1999;40:5945–8. d) Kiyokawa K, Yahata S, Kojima T, Minakata S. Hypervalent iodine (III)-mediated oxidative decarboxylation of β, γ-unsaturated carboxylic acids. Org Lett 2014;16:4646–9.
- a) Aurelio L, Scullino CV, Pitman MR, et al. Development of 3,5-dinitrobenzylsulfanyl-1,3,4-oxadiazoles and thiadiazoles as selective antitubercular agents active against replicating and nonreplicating mycobacterium tuberculosis. J Med Chem 2016;59:2362–80. b) Johansson A, Lofberg C, Antonsson M, et al. Discovery of (3-(4-(2-Oxa-6-azaspiro[3.3]heptan-6-ylmethyl)phenoxy)azetidin-1-yl)(5-(4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)methanone (AZD1979), a melanin concentrating hormone receptor 1 (MCHr1) antagonist with favorable physicochemical properties. J Med Chem 2016;59:2497–511. c) Nieddu V, Pinna G, Marchesi I, et al. Synthesis and antineoplastic evaluation of novel unsymmetrical 1, 3, 4-oxadiazoles. J Med Chem 2016;59:10451–69.
- a) El-Emam AA, Al-Deeb OA, Al-Omar M, Lehmann J. Synthesis, antimicrobial, and anti-HIV-1 activity of certain 5-(1-adamantyl)-2-substituted thio-1,3,4-oxadiazoles and 5-(1-adamantyl)-3-substituted aminomethyl-1,3,4-oxadiazoline-2-thiones. Bioorg Med Chem 2004;12:5107–13. b) Mullican MD, Wilson MW, Conner DT, et al. Design of 5-(3,5-di-tert-butyl-4-hydroxyphenyl)-1,3,4-thiadiazoles, -1,3,4-oxadiazoles, and -1,2,4-triazoles as orally active, nonulcerogenic antiinflammatory agents. J Med Chem 1993;36:1090–9. c) Mastrolorenzo A, Rusconi S, Scozzafava A, et al. Inhibitors of HIV-1 protease: current state of the art 10 years after their introduction. From antiretroviral drugs to antifungal, antibacterial and antitumor agents based on aspartic protease inhibitors. Curr Med Chem 2007;14:2734–48. d) Chohan ZH, Supuran CT, Scozzafava A. Metal binding and antibacterial activity of ciprofloxacin complexes. J Enzyme Inhib Med Chem 2005;20:303–7. e) Supuran CT, Scozzafava A, Mastrolorenzo A. Bacterial proteases: current therapeutic use and future prospects for the development of new antibiotics. Expert Opin Ther Pat 2001;11:221–59.
- a) He GS, Tan LS, Zheng Q, Prasad PN. Multiphoton absorbing materials: molecular designs, characterizations, and applications. Chem Rev 2008; 108:1245–330. b) Rehmann N, Ulbricht C, Köhnen A, et al. Advanced device architecture for highly efficient organic light‐emitting diodes with an orange‐emitting cross linkable Iridium (III) complex. Adv Mater 2008;20:129–33. c) Paraschivescu CC, Matache M, Dobrotă C, et al. Unexpected formation of N-(1-(2-Aryl-hydrazono) isoindolin-2-yl) benzamides and their conversion into 1, 2-(Bis-1, 3, 4-oxadiazol-2-yl) benzenes. J Org Chem 2013;78:2670–9.
- a) Jedlovska E, Lesko J. A simple one-pot procedure for the synthesis of 1, 3, 4-oxadiazoles. Synth Commun 1994;24:1879–85. b) Rostamizadeh S, Housaini SAG. Microwave assisted syntheses of 2,5-disubstituted 1,3,4-oxadiazoles. Tetrahedron Lett 2004;34:8753–6. c) Flidallah HM, Sharshira EM, Basaif SA, A-Ba-Oum AEK. Synthesis and spectral characterization of novel 1, 3, 4-oxadiazole and 1, 2, 4-triazole derivatives: synthesis for potential pharmacological activities. Phosphorus Sulfur Silicon Relat Elem 2002;177:67–79.
- a) Al-Talib M, Tashtoush H, Odeh N. A convenient synthesis of alkyl and aryl substituted bis-1, 3, 4-oxadiazoles. Synth Commun 1990;20:1811–7. b) Kerr VN, Ott DG, Hayes FN. Quaternary salt formation of substituted oxazoles and thiazoles. J Am Chem Soc 1960;82:186–9. c) Short FW, Long LM. Synthesis of 5‐aryl‐2‐oxazolepropionic acids and analogs. Antiinflammatory agents. J Heterocycl Chem 1969;6:707–12. d) Klingsberg E. Synthesis of carboxylic acid hydrazides and s-triazoles of the anthraquinone series. J Am Chem Soc 1958;80:5786–9. e) Reddy CK, Reddy PSN, Ratnam CV. A facile synthesis of 2-Aryl-3,4-dihydro-5H-1,3,4-benzotriazepin-5-ones. Synthesis 1983;842–4.
- a) Guin S, Rout SK, Ghosh T, et al. A one pot synthesis of [1,3,4]-oxadiazoles mediated by molecular iodine. RSC Adv 2012;2:3180–3. b) Fan Y, He Y, Liu X, et al. Iodine-mediated domino oxidative cyclization: one-pot synthesis of 1,3,4-oxadiazoles via oxidative cleavage of C(sp2)–H or C(sp)–H bond. J Org Chem 2016;81:6820–5. c) Gao Q, Liu S, Wu X, et al. Direct annulation of hydrazides to 1,3,4-oxadiazoles via oxidative C (CO)–C (methyl) bond cleavage of methyl ketones. Org Lett 2015;17:2960–3. d) Majji G, Rout SK, Guin S, et al. Iodine-catalysed oxidative cyclisation of acylhydrazones to 2, 5-substituted 1, 3, 4-oxadiazoles. RSC Adv 2014;4:5357–62.
- Kawano T, Yoshizumi T, Hirano K, et al. Copper-mediated direct arylation of 1,3,4-oxadiazoles and 1,2,4-triazoles with aryl iodides. Org Lett 2009;11:3072–5.
- Guin S, Ghosh T, Rout SK, et al. Cu(II) catalyzed imine C-H functionalization leading to synthesis of 2,5-substituted 1,3,4-oxadiazoles. Org Lett 2011;13:5976–9.
- Xu C, Jia F, Cai Q, et al. Intramolecular decarboxylative coupling as the key step in copper-catalyzed domino reaction: facile access to 2-(1,3,4-oxadiazol-2-yl)aniline derivatives. Chem Comm 2015;51:6629–32.
- Yan Y, Pan W, Song H. The synthesis and optical properties of novel 1, 3, 4-oxadiazole derivatives containing an imidazole unit. Dyes Pigments 2010;86:249–58.
- a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nature Rev Drug Discov 2008;7:168–81. b) Capasso C, Supuran CT. An overview of the α-, β-and γ-carbonic anhydrases from bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32. c) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nature Rev Drug Discov 2011;10:767–77.
- a) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72. b) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. c) Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30. d) Ward C, Langdon SP, Mullen P, et al. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat Rev 2013;39:171–9.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- a) Göçer H, Akincioğlu A, Göksu S, et al. Carbonic anhydrase and acetylcholinesterase inhibitory effects of carbamates and sulfamoylcarbamates. J Enzyme Inhib Med Chem 2015;30:316–20. b) Ceruso M, Bragagni M, AlOthman Z, et al. New series of sulfonamides containing amino acid moiety act as effective and selective inhibitors of tumor-associated carbonic anhydrase XII. J Enzyme Inhib Med Chem 2015;30:430–4. c) Zolfaghari Emameh R, Syrjänen L, Barker H, et al. Drosophila melanogaster: a model organism for controlling dipteran vectors and pests. J Enzyme Inhib Med Chem 2015;30:505–13. d) Le Darz A, Mingot A, Bouazza F, et al. Fluorinated pyrrolidines and piperidines incorporating tertiary benzenesulfonamide moieties are selective carbonic anhydrase II inhibitors. J Enzyme Inhib Med Chem 2015;30:737–45. e) Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91.
- a) Supuran CT, Barboiu M, Luca C, et al. Carbonic anhydrase activators. Part 14. Syntheses of mono and bis pyridinium salt derivatives of 2-amino-5-(2-aminoethyl)-and 2-amino-5-(3-aminopropyl)-1, 3, 4-thiadiazole and their interaction with isozyme II. Eur J Med Chem 1996;31:597–606. b) Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates strongly inhibit carbonic anhydrases and show antiglaucoma action in vivo. J Med Chem 2012;55:1721–30. c) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8. d) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3.
- Menchise V, De Simone G, Alterio V, et al. Carbonic anhydrase inhibitors: stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J Med Chem 2005;48:5721–7. b) Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors—part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents in rabbits. Eur J Med Chem 1998;33:247–54. c) Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35. d) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8. e) Şentürk M, Gülçin İ, Beydemir Ş, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9. f) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47. g) Dogne JM, Hanson J, Supuran C, Pratico D. Coxibs and cardiovascular side-effects: from light to shadow. Curr Pharm Des 2006;12:971–5.
- a) Krall N, Pretto F, Decurtins W, et al. A small‐molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5. b) Rehman SU, Chohan ZH, Gulnaz F, Supuran CT. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzyme Inhib Med Chem 2005;20:333–40. c) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: structure‐activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73. d) Dubois L, Peeters S, Lieuwes NG, et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 2011;99:424–31. e) Carta F, Scozzafava A, Supuran CT. Sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2012;22:747–58. f) Chohan ZH, Munawar A, Supuran CT. Transition metal ion complexes of Schiff-bases. Synthesis, characterization and antibacterial properties. Met Based Drugs 2001;8:137–43. g) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal β-class (Cab) and γ-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–8. h) Maresca A, Carta F, Vullo D, Supuran CT. Dithiocarbamates strongly inhibit the β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:407–11. i) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.