
Abstract
Cisplatin is the most widely used chemotherapeutic drug for the treatment of various types of cancer; however, its administration brings also numerous side effects. It was demonstrated that cisplatin can inhibit the Na+/K+-ATPase (NKA), which can explain a large part of the adverse effects. In this study, we have identified five cysteinyl residues (C452, C456, C457, C577, and C656) as the cisplatin binding sites on the cytoplasmic loop connecting transmembrane helices 4 and 5 (C45), using site-directed mutagenesis and mass spectrometry experiments. The identified residues are known to be susceptible to glutathionylation indicating their involvement in a common regulatory mechanism.
Introduction
Cisplatin (cis-diamminedichloroplatinum(II)) is the oldest-approved and still the most widely used chemotherapeutic drug, utilised namely in the treatment of testicular, ovarian, bladder, head, neck, and small cell lung cancerCitation1. The mechanism of its function is based on the formation of bifunctional adducts with nuclear DNA that impairs the processes of DNA repair and cell division. Unfortunately, patients receiving cisplatin suffer from numerous adverse effects, such as nephrotoxicity, neuropathies, or hearing lossCitation2,Citation3. During the initial phase of the chemotherapy, the risk of a kidney damage caused by cisplatin is the main dose-limiting factorCitation4,Citation5.
It has recently been demonstrated that cisplatin can inhibit the Na+/K+-ATPase (NKA) and proposed that this interaction can explain a large portion of the cisplatin adverse effectsCitation6,Citation7. NKA is present in the plasma membrane of every animal cell and has a prominent role in the cellular metabolismCitation8. First, it is responsible for maintaining the resting plasma membrane potential, which is of particular importance in neurons. Second, it is the only enzyme that actively exports sodium ions from the cytoplasm, thus, creating a steep gradient of sodium concentrations over the plasma membrane. This gradient is utilised by numerous secondary active transporters of other solutes, which is particularly critical in the tissues with rapid metabolisms, such as kidney. The impairment of the NKA function results in a failure of these secondary active transporters, and consequent failure of H+, Ca2+, glucose, amino acids, and other solutesCitation7.
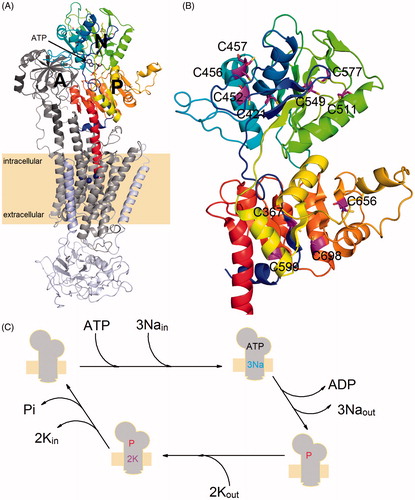
A functional NKA molecule is composed of two main subunits (denoted as α and β), which are often associated with a tissue-specific protein from FXYD family ()Citation8. The transmembrane domain of the catalytic α-subunit contains the binding sites for the transported cations and its large cytoplasmic part contains both ATP-binding and phosphorylation sites. Three large domains (named as A, P, and N) could be identified on the cytoplasmic side of the membrane (see ). The A-domain is formed by the N-terminus and loops between the transmembrane helices M2 and M3 (loop C23). The two other domains (N- and P-domain) are formed by a large cytoplasmic loop connecting the helices M4 and M5 (loop C45). The C45 loop plays a significant role in the NKA function as the nucleotide binding- and phosphorylation sites are localised within the N- and P-domain, respectively. The β-subunit has a single transmembrane helix and a large glycosylated ectodomain. It serves as a molecular chaperone assisting the correct α-subunit folding, and it probably also plays a role in the K+ countertransport or in the cell-cell adhesion processesCitation9.
Figure 1. (A) An overall view of the crystal structure of Na+/K+-ATPase in its closed conformation (PDB code: 4HQJ). The protein is composed of three separate chains denoted as α-subunit (gray transmembrane region, dark gray A-domain, color scale P-domain as well as N-domain), β-subunit (light blue) and FYXD protein (light purple). The C45 loop of the α-subunit is composed of N- and P-domain. (B) A close-up view of the C45 loop structure with the cysteine residues (magenta) mutated in this study. (C) Simplified scheme of reactions forming the NKA catalytic cycle, sign “P” denotes the transiently autophosphorylated state of the enzyme.
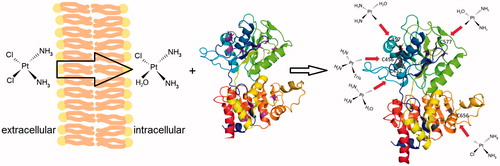
Soaking of the NKA crystals by cisplatin revealed that cisplatin is bound to the cytoplasmic part of the enzymeCitation10. This observation is in line with the cisplatin therapeutic action in vivo, which takes the advantage of a low reactivity of its diamminodichloro form. However, after passing into the cytoplasm, where the chloride concentration is lower compared to that in the blood, the chlorines can dissociate, yielding more reactive monoaqua or even diaqua formsCitation11.
Therefore, our experiments focused on the detailed identification of the cisplatin binding sites were performed using the isolated large cytoplasmic loop C45 (). This construct has been isolated in several laboratoriesCitation12–14, and so far, all the experiments suggest that it retains its structure as when being part of the entire enzymeCitation15: the ability to bind ATP and its analoguesCitation16–20 and to change its conformation on nucleotide bindingCitation14,Citation21. It can be expressed with high yields in bacteria, and its solubility greatly facilitates the subsequent experiments. Based on previous electrochemistry experiments, which suggested that cisplatin was bound to the cysteine residues of C45Citation6, we have prepared 11 constructs, where the C45 cysteines were mutated to serines. Consequently, the interaction of the mutant proteins with cisplatin was evaluated.
Material and methods
Chemicals
All used chemicals were from Sigma-Aldrich Chemie (Steinheim, Germany), except for cysteine purchased from Roth (Overland Park, KS, USA) and methionine from Fluka (Milwaukee, WI, USA).
Preparation of the set of cysteine mutants
The C45 loop of the mouse brain NKA contains 11 cysteine residues at the positions 367, 421, 452, 456, 457, 511, 549, 577, 599, 656, and 698 (human α1 sequence numbering is used throughout the text). To create individual mutants, the cysteine residues were replaced by a serine (C367S, C421S, C452S, C456S, C457S, C511S, C549S, C577S, C599S, C656S, and C698S). A double mutant (C456S + C457S) was prepared by an additional replacement of the residue C457 in the C456S mutant.
All point mutations were carried out by QuikChange Lightning site-directed mutagenesis kit (Agilent, Santa Clara, CA, USA) and verified by DNA sequencing (Seqme, Dobris, Czech Republic).
Mouse C45 loop expression and purification
The expression and purification of (His)10-tagged proteins were done as described previouslyCitation22. The total protein concentration was assayed using Bradford’s method with BSA as a standard. The purity of the recombinant proteins was determined by 10% Tris–Tricine SDS-PAGECitation23 (the molecular mass of the C45 loop is 48 kDa). The single amino acid replacements did not alter the overall protein yield and purity as revealed by SDS-PAGE. Only the mutant C698S had to be excluded from further experiments because it could not be purified as an intact protein.
Intact mass determination
MALDI-TOF analysis for intact mass determination of the C45 loop and its mutants in a native form or after the incubation with a 20-fold molar excess of cisplatin was performed on a Microflex LRF20 instrument as described previouslyCitation6. The final intact mass values were averaged from 10 technical replicates and calculated from the acquired m/z values of pseudo-molecular ions [M + H]+.
Chemical modification of cysteine residues
This chemical modification was performed according to previous protocolsCitation24,Citation25. The C45 WT proteins were diluted to a concentration of 1 mg/ml and alkylated by 330 mM iodoacetate in 100 mM ammonium bicarbonate (the final concentration of iodoacetate was 55 mM) at room temperature for 30 min. The reaction was stopped by adding 10 mM β-mercaptoethanol. The modified C45 WT was dialysed against 2 l of dialysis buffer (20 mM Tris–HCl, pH 7.5) at 4 °C overnight.
The intact molecular masses of the alkylated C45 WT proteins before and after the cisplatin treatment were measured using the same setup as described in Section 2.4.
Molecular dynamics
A model of the C45 loop in the closed conformation was created based on the 4HQJ crystal structureCitation26. Point mutations were introduced manually using PyMolCitation27 (Schrödinger, New York City, NY, USA). The system was inserted into a 9 × 9×9 nm3 fully hydrated box including 21 Na+ ions for charge neutralisation, and minimised. The molecular dynamics (MD) simulation was performed using GROMACS version 5.1.1Citation28 (GROMACS, Arlington, VA, USA) and GROMOS96 54A7 force field. The system was simulated for 10 ns with the step of 2 fs, using a velocity rescaled thermostat set to 298.5 K and Berendsen barostatCitation29 at 1 bar. The simulations were performed in two copies, denoted sim_WT1 and sim_WT2. The radial distribution function was calculated using the gmx rdf (GROMACS, Arlington, VA, USA) programme with respect to cysteine sulphur and water oxygen, with the first frame of analysis at 1 ns.
Results
Cysteines are exclusive binding sites for cisplatin on C45
Mass spectrometry estimated that the intact mass of C45 (wild-type, including His-tag) is 48,316 ± 31 Da, which is in a good agreement with the calculation based on the amino acid sequence and with the previously published dataCitation6. Cisplatin can form variety () of mono-, di-, tri-, or even tetravalent complexes causing a molecular mass increase in the range of 200–350 Da per one cisplatin adductCitation30–34. The intact mass of cisplatin-treated C45 protein was estimated as 49,490 ± 20 Da ( and Figure S1 in Supplementary Material), suggesting the formation of 4–5 adducts. It should be emphasised that cisplatin forms covalent adducts with proteins, and, hence, the binding stoichiometry is more proper interaction descriptor than the equilibrium binding constant used in some previous studiesCitation35. Sulfhydryl groups of cysteines are the most reactive functional groups of amino acid residues towards cisplatin, and also previous electrochemistry data indicated that cysteines within the C45 interact with cisplatinCitation6.
Figure 2. Schematic explanation of the cisplatin interaction with C45. In extracellular milieu (left), the unreactive diamminodichlo-form of cisplatin prevails. After passing into cytoplasm (middle) with lower chloride concentration, cisplatin is transformed to more reactive diamminomonochloromonoaqua-form, which can interact with the cytoplasmic part of NKA. Examples of the monovalent adducts with cysteine on C45 are shown (right), moreover, numerous bi- tri- or tetra-functional adducts are also possible (not shown).
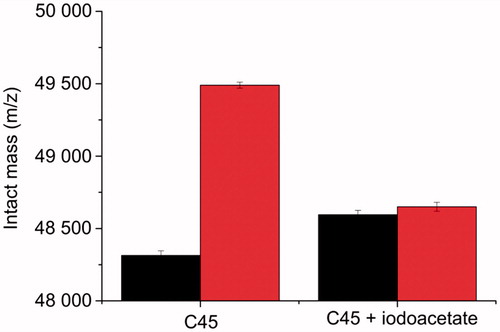
Chemical modification by iodoacetate is based on the alkylation of available cysteine residues (carboxymethylation), which increases the protein intact mass by 58 Da per one modified residueCitation24. For C45 WT treated by iodoacetate, we detected intact mass of 48,595 ± 31 Da, again, suggesting the modification of four to five cysteine residues accessible from the solvent. The treatment by cisplatin of this iodoacetate-labelled C45 did not virtually change the intact mass (48,650 ± 31 Da), providing an evidence that cysteines are indeed the interaction sites for cisplatin on C45, and moreover, no other amino acids interacted with cisplatin under given experimental conditions (). This is also a confirmation of the expected binding specificity.
Figure 3. Intact mass of C45 without or after the chemical modification of cysteine residues by iodoacetate (black) and after the treatment by cisplatin (red).
Cysteine mutants
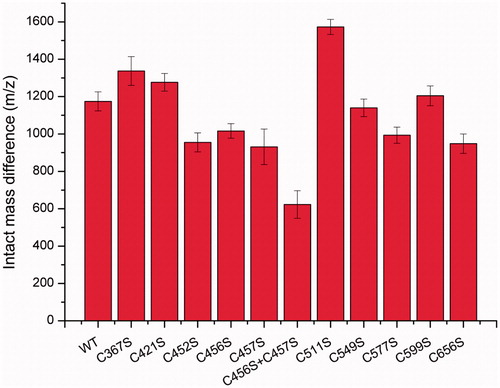
In order to identify the cisplatin binding sites on C45, we prepared a set of mutants, where cysteines were replaced by serine residues. The differences in the intact mass as for wild-type between untreated and cisplatin-treated proteins were rather similar for the mutants C367S, C421S, C549S, and C599S. On the other hand, a lower value of molecular mass difference (approximately about 250 Da) was detected for the mutants C452S, C456S, C457S, C577S, and C656S (), suggesting that one cisplatin binding site might be lost in these mutants. The molecular mass difference for the cisplatin complex with double mutant C456S + C457S decreased even more compared to that for the wild type indicating that despite their close proximity, both of these cysteine residues can be modified by cisplatin at the same time. An abnormal mass difference, higher than that for the wild-type, was detected for the C511S mutant.
Figure 4. Differences in the intact mass of untreated and cisplatin-treated cysteine mutants.
Simulation results
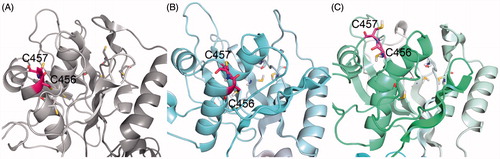
In both simulations replicates, the structure of the C45 loop remained stable, with a RMSD of about 4 Å compared to the starting structure. There were small differences in the position of the free loops between N-domain β-sheets, but overall, the main features of the C45 loop were identical. An interesting difference appeared at the α-helix containing C452, C456, and C457. It was quite dynamic during the simulation, and while the end of the helix stayed regular in the first simulation, it was unwound in second one, allowing both C456 and C457 to point to the bulk solution (). In a good agreement with the experimental data, the cysteines C452, C457, C577, and C656 were all found on the surface of the protein in both simulations, which was confirmed both visually and by the radial distribution function (Supplementary Material, Figure S2).
Figure 5. The positions of C456 and C457 (highlighted in pink) in the N-domain, the starting crystal structure (A) and two final frames of MD simulation – sim_WT1 (B) and sim_WT2 (C).
Discussion
Understanding the structural details of the cisplatin interaction with NKA can provide fundamentals for the attempts to eliminate the adverse effects of cisplatin. The adverse effects were proposed to be associated to the cisplatin interaction with proteins, and recently, it was demonstrated that cisplatin can interact e.g. with human serum albuminCitation36, ribonuclease ACitation37, lysozymeCitation38, and other proteinsCitation39. The most reactive functional groups in proteins towards cisplatin seem to be the sulfhydryl groups of cysteinesCitation30,Citation34. Indeed, such an interaction of cisplatin has been reported for other proteinsCitation31,Citation32 and suggested also for the C45 loopCitation6. On the other hand, low-resolution crystallographic data revealed that only one cisplatin-binding site was located in the proximity of the segment with C452, C456, and C457, and other binding sites were located in the proximity of other residues, namely methioninesCitation10.
Our experiments revealed that the blocking of the cysteine sulfhydryl groups by their interaction with iodoacetate almost completely disabled a further interaction of the C45 with cisplatin, indicating that cysteines could, therefore, represent the exclusive reaction partners for cisplatin under given experimental conditions. We propose that the other binding sites detected by crystallographyCitation10 were found in consequence of the different buffer composition used. Particularly, a lower pH value of the crystallographic buffer disfavours interaction with cysteines and increases the probability of interaction with methionines.
The C45 contains 11 cysteinyl residues, however, some of them are buried under the protein surface, and, hence, they are not accessible to chemical reactions with species diluted in the solvent. The mass spectrometry analyses of C45 modified either by iodoacetamide or cisplatin revealed that it is possible to modify 4–5 cysteinyl residues, which is in a good agreement with previous reportsCitation7,Citation6 as well as with the predictions from molecular models. An abnormally high mass difference was detected for the C511S mutant. According to the model, C511 should be buried residue. We hypothesise that this mutant may not be correctly folded, and more cysteines become exposed to the solvent in this case. Hence, the data for this mutant are of little structural relevance.
The mutagenesis revealed that one cisplatin binding site was lost after mutation of the cysteine residues at the positions 452, 456, 457, 577, or 656. Furthermore, two binding sites were lost in the double-mutant C456S + C457S, indicating that both these adjacent residues reacted simultaneously without any steric restrictions.
The segment containing C452, C456, and C457, as well as Cys577, were recently identified as glutathionylation sites in NKACitation40. Glutathionylation resulted in a suppression of the enzyme affinity to the nucleotide and consequent decrease in its activityCitation41. One can expect that the modification of these sites by cisplatin can have a similar effect. However, it should be emphasised that the cisplatin binding results in an irreversible NKA inhibition, which can be responsible for the large part of adverse effects observed during chemotherapy. It is in contrast to glutathionylation, which is reversible and can be considered as a natural transient regulatory event.
Conclusion
Our data represent a solid structural background for the elucidation of the cisplatin adverse effects that are consequence of NKA inhibition. We have identified five cysteinyl residues on the cytoplasmic part of NKA that are able to interact with cisplatin. These sites are identical to the sites that are responsible for NKA regulation by glutathionylation, indicating a common regulatory mechanism. Our experiments revealed that under physiological pH values, these cysteines are the predominant interaction sites for cisplatin in this structural part of the enzyme.
IENZ_1445735_Supplementary_Material.pdf
Download PDF (228 KB)Acknowledgements
J. Š. acknowledges Martin Šefl from the Faculty of Nuclear Sciences and Physical Engineering, Czech Technical University in Prague for his help with the Matlab data processing. P. Č. would like to acknowledge that the computational resources were provided by the CESNET LM2015042 and the CERIT Scientific Cloud LM2015085, provided under the programme “Projects of Large Research, Development, and Innovations Infrastructures”.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Cepeda V, Fuertes M, Castilla J, et al. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med Chem 2007;7:3–18.
- Barabas K, Milner R, Lurie D, Adin C. Cisplatin: a review of toxicities and therapeutic applications. Vet Comp Oncol 2008;6:1–18.
- Dzagnidze A, Katsarava Z, Makhalova J, et al. Repair capacity for platinum-DNA adducts determines the severity of cisplatin-induced peripheral neuropathy. J Neurosci 2007;27:9451–7.
- Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective trategies. Kidney Int 2008;73:994–1007.
- Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel) 2010;2:2490–518.
- Huličiak M, Vacek J, Šebela M, et al. Covalent binding of cisplatin impairs the function of Na+/K+-ATPase by binding to its cytoplasmic part. Biochem Pharmacol 2012;83:1507–13.
- Kubala M, Geleticova J, Huliciak M, et al. Na+/K+-ATPase inhibition by cisplatin and consequences for cisplatin nephrotoxicity. Biomed Pap 2014;158:194–200.
- Kaplan JH. Biochemistry of Na,K-ATPase. Annu Rev Biochem 2002;71:511–35.
- Geering K. The functional role of beta subunits in oligomeric P-type ATPases. J Bioenerg Biomembr 2001;33:425–38.
- Huliciak M, Reinhard L, Laursen M, et al. Crystals of Na+/K+-ATPase with bound cisplatin. Biochem Pharmacol 2014;92:494–8.
- Jamieson ER, Lippard SJ. Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev 1999;99:2467–98.
- Tran CM, Farley RA. Catalytic activity of an isolated domain of Na, K-ATPase expressed in Escherichia coli. Biophys J 1999;77:258–66.
- Gatto C, Wang AX, Kaplan JH. The M4M5 cytoplasmic loop of the Na, K-ATPase, overexpressed in Escherichia coli, binds nucleoside triphosphates with the same selectivity as the intact native protein*. J Biol Chem 1998;273:10578–85.
- Grycova L, Sklenovsky P, Lansky Z, et al. ATP and magnesium drive conformational changes of the Na+/K+-ATPase cytoplasmic headpiece. Biochim Biophys Acta Biomembr 2009;1788:1081–91.
- Kubala M, Obsil T, Obsilová V, et al. Protein modeling combined with spectroscopic techniques: an attractive quick alternative to obtain structural information. Physiol Res 2004;53:S187–S97.
- Kubala M, Plášek J, Amler E. Fluorescence competition assay for the assessment of ATP binding to an isolated domain of Na+,K+-ATPase. Physiol Res 2004;53:109–13.
- Kubala M, Plášek J, Amler E. Limitations in linearized analyses of binding equilibria: binding of TNP-ATP to the H 4-H 5 loop of Na/K-ATPase. Eur Biophys J 2003;32:363–9.
- Lánský Z, Kubala M, Ettrich R, et al. The hydrogen bonds between Arg423 and Glu472 and other key residues, Asp443, Ser477, and Pro489, are responsible for the formation and a different positioning of TNP-ATP and ATP within the nucleotide-binding site of Na+/K+-ATPase. Biochemistry 2004;43:8303–11.
- Kubala M, Teisinger J, Ettrich R, et al. Eight amino acids form the ATP recognition site of Na(+)/K(+)-ATPase. Biochemistry 2003;42:6446–52.
- Kubala M, Krumscheid R, Schoner W. Phe475 and Glu446 but not Ser445 participate in ATP-binding to the α-subunit of Na+/K+-ATPase. Biochem Biophys Res Commun 2002;297:154–9.
- Kubala M, Grycova L, Lansky Z, et al. Changes in electrostatic surface potential of Na+/K+-ATPase cytoplasmic headpiece induced by cytoplasmic ligand(s) binding. Biophys J 2009;97:1756–64.
- Biler M, Štenclová T, Trouillas P, Biedermann D. Flavonolignans as a novel class of sodium pump inhibitors. Front Physiol 2016;7:115.
- Schägger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 1987;166:368–79.
- Lundblad RL, Noyes CM. Chemical reagents for protein modification. Boca Ralton (FL): CRC Press; 1984. 192 p.
- Beinhauer J, Lenobel R, Loginov D, et al. Identification of Bremia lactucae and Oidium neolycopersici proteins extracted for intact spore MALDI mass spectrometric biotyping. Electrophoresis 2016;37:2940–52.
- Nyblom M, Reinhard L, Andersson M, et al. Crystal structure of Na+,K+-ATPase in the Na+-bound state. Science 2013;342:123–7.
- DeLano WL. The PyMOL molecular graphics system. New York: Schrödinger, LLC; 2013.
- Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015;1–2:19–25.
- Berendsen HJC, Postma JPM, van Gunsteren WF, et al. Molecular dynamics with coupling to an external bath. J Chem Phys 1984;81:3684–90.
- Dabrowiak JC, Metals in medicine. Weinheim, Germany: Wiley-VCH; 2009. 334 p.
- Hartinger CG, Tsybin YO, Fuchser J, Dyson PJ. Characterization of platinum anticancer drug protein-binding sites usin a top-down mass spectrometric approach. Inorg Chem 2008;47:17–19.
- Hartinger CG, Ang WH, Casisni A, et al. Mass spectroscopic analysis of ubiquitin-platinum interactions of leading anticancer drugs: MALDI versus ESI. J Anal at Spectrom 2007;22:960–7.
- Ishikawa T, Aliosman F. Glutathione-associated cis-diamminodichloroplatinum(II) metabolism and ATP-dependent efflux from leukemia-cells - molecular characterization of glutathione-platinum complex and its biological significance. J Biol Chem 1993;268:20116–25.
- Odenheimer B, Wolf W. Reactions of cisplatin with sulfur-containing amino-acids and peptides. 1. Cysteine and glutathione. Inorganica Chim Acta 1982;66:L41–3.
- Neault JF, Benkirane A, Malonga H, Tajmir-Riahi HA. Interaction of cisplatin drug with Na,K-ATPase: drug binding mode and protein secondary structure. J Inorg Biochem 2001;86:603–9.
- Ferraro G, Massai L, Messori L, Merlino A. Cisplatin binding to human serum albumin: a structural study. Chem Commun 2015;51:9436–9.
- Messori L, Merlino A. Cisplatin binding to proteins: molecular structure of the ribonuclease a adduct. Inorg Chem 2014;53:3929–31.
- Marasco D, Messori L, Marzo T, Merlino A. Oxaliplatin vs. cisplatin: competition experiments on their binding to lysozyme. Dalt Trans 2015;44:10392–8.
- Messori L, Merlino A. Cisplatin binding to proteins: a structural perspective. Coord Chem Rev 2016;315:67–89.
- Bogdanova A, Petrushanko IY, Hernansanz-Agustín P. “Oxygen Sensing” by Na, K-ATPase: these miraculous thiols. Front Physiol 2016;7:314.
- Petrushanko IY, Yakushev S, Mitkevich VA, et al. S-glutathionylation of the Na,K-ATPase catalytic α subunit is a determinant of the enzyme redox sensitivity. J Biol Chem 2012;287:32195–205.