
Abstract
Being the primary sulfonamide among the most efficient zinc binding group (ZBG) to design inhibitors for the metallo-enzymes carbonic anhydrases (CA, EC 4.2.1.1), herein, we propose an investigation on four physiologically important human (h) CAs (hCA I, II, IV, and IX) with N1-substituted secondary sulfonamides incorporating thiazolinone or imidazolone-indole tails. The effect of the functionalisation of the sulfonamide group with five different substitution patterns, namely acetyl, pyridine, thiazole, pyrimidine, and carbamimidoyl, was evaluated in relation to the inhibition profile of the corresponding primary sulfonamide analogues. With most of these latter being nanomolar inhibitors of all four considered isoforms, a totally counterproductive effect on the inhibition potency can be ascribed to N1-functionalisations of the ZBG primary sulfonamide structure with pyridine, thiazole, and pyrimidine moieties. On the other hand, incorporation of less hindered groups, such as sulfonylacetamides and sulfonylguanidines, maintained a certain degree of activity dependent on the tailing moiety, with KIs spanning in the low micromolar range.
1. Introduction
Primary sulfonamide is the most efficient zinc binding group (ZBG) to design inhibitors for the metallo-enzymes carbonic anhydrases (CA, EC 4.2.1.1), being its structural features ideal for the binding to the Zn2+ ion present at the bottom of the active site cavity and the residues nearbyCitation1–3. The negatively charged nitrogen of SO2NH− coordinates the positively charged metal ion replacing the physiological zinc-bound nucleophile, with the proton on the coordinated nitrogen atom being at H-bond distance to Thr199 OG1 atom, which act as acceptor ()Citation1.
Figure 1. Schematic representation of the binding mode of benzenesulfonamide within the hCA II active site.
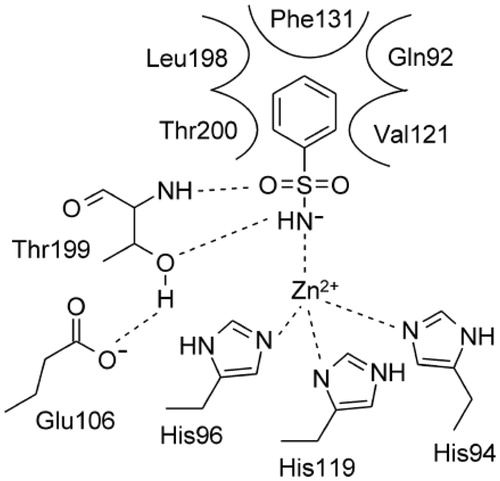
Benzenesulfonamides constitute the most common and best characterised class of CA inhibitors (CAIs)Citation3. The presence of an aromatic/heteroaromatic scaffold bearing the sulfonamide group further stabilise the enzyme-ligand complex, by several Van der Waals interactions taking place with residues Gln92, Val121, Phe131, Leu198, and Thr200 (hCA II binding site ())Citation1,Citation3.
All CAs found in humans belong to the α-class (α-CA) and are characterised as sixteen isoforms, which differ by molecular features, oligomeric arrangement, cellular localisation, distribution in organs and tissues, expression levels, and kinetic propertiesCitation1,Citation2. Abnormal levels or activities of most sixteen hCA isoforms have been often associated with different human diseases, making these isozymes of great interest for the design of inhibitors which selectively target specific isoforms, in order to reduce the overall side effects exhibited by most non-isoform selective CAIs clinically used up to nowCitation1–4. Most efforts to design isoform selective CAIs have been pursued by modulating the ring directly linked to the ZBG (ring approach)Citation3,Citation5 or appending different tails to the aromatic ring just mentioned (tail approach)Citation3,Citation5–7.
The previously accepted ideaCitation8 that only primary sulfonamides may act as effective inhibitors has been shown to be incorrect, but only few modifications were carried out on the sulfonamide moietyCitation1a, in comparison to the high number of tails appended at the aromatic ring incorporating this ZGBCitation6,Citation3,Citation9,Citation10. The rational of this choice could be that of not losing the ideal features of primary sulfonamides, i.e. the negative charge on nitrogen and the presence of a proton on it, which are both important for the inhibition mechanismCitation1–3. Conversely, it should be taken into account that N1-substitution with proper functional moieties could also elicit novel inhibition mechanismsCitation1.
Secondary sulfonamides maintain the possibility to coordinate the Zn ion in the deprotonated form, as confirmed by X-ray crystallography. Di Fiore et al. demonstrated that N-hydroxy and N-methoxy sulfonamides are able to bind to the Zn(II) ion in the deprotonated formCitation11. An analogous inhibition mechanism has been identified for the cyclic secondary sulfonamide saccharin and its derivativesCitation12. Likewise, we previously reported that the incorporation of a nitro-group at the N1-atom of aromatic sulfonamide moieties does not deprive them of CA inhibitory efficacy, although the precise inhibitory mode has not been clarified yetCitation13.
Tertiary sulfonamides lose their acid character, paving the way to a totally new inhibition mechanism. Carradori et al.Citation14,Citation15 recently synthesised different series of tertiary sulfonamides, namely probenecid and saccharin derivatives, obtaining interesting and selective inhibition profile. However, the inhibition mechanism with such compounds was not yet been elucidated. A plethora of series of benzenesulfonamide bearing indole or thiazolidinone moieties have also been reported so far and demonstrated to possess an effective inhibitory profile against a wide panel of hCAsCitation16–20.
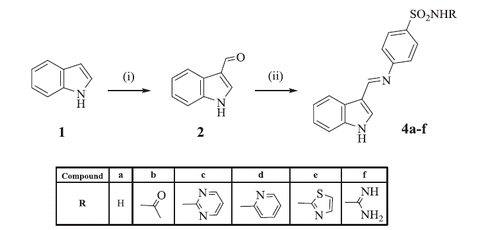
In an earlier investigation of some of us, a series of benzenesulfonamides linked to a chromone nucleus through thiazolinone and imidazolone spacers demonstrated excellent inhibitory activity against a panel of hCAs in a nanomolar rangeCitation21. In continuation of the previous work, and with the aim to pursue the identification of new potent and selective CAIs, herein we explore the effect of the functionalisation of the sulfonamide group on the inhibitory properties by appending several moieties, namely acetyl, pyridine, thiazole, pyrimidine, and carbamimidoyl, at the N1-atom. A consistent series of imine-, thiazolinone-, and imidazolone benzenesulfonamides appending an indole ring instead of the chromone nucleus (from our earlier work) was thus obtained. All the synthesised derivatives were tested in vitro in order to evaluate their inhibition profiles against four physiologically important hCAs, i.e. hCA I, II, IV, and IX. Comparison of the inhibitory efficacy of primary sulfonamide derivatives with the N1-substituted analogues further strengthened the role of the sulfonamide in the pharmaceutical field related to CAs.
Materials and methods
Chemistry
Melting points were recorded on a Stuart SMP10 digital melting point apparatus and were uncorrected. Infrared (IR) spectra were recorded as KBr disks using a Shimadzu FT-IR 8400S infrared spectrophotometer. Mass spectral data are given as m/z (intensity %). 1H NMR spectra were recorded on either on a Varian Mercury VX-300 MHz spectrophotometer or Bruker AVANCE III Nano Bay 400 MHz FT-NMR spectrophotometer. 13C NMR spectra were run at 100 MHz in deuterated dimethylsulfoxide (DMSO-d6) on Bruker AVANCE III Nano Bay 400 MHz FT-NMR spectrophotometer. Chemical shifts are expressed in δ values (ppm) using the solvent peak as internal standard. All coupling constant (J) values are given in Hz. The abbreviations used are as follows: s, singlet; d, doublet; t, triplet; m, multiplet. Elemental microanalyses were carried out at the Regional Center for Mycology and Biotechnology, Al-Azhar University, Egypt. Reaction were routinely monitored by Thin Layer Chromatography (TLC) on silica gel precoated F254 Merck plates. Unless otherwise noted, all solvents and reagents were commercially available and used without further purification. Compounds 2Citation22, 4a–fCitation23,Citation24, 6a–fCitation23,Citation24, 7aCitation25, 8a,bCitation26,Citation27 and 9aCitation28 were prepared according to the reported procedures.
General procedure for the preparation of compounds 4a-f
A mixture of indole-3-carboxaldehyde 2 (1.45 g, 10 mmol) and the appropriate sulfonamide 3a–f (10 mmol) were grinded and mixed together using a mortar and a pestle. The mixture was fused on a watch glass using drops of glacial acetic acid. The resulted paste was washed with ethanol/water mixture then crystallised from ethanol to afford compounds 4a–f.
4-{[(1H-indol-3-yl)methylene]amino}benzenesulfonamide (4a)
Yellowish brown powder, (yield 60%), m.p. 180–182 °C; IR (KBr, ν cm−1): 3479–3244 (NHs), 1311 and 1149 (SO2); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 5.80 (s, 2H, NH2, D2O exchangeable), 6.58 (d, 2H, Ar–H, J = 8.6), 6.88 (s, 1H, NH, D2O exchangeable), 7.21–7.26 (m, 2H, Ar–H), 7.45 (d, 2H, Ar–H, J = 8.6), 7.51 (d, 1H, Ar–H, J = 7.7), 8.09 (d, 1H, Ar–H, J =7.3), 8.28 (s, 1H, Ar–H), 9.93 (s, 1H, Ar–H); MS m/z: 299.0 [M]+; Anal. Calcd. for C15H13N3O2S (299.35): C, 60.18; H, 4.38; N, 14.04; Found C, 60.41; H, 4.28; N, 13.89.
N-{[4-(((1H-indol-3-yl)methylene)amino)phenyl]sulfonyl}acetamide (4 b)
Off-white powder, (yield 70%), m.p. 170–172 °C; IR (KBr, ν cm−1): 3379–3200 (NHs), 1635 (C = O), 1334 and 1127 (SO2); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 2.06 (s, 3H, CH3), 5.38 (s, 2H, 2NH, D2O exchangeable), 6.42–6.44 (m, 2H, Ar–H), 6.56 (d, 1H, Ar–H), 7.36 (d, 2H, Ar–H, J = 8.8), 7.52 (d, 2H, Ar–H, J = 8.8), 7.61 (d, 1H, Ar–H, J =8.8), 8.46 (s, 1H, Ar–H), 9.90 (s, 1H, Ar–H); 13 C NMR (DMSO-d6) δ ppm: 27.1, 110.0, 112.2, 112.8, 121.0, 122.5, 123.5, 127.8, 128.4, 133.4, 150.6, 173.9 175.3; MS m/z: 341.42 [M]+; Anal. Calcd. for C17H15N3O3S (341.38): C, 59.81; H, 4.43; N, 12.31; Found C, 59.63; H, 4.74; N, 12.52.
4-[((1H-indol-3-yl)methylene)amino)-N-(pyrimidin-2-yl)]benzenesulfonamide (4c)
Brown powder, (yield 68%), m.p. 217–219 °C; IR (KBr, ν cm−1): 3230–3147 (NHs), 1334 and 1126 (SO2); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 5.68 (s, 1H, NH, D2O exchangeable), 6.54 (d, 2H, Ar–H, J = 8.6), 7.20–7.29 (m, 4H, Ar–H), 7.39 (d, 2H, Ar–H, J = 7.6), 7.51 (d, 1H, Ar–H, J = 7.6), 8.09 (d, 2H, Ar–H, J = 7.6), 8.29 (s, 1H, Ar–H), 9.86 (s, 1H, Ar–H), 12.13 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6) δ ppm: 112.5, 112.8, 115.8, 118.6, 121.2, 122.5, 123.9, 124.5, 125.5, 130.2, 137.4, 138.9, 153.3, 158.6, 159.8, 185.4; MS m/z: 377.12 [M]+; Anal. Calcd. for C19H19N5O2S (377.42): C, 60.46; H, 4.01; N, 18.56; Found C, 60.29; H, 3.97; N, 18.64.
4-[((1H-indol-3-yl)methylene)amino)-N-(pyridin-2-yl)]benzenesulfonamide (4d)
Yellowish brown powder, (yield 55%), m.p. 144–146 °C; IR (KBr, ν cm−1): 3417–3207 (NHs), 1323 and 1126 (SO2); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 5.91 (s, 1H, NH, D2O exchangeable), 6.53 (d, 1H, Ar–H, J = 9.6), 6.86 (t, 1H, Ar–H, J = 6.2), 7.04 (d, 2H, Ar–H, J = 8.4), 717–7.24 (m, 3H, Ar–H), 7.49 (d, 2H, Ar–H, J = 8), 7.62 (t, 1H, Ar–H, J = 8.8), 8.07 (d, 2H, Ar–H, J = 6.8), 8.26 (s, 1H, Ar–H), 9.91 (s, 1H, Ar–H), 12.10 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6) δ ppm: 112.8, 118.6, 121.2, 122.5, 123.9, 124.5, 129.3, 137.4, 138.9, 153.1, 185.4; Anal. Calcd. for C19H19N5O2S (376.43): C, 63.81; H, 4.28; N, 14.88; Found C, 63.97; H, 4.19; N, 14.52.
4-[((1H-indol-3-yl)methylene)amino)-N-(thiazol-2-yl)]benzenesulfonamide (4e)
Yellowish brown powder, (yield 50%), m.p. 148–150 °C; IR (KBr, ν cm−1): 3367–3209 (NHs), 1334 and 1138 (SO2). 1H NMR (DMSO-d6, 400 MHz) δ ppm: 5.18 (s, 1H, NH, D2O exchangeable), 6.52 (t, 1H, Ar–H, J = 8), 7.15–7.26 (m, 2H, Ar–H), 7.35 (d, 1H, Ar–H, J = 8.8), 7.38 (d, 2H, Ar–H, J = 9.2), 7.49 (d, 2H, Ar–H, J = 7.6), 8.07 (d, 2H, Ar–H, J = 7.2), 8.26 (s, 1H, Ar–H), 9.91 (s, 1H, Ar–H), 12.10 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6) δ ppm: 112.8, 112.9, 118.6, 121.2, 122.5, 123.9, 124.5, 128.0, 128.1, 137.4, 138.9, 152.6, 185.4; Anal. Calcd. for C18H14N4O2S2 (382.46): C, 56.53; H, 3.69; N, 14.65; Found C, 56.40; H, 3.62; N, 14.37.
4-{[(1H-indol-3-yl)methylene]amino}-N-carbamimidoylbenzenesulfonamide (4f)
Brown powder, (yield 68%), m.p. 154–156 °C; IR (KBr, ν cm−1): 3433–3332 (NHs), 1334 and 1126 (SO2); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 5.65 (s, 3H, 3NHs, D2O exchangeable), 6.51 (d, 2H, Ar–H, J = 8.8), 7.19–7.24 (m, 2H, Ar–H), 7.35 (d, 1H, Ar–H, J = 8.4), 7.49 (d, 2H, Ar–H, J = 7.6), 8.06 (d, 1H, Ar–H, J = 8.0), 8.26 (s, 1H, Ar–H), 9.91 (s, 1H, Ar–H), 12.05 (s, 2H, 2NHs, D2O exchangeable); 13 C NMR (DMSO-d6) δ ppm: 112.7, 112.8, 118.6, 121.2, 122.5, 123.9, 124.5, 127.6, 131.2, 137.4, 138.9, 151.8, 159.1, 185.4; Anal. Calcd. for C16H15N5O2S (341.39): C, 56.29; H, 4.43; N, 20.51; Found C, 56.01; H, 4.37; N, 20.79.
General procedure for the preparation of compounds 7a–f
A mixture of indole-3-carboxaldehyde 2 (1.45 g, 10 mmol), the appropriate thiazolinone derivative 6a–f (10 mmol) and fused sodium acetate (0.82 g, 10 mmol) in glacial acetic acid (50 ml) was refluxed for 12 h. The separated solid was filtered off while hot, washed with hot water and crystallised from DMF/H2O.
4-{[5-((1H-indol-3-yl)methylene)-4-oxo-4,5-dihydrothiazol-2-yl]amino}benzenesulfonamide (7a)
Yellowish brown powder, (yield 60%), m.p. 230–232 °C; IR (KBr, ν cm−1): 3360–3199 (NHs), 1674 (C = O), 1334 and 1151 (SO2); 1H NMR (DMSO-d6, 300 MHz) δ ppm: 7.14–7.25 (m, 4H, Ar–H), 7.34 (s, 1H, NH, D2O exchangeable), 7.48 (d, 2H, Ar–H, J = 7.5), 7.61 (s, 1H, Ar–H), 7.48 (d, 2H, Ar–H, J = 8.4), 7.94 (s, 1H, Ar–H), 11.89 (s, 2H, NH2, D2O exchangeable), 12.23 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6) δ ppm: 115.1, 116.4, 119.8, 120.4, 124.7, 125.9, 126.9, 127.6, 129.6, 133.8, 137.8, 142.7, 144.0, 159.1, 169.7, 170.2; Anal. Calcd. for C18H14N4O3S2 (398.46): C, 54.26; H, 3.54; N, 14.06; Found C, 54.01; H, 3.75; N, 13.89.
N-{[4-((5-((1H-indol-3-yl)methylene)-4-oxo-4,5-dihydrothiazol-2-yl)amino)phenyl]sulfonyl} acetamide (7 b)
Green powder, (yield 55%), m.p. 242–244 °C; IR (KBr, ν cm−1): 3371–3213 (NHs), 1701 and 1658 (2 C = Os), 1311 and 1149 (SO2); 1H NMR (DMSO-d6, 300 MHz) δ ppm: 1.94 (s, 3H, CH3), 7.18–7.27 (m, 4H, Ar–H), 7.49 (d, 1H, Ar–H, J = 9), 7.51 (s, 1H, NH, D2O exchangeable), 7.88 (d, 1H, Ar–H, J = 8.1), 8.07–8.09 (m, 3H, Ar–H), 8.26 (s, 1H, Ar–H), 12.10 (s, 2H, 2NHs, D2O exchangeable); Anal. Calcd. for C20H16N4O4S2 (440.50): C, 54.53; H, 3.66; N, 12.72; Found C, 54.62; H, 3.92; N, 12.93.
4-{[5-((1H-indol-3-yl)methylene)-4-oxo-4,5-dihydrothiazol-2-yl)amine]-N-(pyrimidin-2-yl)} benzenesulfonamide (7c)
Off-white powder, (yield 60%), m.p. 228–230 °C; IR (KBr, ν cm−1): 3317–3199 (NHs), 1681 (C = O), 1328 and 1150 (SO2); 1H NMR (DMSO-d6, 300 MHz) δ ppm: 7.03 (t, 2H, Ar–H, J = 4.8), 7.31–7.78 (m, 3H, Ar–H), 7.87 (d, 1H, J = 9), 7.89 (s, 2H, Ar–H), 8.07 (d, 3H, Ar–H, J = 8.7), 9.06 (d, 2H, Ar–H, J = 5.1), 10.84 (s, 3H, 3NHs, D2O exchangeable); Anal. Calcd. for C22H16N6O3S2 (476.53): C, 55.45; H, 3.38; N, 17.64; Found C, 55.37; H, 3.76; N, 17.41.
4-{[5-((1H-indol-3-yl)methylene)-4-oxo-4,5-dihydrothiazol-2-yl)amino]-N-(pyridin-2-yl)] benzenesulfonamide (7d)
Yellow powder, (yield 50%), m.p. 245–247 °C; IR (KBr, ν cm−1): 3350–3200 (NHs), 1697 (C = O), 1334 and 1138 (SO2); 1H NMR (DMSO-d6, 300 MHz) δ ppm: 7.37–7.45 (m, 5H, 4 Ar–H +1H, NH, D2O exchangeable), 7.63–7.45 (m, 4H, 2 Ar–H + 2H, 2NHs, D2O exchangeable), 8.13–8.17 (m, 3H, Ar–H), 8.34–8.43 (m, 3H, Ar–H), 8.87 (s, 2H, Ar–H); Anal. Calcd. for C23H17N5O3S2 (475.54): C, 58.09; H, 3.60; N, 14.73; Found C, 57.96; H, 3.42; N, 14.94.
4-{[(5-((1H-indol-3-yl)methylene)-4-oxo-4,5-dihydrothiazol-2-yl)amino]-N-(thiazol-2-yl)] benzenesulfonamide (7e)
Grey powder, (yield 60%), m.p. 218–220 °C; IR (KBr, ν cm−1): 3327–3207 (NHs), 1687 (C = O), 1317 and 1153 (SO2); 1H NMR (DMSO-d6, 300 MHz) δ ppm: 6.80–6.83 (m, 2H, Ar–H), 7.18–7.27 (m, 4H, Ar–H), 7.48 (t, 1H, Ar–H, J = 7.2), 7.81 (d, 2H, Ar–H, J = 9), 8.07 (d, 1H, Ar–H, J = 7,2), 8.26 (s, 1H, Ar–H), 8.27 (s, 1H, Ar–H), 11.95 (s, 1H, NH, D2O exchangeable), 12.10 (s, 2H, 2NHs, D2O exchangeable); Anal. Calcd. for C21H15N5O3S3 (481.57): C, 52.38; H, 3.14; N, 14.54; Found C, 52.53; H, 3.33; N, 14.71.
4-{[5-((1H-indol-3-yl)methylene)-4-oxo-4,5-dihydrothiazol-2-yl]amino}-N-carbamimidoyl benzenesulfonamide (7f)
Brown powder, (yield 65%), m.p. 227–229 °C; IR (KBr, ν cm−1): 3433–3209 (NHs), 1627 (C = O), 1334 and 1126 (SO2); 1H NMR (DMSO-d6, 300 MHz) δ ppm: 6.88 (s, 1H, Ar–H), 7.16–7.27 (m, 4H, Ar–H), 7.48 (t, 1H, Ar–H, J = 6.3), 7.89 (d, 2H, Ar–H, J = 8.7), 8.08 (d, 1H, Ar–H, J = 7.2), 8.27 (s, 1H, Ar–H), 11.91 (s, 3H, NHs, D2O exchangeable), 12.11 (s, 3H, NHs, D2O exchangeable); 13 C NMR (DMSO-d6) δ ppm: 112.8, 114.4, 118.6, 118.7, 121.2, 122.5, 123.0, 123.9, 124.5, 127.2, 128.6, 136.7, 137.5, 138.9, 140.9, 170.9, 185.4; Anal. Calcd. for C19H16N6O3S2 (440.50): C, 51.81; H, 3.66; N, 19.08; Found C, 52.13; H, 3.90; N, 19.23.
4-[(1-Acetyl-1H-indol-3-yl)methylene]-2–(4-methoxyphenyl)oxazol-5(4H)-one (9 b)
A mixture of indole-3-carboxaldehyde 2 (1.45 g, 10 mmol), N-(4-methoxybenzoyl)glycine 8 b (2.09 gm, 10 mmol) and fused sodium acetate (0.5 g, 6 mmol) in acetic anhydride (20 ml) was heated in a boiling water bath for 5 h. The mixture was cooled and ethanol (20 ml) was added slowly and allowed to stand overnight in the refrigerator. The crystalline product was filtered off, washed with hot water and recrystallised from benzene to give compound 9 b.
Buff powder, (yield 60%), m.p. 149–151 °C; IR (KBr, ν cm−1): 1743 and 1689 (2 C = Os); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 1.91 (s, 3H, CH3), 3.88 (s, 3H, OCH3), 7.17 (d, 2H, Ar–H, J = 8), 7.39–7.46 (m, 2H, Ar–H), 7.50 (s, 1H, Ar–H), 8.07 (d, 2H, Ar–H, J = 8), 8.35 (d, 2H, Ar–H, J = 8), 8.79 (s, 1H, Ar–H); Anal. Calcd. for C21H16N2O4 (360.36): C, 69.99; H, 4.48; N, 7.77; Found C, 70.23; H, 4.65; N, 7.91.
General procedure for the preparation of compounds 10a–l
An equimolar amount of compound 9a,b (10 mmol) and the appropriate sulfonamide 3a–f (10 mmol) in glacial acetic acid (20 ml) containing freshly fused sodium acetate (0.03 g, 0.36 mmol) was heated in a boiling water bath with constant stirring for 10–14 h. The separated solid was filtered off, washed with water, and crystallised from DMF/water to give compounds 10a–l.
4-[4-((1-Acetyl-1H-indol-3-yl)methylene)-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl]benzenesulfonamide (10a)
Grey powder, (yield 60%), m.p. 240–242 °C; IR (KBr, ν cm−1): 3371, 3305 (NH2), 1708 and 1650 (2 C = Os), 1338 and 1157 (SO2); 1H NMR (DMSO-d6, 300 MHz) δ ppm: 1.90 (s, 3H, CH3), 7.26 (s, 2H, NH2, D2O exchangeable), 7.31–7.42 (m, 2H, Ar–H), 7.45 (s, 1H, Ar–H), 7.51–7.61 (m, 3H, Ar–H), 7.79 (d, 1H, Ar–H, J = 9), 7.87 (d, 2H, Ar–H, J = 6.9), 7.93 (d, 1H, Ar–H, J = 9), 8.06 (d, 2H, Ar–H, J = 7.2), 8.16 (s, 1H, Ar–H), 8.33 (d, 2H, Ar–H, J = 6.9); 13 C NMR (DMSO-d6) δ ppm: 24.1, 115.1, 116.4, 119.9, 120.4, 124.7, 125.9, 126.9, 127.6, 128.6, 128.9, 129.1, 131.2 132.3, 133.6, 137.5 138.9, 142.7 144.0, 168.4, 169.7, 170.2; Anal. Calcd. for C26H20N4O4S (484.53): C, 64.45; H, 4.16; N, 11.56; Found C, 64.18; H, 4.19; N, 11.48.
N-[(4–(4-((1-acetyl-1H-indol-3-yl)methylene)-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl)phenyl)sulfonyl]acetamide (10 b)
Brown powder, (yield 55%), m.p. 237–239 °C; IR (KBr, ν cm−1): 3390 (NH), 1730, 1701 and 1635 (3 C = Os), 1327 and 1165 (SO2); 1H NMR (DMSO-d6, 300 MHz) δ ppm: 1.91 (s, 3H, CH3), 1.92 (s, 3H, CH3), 7.15–7.26 (m, 2H, Ar–H), 7.31–7.73 (m, 3H, Ar–H), 7.88 (d, 2H, Ar–H, J = 8.7), 7.93–7.99 (m, 2H, Ar–H), 8.05 (d, 2H, Ar–H, J = 6.9), 8.18 (s, 1H, Ar–H), 8.36 (d, 2H, Ar–H, J = 6.9), 8.81 (s, 1H, Ar–H), 12.51 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6) δ ppm: 23.6, 24.1, 115.0, 116.3, 119.9, 124.2, 124.7, 125.9, 127.7, 128.3, 128.5, 128.8, 129.1, 129.3, 129.6, 131.1, 132.0, 132.3, 133.8, 135.0, 144.4, 164.9, 169.1, 169.7; Anal. Calcd. for C28H22N4O5S (526.56): C, 63.87; H, 4.21; N, 10.64; Found C, 63.81; H, 4.27; N, 10.97.
4-[4-((1-Acetyl-1H-indol-3-yl)methylene)-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl]-N-(pyrimidin-2-yl)benzenesulfonamide (10c)
Brownish red powder, (yield 55%), m.p. 248–250 °C; IR (KBr, ν cm−1): 3383 (NH), 1689 and 1643 (2 C = Os), 1323 and 1153 (SO2); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 1.96 (s, 3H, CH3), 7.06 (t, 1H, Ar–H, J = 4.2), 7.37–7.48 (m, 3H, Ar–H), 7.59 (t, 2H, Ar–H, J = 7.3), 7.65 (d, 2H, Ar–H, J = 6.8), 7.92 (d, 2H, Ar–H, J = 7.8), 8.00 (s, 1H, Ar–H), 8.11 (d, 2H, Ar–H, J = 7.2), 8.23 (s, 1H, Ar–H), 8.39 (d, 2H, Ar–H, J = 8), 8.54 (d, 2H, Ar–H, J = 4.6), 11.91 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6) δ ppm: 24.1, 115.0, 116.3, 119.7, 119.9, 124.2, 125.8, 127.7, 128.3, 128.5, 128.8, 129.0, 129.7, 131.2, 132.3, 133.8, 134.9, 144.4, 158,7, 164.8, 164.8, 169.7; Anal. Calcd. for C30H22N6O4S (562.60): C, 64.05; H, 3.94; N, 14.94; Found; C, 64.39; H, 4.20; N, 15.21.
4-[4-((1-Acetyl-1H-indol-3-yl)methylene)-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-y])-N-(pyridin-2-yl)benzenesulfonamidee (10d)
Yellow powder, (yield 60%), m.p. 248–250 °C; IR (KBr, ν cm−1): 3325 (NH), 1730 and 1631 (2 C = Os), 1327 and 1136 (SO2); 1H NMR (DMSO-d6, 300 MHz) δ ppm: 2.05 (s, 3H, CH3), 6.86 (t, 1H, Ar–H, J = 6.3), 7.11 (d, 1H, Ar–H, J = 8.7), 7.18–7.28 (m, 5H, Ar–H), 7.50 (d, 1H, Ar–H, J = 6.6), 7.65–7.69 (m, 4H, Ar–H), 7.70 (s, 1H, Ar–H), 7.79 (d, 2H, Ar–H, J = 8.7), 8.01 (s, 1H, Ar–H), 8.08 (d, 2H, Ar–H, J = 7.2), 8.28 (d, 1H, Ar–H, J = 6.6), 12.10 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C31H23N5O4S (561.61): C, 66.30; H, 4.13; N, 12.47; Found C, 66.23; H, 4.38; N, 12.68.
4-[4-((1-Acetyl-1H-indol-3-yl)methylene)-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl]-N-(thiazol-2-yl)benzenesulfonamide (10e)
Green powder, (yield 65%), m.p. 265–267 °C; IR (KBr, ν cm−1): 3251 (NH), 1703 and 1627 (2 C = Os), 1328 and 1147 (SO2); 1H NMR (DMSO-d6, 300 MHz) δ ppm: 1.89 (s, 3H, CH3), 6.80–6.87 (m, 2H, Ar–H), 7.23–7.37 (m, 2H, Ar–H), 7.42 (t, 1H, Ar–H, J = 6.9), 7.53 (t, 2H, Ar–H, J = 7.3), 7.77 (d, 2H, Ar–H, J = 8.7), 7.85–7.80 (m, 2H, Ar–H), 7.92 (s, 1H, Ar–H), 8.05 (d, 2H, J = 6.9), 8.16 (s, 1H, Ar–H), 8.33 (d, 2H, Ar–H, J = 8.1), 12.72 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6) δ ppm: 24.3, 115.1, 116.4, 120.3, 123.9, 125.8, 126.8, 127.7, 128.9, 129.0, 129.7, 131.2, 133.9, 134.9, 137.2, 139.0, 142.2, 144.5, 158.6, 159.2, 168.9, 170.1; Anal. Calcd. for C29H21N5O4S2 (567.64): C, 61.36; H, 3.73; N, 12.34; Found C, 61.08; H, 3.44; N, 12.59.
4-[4-((1-Acetyl-1H-indol-3-yl)methylene)-5-oxo-2-phenyl-4,5-dihydro-1H-imidazol-1-yl]-N-carbamimidoylbenzenesulfonamide (10f)
Brown powder, (yield 65%), m.p. 252–254 °C; IR (KBr, ν cm−1): 3217 (NH), 1708 and 1635 (2 C = Os), 1315 and 1138 (SO2); 1H NMR (DMSO-d6, 300 MHz) δ ppm: 1.90 (s, 3H, CH3), 7.14–7.25 (m, 4H, Ar–H), 7.34 (s, 2H, NH2, D2O exchangeable), 7.61 (s, 1H, Ar–H), 7.47–7.61 (m, 5H, Ar–H), 7.83–7.94 (m, 5H, Ar–H), 11.89 (s, 2H, 2NHs, D2O exchangeable); 13 C NMR (DMSO-d6) δ ppm: 24.4, 108.5, 115.0, 116.6, 119.8, 120.4, 124.6, 125.8, 127.2, 127.6, 128.5, 129.0, 129.7, 131.2, 132.3, 134.9, 135.7, 136.8, 142.3, 143.0, 164.9, 168.9, 169.7; Anal. Calcd. for C27H22N6O4S (526.57): C, 61.59; H, 4.21; N, 15.96; Found C, 61.31; H, 4.50; 16.04.
4-[4-((1-Acetyl-1H-indol-3-yl)methylene)-2–(4-methoxyphenyl)-5-oxo-4,5-dihydro-1H-imidazol-1-yl]benzenesulfonamide (10 g)
Off-white powder, (yield 70%), m.p. 226–228 °C; IR (KBr, ν cm−1): 3479, 3375 (NH2), 1627 and 1597 (2 C = Os), 1315 and 1145 (SO2); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 2.08 (s, 3H, CH3) , 3.80 (s, 3H, OCH3), 5.77 (s, 2H, NH2, D2O exchangeable), 7.5–6.85 (m, 3H, Ar–H), 6.85 (s, 1H, Ar–H), 6.99 (d, 1H, J = 7.2), 7.17–7.26 (m, 3H, Ar–H), 7.42 (d, 1H, Ar–H, J = 6.8), 7.86 (d, 2H, Ar–H, J = 7.2), 8.07 (d, 2H, Ar–H, J = 8.4), 8.26 (s, 1H, Ar–H); 13 C NMR (DMSO-d6) δ ppm: 24.5, 55.7, 112.8, 113.9, 114.2, 118.9, 124.0, 127.0, 127.3, 127.8, 129.3, 133.0, 138.4, 140.9, 142.7, 152.4, 161.8, 162.9, 165.6, 169.5, 173.1, 173.9; Anal. Calcd. for C27H22N4O5S (514.55): C, 63.02; H, 4.31; N, 10.89; Found C, 63.09; H, 4.64; N, 11.07.
N-[(4–(4-((1-acetyl-1H-indol-3-yl)methylene)-2–(4-methoxyphenyl)-5-oxo-4,5-dihydro-1H-imidazol-1-yl)phenyl)sulfonyl]acetamide (10 h)
Brown powder, (yield 70%), m.p. 234–236 °C; IR (KBr, ν cm−1): 3367 (NH), 1685 (br.) and 1604 (3 C = Os), 1330 and 1168 (SO2); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 2.02 (s, 3H, CH3), 2.06 (s, 3H, CH3), 3.80 (s, 3H, OCH3), 7.21–7.33 (m, 3H, Ar–H), 7.42–7.44 (m, 2H, Ar–H), 7.51 (d, 1H, Ar–H, J = 8.8), 7.61 (d, 2H, Ar–H, J = 8.8), 7.72 (s, 1H, Ar–H), 7.84 (s, 1H, Ar–H), 7.85 (d, 2H, Ar–H, J = 7.2), 7.89 (d, 2H, Ar–H, J = 6.8), 10.36 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6) δ ppm: 22.0, 24.5, 55.7, 112.9, 113.5, 113.8, 118.2, 118.5, 118.9, 121.2, 122.5, 123.8, 125.9, 127.1, 128.0, 131.3, 131.6, 138.4, 142.6, 162.5, 168.4, 169.5, 173.1, 185.5; Anal. Calcd. for C29H24N4O6S (556.59): C, 62.58; H, 4.35; N, 10.07; Found C, 62.24; H, 4.67; N, 10.03.
4-[4-((1-Acetyl-1H-indol-3-yl)methylene)-2–(4-methoxyphenyl)-5-oxo-4,5-dihydro-1H-imidazol-1-yl]-N-(pyrimidin-2-yl)benzenesulfonamide (10i)
Off-white powder, (yield 65%), m.p. 178–180 °C; IR (KBr, ν cm−1): 3356 (NH), 1651 and 1597 (2 C = Os), 1327 and 1153 (SO2); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 2.07 (s, 3H, CH3), 3.81 (s, 3H, OCH3), 6.00 (s, 1H, NH, D2O exchangeable), 6.56 (d, 2H, Ar–H, J = 8.8), 6.99–7.03 (m, 3H, Ar–H), 7.61 (d, 2H, Ar–H, J = 8.7), 7.73 (d, 2H, Ar–H, J = 8.8), 7.84 (d, 2H, Ar–H, J = 8.8), 7.89 (d, 2H, Ar–H, J = 8.8), 7.95 (s, 1H, Ar–H), 8.25 (s, 1H, Ar–H), 8.47 (d, 2H, Ar–H, J = 8.8); Anal. Calcd. for C31H24N6O5S (592.62): C, 62.83; H, 4.08; N, 14.18; Found; C, 63.17; H, 4.40; N, 14.56.
4-[4-((1-Acetyl-1H-indol-3-yl)methylene)-2–(4-methoxyphenyl)-5-oxo-4,5-dihydro-1H-imidazol-1-yl]-N-(pyridin-2-yl)benzenesulfonamide (10j)
Dark brown powder, (yield 75%), m.p. 198–200 °C; IR (KBr, ν cm−1): 3244 (NH), 1660 and 1635 (2 C = Os), 1320 and 1126 (SO2); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 2.04 (s, 3H, CH3), 3.80 (s, 3H, OCH3), 6.51–6.54 (m, 3H, Ar–H), 6.58–6.98 (m, 3H, Ar–H), 7.04 (t, 1H, Ar–H, J =10.6), 7.11–7.26 (m, 2H, Ar–H), 7.50 (d, 2H, Ar–H, J = 8.8), 7.71–7.64 (m, 4H, Ar–H), 8.07 (d, 2H, Ar–H, J = 8.4), 8.25 (s, 1H, Ar–H), 11.01 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C32H24N5O5S (591.64): C, 64.96; H, 4.26; N, 11.84; Found C, 65.19; H, 4.62; N, 11.88.
4-[4-((1-Acetyl-1H-indol-3-yl)methylene)-2–(4-methoxyphenyl)-5-oxo-4,5-dihydro-1H-imidazol-1-yl]-N-(thiazol-2-yl)benzenesulfonamide (10k)
Light brown powder, (yield 65%), m.p. 206–208 °C; IR (KBr, ν cm−1): 3294 (NH), 1670 and 1639 (2 C = Os), 1330 and 1145 (SO2); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 2.06 (s, 3H, CH3), 3.80 (s, 3H, OCH3), 5.80 (d, 1H, Ar–H, J = 4.8), 6.49–6.55 (m, 3H, Ar–H), 6.72 (d, 1H, Ar–H, J = 4.4), 6.95–7.00 (m, 2H, Ar–H), 7.18 (d, 1H, Ar–H, J = 6.4), 7.35 (d, 1H, Ar–H, J = 8.4), 7.37 (s, 1H, Ar–H), 7.41 (d, 2H, Ar–H, J = 8.8), 7.73 (s, 1H, Ar–H), 7.55 (s, 1H, Ar–H), 7.87 (d, 2H, Ar–H, J = 8.8), 11.15 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C30H23N5O5S2 (597.66): C, 60.29; H, 3.88; N, 11.72; Found C, 60.13; H, 4.20; N, 11.70.
4-[4-((1-Acetyl-1H-indol-3-yl)methylene)-2–(4-methoxyphenyl)-5-oxo-4,5-dihydro-1H-imidazol-1-yl]-N-carbamimidoylbenzenesulfonamide (10 l)
Brown powder, (yield 70%), m.p. 222–224 °C; IR (KBr, ν cm−1): 3433–3213 (NHs), 1635 and 1608 (2 C = Os), 1300 and 1126 (SO2); 1H NMR (DMSO-d6, 400 MHZ) δ ppm: 1.90 (s, 3H, CH3), 3.80 (s, 3H, OCH3), 5.68 (s, 3H, NHs, D2O exchangeable), 6.54 (d, 2H, Ar–H, J = 8.6), 7.02 (d, 1H, Ar–H, J = 8.8), 7.20–7.28 (m, 2H, Ar–H), 7.39 (d, 2H, Ar–H, J = 8.6), 7.51 (d, 2H, Ar–H, J = 7.6), 7.67 (s, 1H, Ar–H), 7.90 (d, 1H, Ar–H, J = 8.8), 8.10 (d, 2H, Ar–H, J = 7.2), 8.29 (s, 1H, Ar–H), 12.13 (s, 1H, NH, D2O exchangeable); Anal. Calcd. for C28H24N6O5S (556.59): C, 60.42; H, 4.35; N, 15.10; Found C, 60.72; H, 4.62; N, 15.39.
Carbonic anhydrase inhibition assay
An applied photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activityCitation29. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 Mm Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial rate. The uncatalysed rates were determine d in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionised water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation30–33, and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation34–36.
Results and discussion
Chemistry
The target compounds 4a–f, 7a–f, and 10a–l were synthesised as illustrated in Schemes 1–3. The key stating material, indole-3-carboxaldehyde 2, was prepared via Vilsemier-Hack formylation reaction, as reported in literatureCitation22. IR spectrum of compound 2 revealed the presence of (NH) stretching band at 3363 cm−1 along with a (C = O) stretching band at 1633 cm−1. In addition, 1H NMR showed a singlet signal at 9.94 ppm attributed to the proton of the (CH = O) group and a D2O exchangeable signal of the (NH) proton at 12.15 ppm. Schiff’s bases 4a–f were obtained via the condensation of the aldehyde compound 2 with different sulfonamide derivatives 3a–f. The reported method for the preparation of these compounds failed to give the expected compounds, and in some derivatives a very poor yield was obtainedCitation37. Searching for an alternative method, Naqvi et al.Citation38 reported an energy efficient greener methodology for the preparation of Schiff’s bases involving a mechano-chemical solvent-free procedure. Applying this new methodology afforded compounds 4a–f in good yields and high purity in a short reaction time. IR spectra of compounds 4a–f were characterised by the disappearance of the (C = O) stretching band of the aldehyde 2 and broadening of the (NH) stretching band at 3350–3200 cm−1 due to the additional (NH) groups. Also, two characteristic stretching bands corresponding to the (SO2) group were identified at 1334–1311 and 1149–1126 cm−1. 1H NMR of compounds 4a–f displayed the presence of sharp singlet signal at 9.86–9.93 ppm attributed to the azomethine proton (CH = N) in addition to the (NH) singlet signals, all disappeared by D2O exchange. As for compound 4 b; a singlet signal at 2.06 ppm integrated for the three protons of the (CH3) group in 1H NMR spectrum and a signal at 27.1 ppm in 13C NMR spectrum confirmed its structure. In addition, 13C NMR of compounds 4c–f adopted the expected pattern of their carbon content. Mass spectra of compounds 4a–c gave m/z at 299.0, 341.42 and 377.42 corresponding to their molecular weights, respectively.
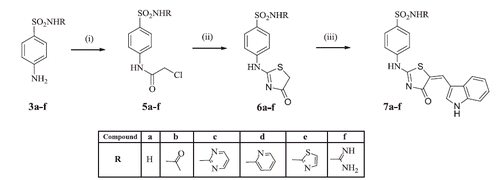
The thiazolinone derivatives 7a–f were synthesised according to Scheme 2. The appropriate sulfonamide derivatives 3a–f were reacted with chloroacetyl chloride at r.t. in DMF to give the intermediate chloroacetyl derivatives 5a–f which were then intramolecularly cyclised by reflux with ammonium thiocyanate in ethanol, to obtain the thiazolinone compounds 6a–fCitation23,Citation24. Knoevenagel condensation of the aldehyde 2 with the active methylene group in compounds 6a–f afforded the newly synthesised compounds 7a–f. IR spectra of compounds 7a–f were consistent with their proposed structures where broad stretching bands appeared at 3433–3199 cm−1 corresponding to the (NH) groups. The (C = O) band of the thiazolinone ring appeared at 1697–1627 cm−1 with an additional (C = O) band at 1701 cm−1 in compound 7 b. The two (SO2) stretching bands of all derivatives of the series were displayed at 1334–1311 and 1151–1126 cm−1. Furthermore, compounds 7a–f showed characteristic 1H NMR signal in the region 7.78–8.87 ppm corresponding to the alkene (CH=) proton accompanied with the absence of the singlet signal corresponding to the aliphatic (CH2) protons with the usual pattern of the protons of both indole and benzene sulfonamide rings. The three protons of the acetamide group of compound 7 b appeared as singlet signal at 1.94 ppm. 13C NMR spectra of compounds 7a and 7f were consistent with their carbon skeleton.
Scheme 1. Synthesis of compounds 4a–f. Reagents and reaction conditions: (i) Phosphorous oxychloride, DMF, 5 °C; (ii) glacial acetic acid.
Scheme 2. Synthesis of compounds 7a–f. Reagents and reaction conditions: (i) Chloroacetyl chloride, DMF, rt; (ii) Ammonium thiocyanate, absolute alcohol; (iii) Indole-3-carboxaldehyde 2, fused sodium acetate, glacial acetic acid, reflux.
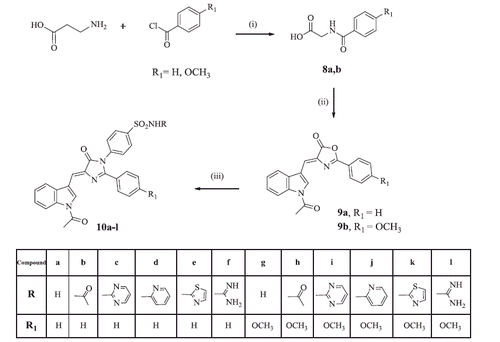
Scheme 3 illustrated the synthesis of the imidazolinone derivatives 10a–l. The oxazolone intermediates 9aCitation28 and 9 b were synthesised by Erlenmeyer reaction of hippuric acid or 4-methoxy hippuric acid with indole-3-carboxaldehyde 2 in acetic anhydride and fused sodium acetate. Interestingly, the IR spectra of compounds 9a,b explored two (C = O) stretching bands for each at 1634, 1689 and 1788, 1743 cm−1, respectively, assigned for the N-acetyl and oxazolone carbonyl groups, respectively. This confirmed that the (NH) group of the indole ring was acetylated under the reaction conditions. 1H NMR of both compounds revealed a singlet signal at 1.90 ppm assigned for the (CH3) protons of the acetyl moiety. Subsequent reaction of compounds 9a,b with the appropriate sulfonamide derivatives 3a–f afforded the expected imidazolinone derivatives 10a–l through a mechanism involving an open intermediate. The IR spectra of compounds 10a–l showed two (C = O) bands in the range of 1730–1635 cm−1 and 1701–1597 cm−1 and two characteristic bands for the (SO2) group at 1338–1300 and 1168–1126 cm−1. 1H NMR demonstrated a singlet signal in the range of 1.90–2.08 ppm assigned for the CH3 protons of the acetyl group, in addition to the (OCH3) group protons in compounds 10 g–l in the range of 3.80–3.81 ppm. Moreover, compounds 10 b and 10 h revealed an additional singlet signal at 1.92 and 2.04 ppm, respectively, assigned for (CH3) protons of the acetamido sulfamoyl moiety. 13C NMR spectra of this series were in accordance with their carbon skeleton where the (CH3) group of the indole acetyl moiety appeared in the range of 24.0–24.4 ppm, in addition to the (CH3) signals of the second acetamide moiety at 23.6 and 22.0 ppm in compounds 10 b and 10 h, respectively. Also, compound 10 h was characterised by another signal corresponding to the (OCH3) group at 55.7 ppm.
Scheme 3. Synthesis of compounds 10a–l. Reagents and reaction conditions: (i) Glycine, 10% sodium hydroxide, ice bath, (0 °C); (ii) Indole-3-carboxaldehyde 2, acetic anhydride, fused sodium acetate, (100 °C); (iii) The appropriate sulfonamide 3a–f, glacial acetic acid, fused sodium acetate, (100 °C).
Carbonic anhydrase inhibitory activity
The CA inhibitory activities of compounds 4a–f, 7a–f, and 10a–l in addition to acetazolamide (AAZ) as standard inhibitor, were measured against hCAI, hCA II, hCA IV and hCA IX by a stopped flow CO2 hydrase assayCitation29. The rational for the choice of these four isoforms is that hCA II and IV are targets for antiglaucoma drugsCitation1–3, whereas hCA IX have been validated as targets for the treatment and prognosis of hypoxic cancersCitation39,Citation40. Otherwise hCA I is one of the main off-target isoforms both for the antiglaucoma or anticancer CAIs therapeutic applicationCitation1,Citation3.
The following structure-activity-relationships (SARs) were obtained from the inhibition data reported in .
Table 1. Inhibition data of human CA isoforms hCA I, II, IV and IX with compounds 4a–f, 7a–f and 10a–l reported here and the standard sulfonamide inhibitor acetazolamide (AAZ) by a stopped flow CO2 hydrase assay.
(i) As expected the inhibitory profile of the tested compounds against the four CA isoforms was strictly dependent on the functionalisation mode of the sulfonamide group, with the primary SO2NH2 bearing derivatives (4a, 7a, 10a, and 10 g) arisen as the best hCA inhibitors herein reported. Sulfonylacetamido (4 b, 7 b, 10 b, and 10 h) and sulfonylguanidino (4c, 7c, 10c, and 10i) compounds maintained a certain degree of activity, dependent on the tailing moiety and the considered isozymes. Conversely, a totally counterproductive effect on the inhibition potency can be ascribed to all other N1-functionalisations, with all following considerations regarding uniquely the active ZBG bearing-derivatives.
(ii) The cytosolic isoform hCA I was significantly inhibited by all primary sulfonamide compounds with KIs in the range of 88.5–96.6 nM. It is pertinent to mention that the corresponding sulfonylguanidine (10f) and sulfonylacetamide (10 h) derivatives lose to a great extent their efficacy, showing a KI of 5764.1 and of 5719.1 nM, respectively.
(iii) hCA II was the most inhibited isoform, among those considered here by the primary sulfonamide derivatives (4a, 7a, 10a, and 10 g) with KIs value 575.8, 76.4, 56.4, 602.3 nM, respectively. In analogy to the SAR reported for the previous isoform, it is interesting to note that the nature of the tail appended at the benzenesulfonamide scaffold greatly affected the inhibition of this isoform in the sulfonylacetamide subset, being the imidazolones (10 b) and (10 h) medium nanomolar inhibitors (KI 487.3 and 548.9 nM), and imine (4 b) and thiazolinone (7 b) devoid of any efficacy.
(iv) Uniquely the primary sulfonamides (4a, 7a, 10a, and 10 g) exhibited significant activity against the membrane-associated isoform IV (KIs spanning between 262.7 and 4429.3 nM), with exception of sulfonylacetamide (10 h) which showed a micromolar inhibition with a KI of 3048.1 nM.
(v) The general tendencies described above were also applicable to the tumour-associated isoform hCA IX, which was moderately inhibited by the primary sulfonamides (4a, 7a, 10a, and 10 g) with binding affinities ranging between 308.0 and 328.0 nM. Sulfonylacetamido (4 b, 7 b, 10 b, and 10 h) and sulfonylguanidino (4c, 7c, 10c, and 10i) compounds were shown to possess comparable effects on hCA IX, inhibiting it in the low micromolar range (KIs of 778.3–1615.9 nM and 1206.6–1636.4 nM).
(vi) The inhibition data reported in unexpectedly highlighted a total loss of efficacy upon functionalisation of the ZBG primary sulfonamide structure by pyridine, thiazolinone and pyrimidine moieties, at least within the present set of imine-, thiazolidinone- and imidazolone-indole benzenesulfamides. On the other hand, incorporation of less hindered groups as in case of sulfonylacetamides and sulfonylguanidines maintained a certain degree of activity dependent on the tailing moiety and the considered isozymes.
(vii) Comparing the results of the current study with the earlier investigation in which a chromone nucleus was used instead of the indole ringCitation21, revealed that the chromone derivatives showed excellent activity profile despite being secondary sulfonamides and bearing similar N1-substitutions. So, it can be claimed that activity is not solely linked to the absence or presence of the N1-substitution, but also affected by other distant fragments in the molecule (chromone vs. indole). In other words, the presence of secondary sulfonamide moiety is not to be exclusively accused of the dramatic decrease in activity of the indole derivatives compared to their chromone counterparts.
Conclusions
Since the primary sulfonamide is the most efficient zinc binding group (ZBG) to design inhibitors for the metallo-enzymes CAs, herein, we propose an investigation on four physiologically important human (h) CAs (I, II, IV, and IX) of N1-substituted secondary sulfonamides incorporating a thiazolinone or imidazolone-indole scaffold. The effect of functionalisation of the sulfonamide group with five different substitution patterns, namely acetyl, pyridine, thiazole, pyrimidine and carbamimidoyl, was evaluated in relation to the inhibition profile of the primary sulfonamide analogues. With most of these latter compounds acting as nanomolar inhibitors of all four considered isoforms, a total loss of inhibitory efficacy upon functionalisation of the ZBG primary sulfonamide structure arose in case of incorporation of pyridine, thiazole and pyrimidine moieties. On the other hand, incorporation of less hindered groups, such as sulfonylacetamide (4 b, 7 b, 10 b, and 10 h) and sulfonylguanidino (4c, 7c, 10c, and 10i) maintained a certain degree of activity dependent on the tailing moiety and the considered isozymes, with KIs spanning in the low micromolar range. Nevertheless, it is worth to be mentioned that the same functionalisation of the sulfonamide group but with chromone scaffolds, led to potent inhibitors, in the nanomolar range as revealed in a previous reportCitation21. This drew our attention to the role of the indole nucleus as a key fragment responsible for switching the activity of this type of compounds.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
Related Research Data
References
- (a) Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95. (b) Carta F, Supuran CT, Scozzafava A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med Chem 2014;6:1149–65.
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discovery 2008;7:168–81. (b) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- (a) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;1:4421–68. (b) Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30.
- (a) Supuran CT, Winum JY. Drug design of zinc-enzyme inhibitors: functional, structural, and disease applications. Hoboken (NJ): John Wiley & Sons; 2009:3–12. Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68.
- Bozdag M, Ferraroni M, Nuti E, et al. Combining the tail and the ring approaches for obtaining potent and isoform-selective carbonic anhydrase inhibitors: solution and X-ray crystallographic studies. Bioorg Med Chem 2014;22:334–40.
- (a) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (b) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring? J Med Chem 1999;42:2641–50.
- Maren TH. Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol Rev 1967;47:595–781.
- Wilkinson BL, Bornaghi LF, Houston TA, et al. A novel class of carbonic anhydrase inhibitors: glycoconjugate benzene sulfonamides prepared by “click-tailing”. J Med Chem 2006;49:6539–48.
- Lopez M, Salmon AJ, Supuran CT, et al. Carbonic anhydrase inhibitors developed through “click tailing”. Curr Pharm Des 2010;16:3277–87.
- Di Fiore A, Maresca A, Alterio V, et al. Carbonic anhydrase inhibitors: x-ray crystallographic studies for the binding of N-substituted benzenesulfonamides to human isoform II. Chem Comm 2011;47:11636–8.
- (a) Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed Engl 2007;4:7697–9. (b) Mahon BP, Hendon AM, Driscoll JM, et al. Saccharin: a lead compound for structure-based drug design of carbonic anhydrase IX inhibitors. Bioorg Med Chem 2015;23:849–54.
- Nocentini A, Vullo D, Bartolucci G, et al. N-Nitrosulfonamides: a new chemotype for carbonic anhydrase inhibition. Bioorg Med Chem 2016;24:3612–7.
- Carradori S, Secci D, De Monte C, et al. A novel library of saccharin and acesulfame derivatives as potent and selective inhibitors of carbonic anhydrase IX and XII isoforms. Bioorg Med Chem 2016;24:1095–105.
- D’Ascenzio M, Carradori S, De Monte C, et al. Design, synthesis and evaluation of N-substituted saccharin derivatives as selective inhibitors of tumor-associated carbonic anhydrase XII. Bioorg Med Chem 2014;22:1821–31.
- Eldehna WM, Abo-Ashour MF, Nocentini A, et al. Novel 4/3-((4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylidene)amino)benzenesulfonamides: synthesis, carbonic anhydrase inhibitory activity, anticancer activity and molecular modelling studies. Eur J Med Chem 2017;139:250–62.
- Eldehna WM, Al-Ansary GH, Bua S, et al. Novel indolin-2-one-based sulfonamides as carbonic anhydrase inhibitors: Synthesis, in vitro biological evaluation against carbonic anhydrases isoforms I, II, IV and VII and molecular docking studies. Eur J Med Chem 2017;127:521–30.
- (a) Ibrahim HS, Abou-Seri SM, Tanc M, et al. Isatin-pyrazole benzenesulfonamide hybrids potently inhibit tumor-associated carbonic anhydrase isoforms IX and XII. Eur J Med Chem 2015;103:583–93. (b) Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91. (c) Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16. (d) Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35.
- Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008–2013). Expert Opin Ther Pat 2013;23:737–49.
- Ekinci D, Cavdar H, Durdagi S, et al. Structure-activity relationships for the interaction of 5,10-dihydroindeno[1,2-b]indole derivatives with human and bovine carbonic anhydrase isoforms I, II, III, IV and VI. Eur J Med Chem 2012;49:68–73.
- Awadallah FM, El-Waei TA, Hanna MM, et al. Synthesis, carbonic anhydrase inhibition and cytotoxic activity of novel chromone-based sulfonamide derivatives. Eur J Med Chem 2015;96:425–35.
- James PN, Snyder HR. Indole-3-aldehyde. Org Synth 1969;4:539.
- Ragab AF, Heiba HI, El-Hazek MR. Anticancer and radio-sensitizing evaluation of some new thiazolopyrane and thiazolopyranopyrimidine derivatives bearing a sulfonamide moiety. Eur J Med Chem 2011;46:5120–6.
- Ghorab MM, Ragab AF, Heiba HI, et al. Synthesis of some novel sulfonamides containing biologically active alkanoic acid, acetamide, thiazole and pyrrole moieties of expected antitumor and radiosensitizing activities. J Basic Appl Chem 2011;1:8–14.
- Song M, Mei W, Weijuan G, et al. 5-(1H-indolyl-3-methylene)-1,3-thiazolidinyl-4-one derivatives, and synthesis method and application thereof. CN 104059060 A. 2014;1–27.
- Crawford M, Little WT. The Erlenmeyer reaction with aliphatic aldehydes, 2-phenyloxazol-5-one being used instead of hippuric acid. J Chem Soc 1959;729–31.
- Achesond RM, Booth DA, Brettle R, et al. The synthesis of some acylglycines and related oxazolone. J Chem Soc 1960;3457–60.
- Daniele AG, Aaron N, Benedetto M, et al. Conformationally constrained tryptophan analogs. Synthesis of (±)-(Z)- and (±)-(E)-2-amino-2,3-methano-3-(indol-3-yl)-propanoic acids. Tetrahedron 1996;52:9901–8.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Akocak S, Lolak N, Nocentini A, et al. Synthesis and biological evaluation of novel aromatic and heterocyclic bis-sulfonamide Schiff bases as carbonic anhydrase I, II, VII and IX inhibitors. Bioorg Med Chem 2017;25:3093–7.
- Entezari Heravi Y, Bua S, Nocentini A, et al. Inhibition of Malassezia globosa carbonic anhydrase with phenols. Bioorg Med Chem 2017;25:2577–82.
- Altug C, Güneş H, Nocentini A, et al. Synthesis of isoxazole-containing sulfonamides with potent carbonic anhydrase II and VII inhibitory properties. Bioorg Med Chem 2017;25:1456–64.
- Zhang Z, Lau J, Zhang C, et al. Design, synthesis and evaluation of 18F labeled cationic carbonic anhydrase IX inhibitors for PET imaging. J Enzyme Inhib Med Chem 2017;32:722–30.
- Nocentini A, Cadoni R, Del Prete S, et al. Benzoxaboroles as efficient inhibitors of the β-carbonic anhydrases from pathogenic fungi: activity and modeling study. ACS Med Chem Lett 2017;8:1194–8.
- Nocentini A, Bua S, Lomelino CL, et al. Discovery of new sulfonamide carbonic anhydrase IX inhibitors incorporating nitrogenous bases. ACS Med Chem Lett 2017;8:1314–19.
- (a) Nocentini A, Vullo D, Del Prete S, et al. Inhibition of the β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa with monothiocarbamates. J Enzyme Inhib Med Chem 2017;32:1064–70. (b) Supuran CT, Capasso C. An overview of the bacterial carbonic anhydrases. Metabolites 2017;7:E56.
- Ebrahimi H, Hadi JS, Al-Ansari HS. A new series of Schiff bases derived from sulfa drugs and indole-3-carboxaldehyde: synthesis, characterization, spectral and DFT computational studies. J Molec Struc 2013;1039:37–45.
- Naqvi A, Shahnawaaz M, Rao AV, et al. Synthesis of Schiff bases via environmentally benign and energy-efficient greener methodologies. El J Chem 2009;6:75–8.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- (a) Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48. (b) Iessi E, Logozzi M, Mizzoni D, et al. Rethinking the combination of proton exchanger inhibitors in cancer therapy. Metabolites 2018;8:E2.