
Abstract
The inhibition of the δ-class carbonic anhydrase (CAs, EC 4.2.1.1) from the diatom Thalassiosira weissflogii, TweCAδ, was investigated using a panel of 36 mono- and di-thiocarbamates chemotypes that have recently been shown to inhibit mammalian and pathogenic CAs belonging to the α- and β-classes. TweCAδ was not significantly inhibited by most of such compounds (KI values above 20 µM). However, some aliphatic, heterocyclic, and aromatic mono and di-thiocarbamates inhibited TweCAδ in the low micromolar range. For some compounds incorporating the piperazine ring, TweCAδ was effectively inhibited (KIs from 129 to 791 nM). The most effective inhibitors identified in this study were 3,4-dimethoxyphenyl-ethyl-mono-thiocarbamate (KI of 67.7 nM) and the R-enantiomer of the nipecotic acid di-thiocarbamate (KI of 93.6 nM). Given that the activity and inhibition of this class of enzyme have received limited attention until now, this study provides new molecular probes and information for investigating the role of δ-CAs in the carbon fixation processes in diatoms, which are responsible for significant amounts of CO2 taken from the atmosphere by these marine organisms.
Introduction
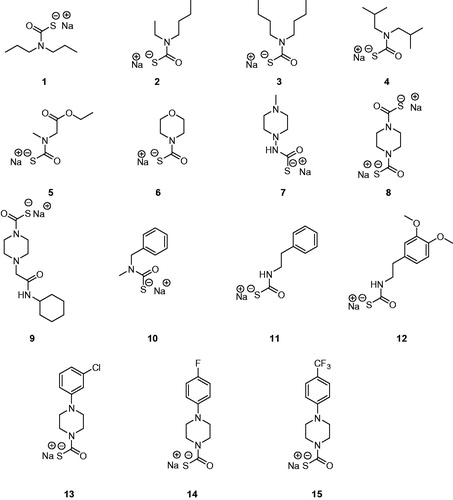
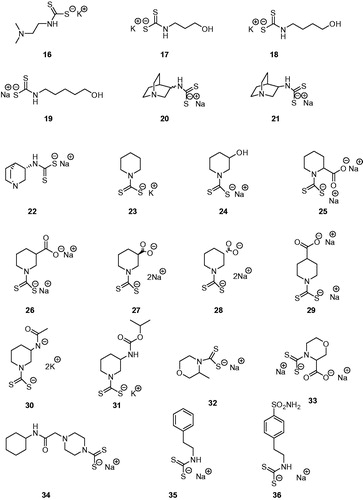
The di-thiocarbamates (DTCs) possessing the general formula RR1NCS2M (where R, R1 may be H, alkyl, cycloalkyl, aryl, hetaryl, etc., and M is a cation) were recently reported as a new class of inhibitors of the metalloenzyme carbonic anhydrase (CA, EC 4.2.1.1)Citation1. Their inhibitory activity was investigated against α- and β-class CAs from various organismsCitation1,Citation2 and they also led to the discovery of two new CA inhibitor (CAI) classes, the xanthatesCitation3 and the mono-thiocarbamates (MTCs)Citation4. Representatives of MTCs and DTCs acting as CAIs are shown in and .
Figure 1. Monothiocarbamates (MTCs) 1–15 investigated as CA inhibitorsCitation4.
Inhibition of CAs belonging to some of the seven genetically distinct families known to dateCitation5–10 has various biomedical applications owing to the fact that these enzymes catalyse a simple but physiologically crucial reaction: the hydration of CO2 to bicarbonate and hydronium ionsCitation5–10. Interference with this process has important physiological and pathological consequences because CAs are involved in pH regulation, biosynthetic processes, metabolism, secretion of electrolytes, transport of CO2/bicarbonate, etc.Citation5–10. Their dysregulated expression or activity leads to various pathologies, and as a consequence, their inhibitors are clinically used as diuretics, antiglaucoma, antiepileptic, anti obesity, and antitumour agentsCitation5–9. Recently, the CAIs were also shown to be effective for the control of neuropathic pain, cerebral ischemia, and some forms of arthritisCitation10. The primary sulphonamides and their isosteres (sulphamides and sulphamates) are the main class of CAIs, but in many cases, they indiscriminately inhibit most of the many CA isoforms known in an organism (e.g. 15 CA isoforms belonging to the α-class are known in humansCitation5,Citation11–16). This is the reason why alternative chemotypes, such as the DTCs and MTCs have recently been exploredCitation1–4. However, this class of CAIs has only been investigated to date for their interaction with human (h), α-class enzymes, and with several CAs from pathogens or model organisms, belonging to the α- and β-CA classesCitation1–4. The δ-CAs were discovered in the diatom Thalassiosira weissflogiiCitation6d, but orthologues of this enzyme have been identified in most diatoms from natural phytoplankton assemblages and are responsible (along with other CAs) for CO2 fixation by marine organismsCitation17. A related species of this diatom, Thalassiosira pseudonana, was shown to possess genes for three α-, five γ-, four δ-, and one ζ-CAsCitation18. However, none of these enzymes have been cloned and characterised in detail to date, except TweCAδCitation11. Diatoms can be considered to be the organisms with the most intricate and poorly understood distribution of CAs, but the roles of these enzymes seem to be crucial for CO2 fixation and photosynthesis in many organisms and are estimated to be responsible for at least 25% of the inorganic carbon fixation in the oceansCitation6,Citation17,Citation18. However, few studies are available for the interaction of δ-CAs with modulators of activity, inhibitors, and activators. TweCAδ was the only representative of the δ-class for which anion and sulphonamide inhibition studies have been reported to dateCitation6d,Citation11. Here we report the first CA inhibition study with MTCs and DTCs of a δ-CA class enzyme, TweCAδ, which was cloned and characterised from the marine diatom T. weissflogiiCitation6d,Citation6e.
Materials and methods
Materials
MTCs 1–15Citation4 and DTCs 16–36Citation1,Citation2 were reported earlier by our group. Reagents/buffers of the highest available purity were obtained from Sigma-Aldrich, Milan, Italy. TweCAδ was a recombinant protein produced as reported earlier by our groupCitation6e,Citation11.
CA enzyme inhibition assay
An Sx.18Mv-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the catalytic activity of various CA isozymes for CO2 hydration reactionCitation12. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.5) as buffer, and 0.1 M Na2SO4 (for maintaining constant ionic strength, which is not inhibitory against TweCAδCitation11), following the CA-catalysed CO2 hydration reaction for a period of 10 s at 25 °C. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and activation constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial rate. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitors (10 mM) were prepared in distilled-deionised diluted to 1 nM using the assay buffer. Inhibitor and enzyme solutions were pre-incubated together for 15 min (standard assay at room temperature) prior to assay, in order to allow for the formation of the enzyme inhibitor complex. The inhibition constant (KI), was obtained by considering the classical Michaelis–Menten equation and the Cheng-Prusoff algorithm by using non-linear least squares fitting as reported earlierCitation13–16.
Results and discussion
TweCAδ is the only CA belonging to the δ-class for which anion and sulphonamide inhibition studies were reported so farCitation6d,Citation11. Here, we investigated the inhibition of this enzyme with the panel of MTCs and DTCs of the types 1–36 shown in and . The results are shown in , where for comparison reasons, the inhibition of the human dominant isoforms hCA I and II with the same compounds are reportedCitation1,Citation2,Citation4.
Table 1. TweCAδ, hCA I, and hCA II Inhibition Data with MTCs 1–15, DTCs 16–36, and acetazolamide (AAZ, 5-acetamido-1,3,4-thiadiazole-2-sulphonamide) as standard drug, by a stopped-flow CO2 hydrase assay.
The following structure-activity relationship (SAR) can be obtained from the data of :
(i) A number of MTCs, including 4–6, 10 and the DTCs 20, 21, 23–25, 32, and 33, did not inhibit TweCAδ up to 20 µM, although many of these compounds were rather effective inhibitors of hCA I and/or hCA II (). Such MTCs/DTCs inhibitors are classified as aliphatic, heterocyclic, aromatic, or polycyclic types. Given the structural diversity of such compounds and high inhibition constants, it is challenging to delineate the SAR.
(ii) The MTCs/DTCs 3, 13–19, 22, 26, 29, and 31 were relatively ineffective inhibitors of TweCAδ with inhibition constants in the micromolar range (KIs ranged between 1142 and 9239 nM; ). These compounds are also highly heterogeneous. The main observation of these data is that the identity of the zinc-binding group, ZBG (MTC or DTC), does not significantly impact the activity of TweCAδ.
(iii) The MTC/DTCs 1, 2, 7–9, 28, 30, and 34–36 were relatively effective inhibitors of TweCAδ, with inhibition constants in the range of 129–997 nM (). Some of the MTC and DTCs incorporate the piperazine ring (7–9, 34). In addition, MTC 9 and DTC 34 have the same scaffold but a different ZBG. In this particular case, MTC 9 inhibited TweCAδ 6.1-times more efficiently than DTC 34. Interestingly, for the β-CAs, the MTCs were usually much weaker inhibitors compared to the structurally similar DTCsCitation4. In addition, the sulphonamide-containing DTC 36 (which contains two potential ZBGs, the sulphonamide and the DTC), there are no net differences of TweCAδ inhibitory activity compared to the structurally similar derivatives (e.g. 35) which probably is due to the fact that the DTC in 36 is primarily binding to the metal ion in the enzyme active site, and not the sulphonamide moiety. However, the heterocyclic sulphonamide acetazolamide (AAZ, 5-acetamido-1,3,4-thiadiazole-2-sulphonamide), a clinically used drugCitation5, is a much more potent inhibitor (KI of 83 nM) of TweCAδ compared to 36 ().
(iv) The most effective TweCAδ inhibitors identified in this MTC/DTC panel were the MTC 12 (KI of 67.7 nM) and the DTC 27 (KI of 93.6 nM). These compounds incorporate scaffolds rather similar to those present in other investigated compounds, which were, however, much less effective as inhibitors of this enzyme. For example, 12 has two methoxy moieties on the scaffold of 11, but there is a difference of activity of 14.7-fold between the two MTCs. The R-enantiomer 27 was on the other hand 5.9 times a more effective inhibitor compared to the S-enantiomer 28. All these data show that small changes in the structure or the stereochemistry of a DTC/MTC lead too dramatic changes of affinity for the target enzyme.
(v) With a few exceptions, TweCAδ was less sensitive to this class of CAIs compared to the α-CAs hCA I and II (). There are several X-ray crystal structures that demonstrate that the DTCs (and presumably also the MTCs) bind to the metal ion in the CA active site by substituting the hydroxide nucleophile that is responsible for the catalytic activity of the enzymeCitation1,Citation2. Most probably, this is also the inhibition mechanism by which DTCs and MTCs interact with δ-CAs. However, this enzyme class is the least studied of the 7 CA genetic families, and there are no X-ray crystal structures or even homology models available for any δ-CAs.
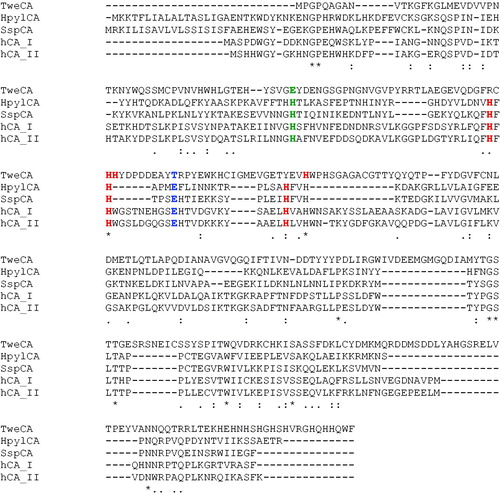
We try to rationalise the obtained inhibition data based on the amino acid sequence of TweCAδ, which has been aligned with that of α-CAs for which the X-ray crystal structure is known, of bacterial (HpylCA, α-CA from Helicobacter pylori, SspCA, α-CA from Sulfurihydrogenibium yellowstonensis) or human origin (hCA I and II) (). Data of show that for the α-CAs, the zinc ligands are three His residues (His94, 96, and119, hCA I numbering system), which align well for the bacterial and human enzymes, whereas the putative zinc ligands of TweCAδ do not align at all with those of the α-class enzyme. The same is true for other amino acid residues from the α-CAs, such as the proton shuttle (His64) which is an Asp residue in TweCAδ, or residues 106 (a conserved Asp residue in all α-CAs), which is a Thr in TweCAδ. Based on these data it is obvious that it is not possible to rationalise the observed SAR with mono- and di-thiocarbamates based only on the sequence of the enzyme, without a homology model or better, an X-ray crystal structure of the diatom enzyme.
Figure 3. Multialignment of the TweCAδ amino acid sequence with those of bacterial (HpylCA, α-CA from Helicobacter pylori, SspCA, α-CA from Sulfurihydrogenibium yellowstonensis) and human (hCA I and II) α-class enzymes. The zinc ligands of the α-CAs and the putative zinc ligands of TweCAδ are evidenced in red, whereas amino acid residues involved in the catalytic inhibition/mechanism (e.g. His64 and Asp106, hCA I numbering) are shown in green and blue, respectively.
Conclusions
The first inhibition study of a δ-CA with mono- and di-thiocarbamates, classes of CAIs recently discovered, was reported. TweCAδ from the marine diatom T. weissflogii was not particularly sensitive to inhibition by these classes of compounds. Many of the mono- and di-thiocarbamates did not show inhibitory action up to 20 µM, whereas some aliphatic, heterocyclic, and aromatic inhibited this enzyme in the low micromolar range. Several MTCs/DTCs incorporating the piperazine ring effectively inhibited TweCAδ with KIs in the range of 129–791 nM. The most effective inhibitors identified were 3,4-dimethoxyphenyl-ethyl-mono-thiocarbamate (KI of 67.7 nM) and the R-enantiomer of the nipecotic acid DTC (KI of 93.6 nM). Such inhibitors can now be used as molecular probes to investigate the role of this enzyme in the carbon fixation processes in diatom marine organisms that are responsible for removing large amounts of CO2 from the atmosphere.
Disclosure statement
The authors do not declare any conflict of interest.
Additional information
Funding
Related Research Data
References
- a) Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun (Camb) 2012;48:1868–70. b) Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates strongly inhibit carbonic anhydrases and show antiglaucoma action in vivo. J Med Chem 2012;55:1721–30. c) Monti SM, Maresca A, Viparelli F, et al. Dithiocarbamates are strong inhibitors of the beta-class fungal carbonic anhydrases from Cryptococcus neoformans, Candida albicans and Candida glabrata. Bioorg Med Chem Lett 2012;22:859–62. d) Maresca A, Carta F, Vullo D, Supuran CT. Dithiocarbamates strongly inhibit the β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:407–11.
- a) Syrjänen L, Tolvanen ME, Hilvo M, et al. Characterization, bioinformatic analysis and dithiocarbamate inhibition studies of two new α-carbonic anhydrases, CAH1 and CAH2, from the fruit fly Drosophila melanogaster. Bioorg Med Chem 2013;21:1516–21. b) Winum JY, Supuran CT. Recent advances in the discovery of zinc-binding motifs for the development of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:321–4. c) Bozdag M, Carta F, Vullo D, et al. Dithiocarbamates with potent inhibitory activity against the Saccharomyces cerevisiae β-carbonic anhydrase. J Enzyme Inhib Med Chem 2016;31:132–6. d) Bozdag M, Carta F, Vullo D, et al. Synthesis of a new series of dithiocarbamates with effective human carbonic anhydrase inhibitory activity and antiglaucoma action. Bioorg Med Chem 2015;23:2368–76. e) Vullo D, Del Prete S, Nocentini A, et al. Dithiocarbamates effectively inhibit the β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa. Bioorg Med Chem 2017;25:1260–5. f) Aspatwar A, Hammarén M, Koskinen S, et al. β-CA-specific inhibitor dithiocarbamate Fc14-584B: a novel antimycobacterial agent with potential to treat drug-resistant tuberculosis. J Enzyme Inhib Med Chem 2017;32:832–40.
- Carta F, Akdemir A, Scozzafava A, et al. Xanthates and trithiocarbonates strongly inhibit carbonic anhydrases and show antiglaucoma effects in vivo. J Med Chem 2013;56:4691–700.
- a) Vullo D, Durante M, Di Leva FS, et al. Monothiocarbamates strongly inhibit carbonic anhydrases in vitro and possess intraocular pressure lowering activity in an animal model of glaucoma. J Med Chem 2016;59:5857–67. b) Nocentini A, Vullo D, Del Prete S, et al. Inhibition of the β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa with monothiocarbamates. J Enzyme Inhib Med Chem 2017;32:1064–70.
- a) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. b) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. c) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. d) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. e) Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30.
- a) Xu Y, Feng L, Jeffrey PD, et al. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008;452:56–61. b) Ferry JG. The gamma class of carbonic anhydrases. Biochim Biophys Acta 2010;1804:374–81. c) Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum–the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96. d) Cox EH, McLendon GL, Morel FM, et al. The active site structure of Thalassiosira weissflogii carbonic anhydrase 1. Biochemistry 2000;39:12128–30. e) Del Prete S, Vullo D, Scozzafava A, et al. Cloning, characterization and anion inhibition study of the δ-class carbonic anhydrase (TweCA) from the marine diatom Thalassiosira weissflogii. Bioorg Med Chem 2014;22:531–7.
- a) Capasso C, Supuran CT. An overview of the alpha-, beta-and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32. b) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72. c) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. d) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68. e) Supuran CT, Capasso C. New light on bacterial carbonic anhydrases phylogeny based on the analysis of signal peptide sequences. J Enzyme Inhib Med Chem 2016;31:1254–60.
- a) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. b) Akocak S, Lolak N, Vullo D, et al. Synthesis and biological evaluation of histamine Schiff bases as carbonic anhydrase I, II, IV, VII, and IX activators. J Enzyme Inhib Med Chem 2017;32:1305–12. c) Angeli A, Vaiano F, Mari F, et al. Psychoactive substances belonging to the amphetamine class potently activate brain carbonic anhydrase isoforms VA, VB, VII, and XII. J Enzyme Inhib Med Chem 2017;32:1253–9. d) Licsandru E, Tanc M, Kocsis I, et al. A class of carbonic anhydrase I: selective activators. J Enzyme Inhib Med Chem 2017;32:37–46.
- a) Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91. b) Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16. c) Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35. d) Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008–2013). Expert Opin Ther Pat 2013;23:737–49. e) Supuran CT, Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48–1248. f) Capasso C, Supuran CT. Inhibition of bacterial carbonic anhydrases as a novel approach to escape drug resistance. Curr Top Med Chem 2017;17:1237. g) Mastrolorenzo A, Rusconi S, Scozzafava A, et al. Inhibitors of HIV-1 protease: current state of the art 10 years after their introduction. From antiretroviral drugs to antifungal, antibacterial and antitumor agents based on aspartic protease inhibitors. Curr Med Chem 2007;14:2734–48.
- a) Carta F, Di Cesare Mannelli L, Pinard M, et al. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg Med Chem 2015;23:1828–40. b) Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother 2016;16:961–8. c) Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. J Enzyme Inhib Med Chem 2016;31:894–9. d) Margheri F, Ceruso M, Carta F, et al. Overexpression of the transmembrane carbonic anhydrase isoforms IX and XII in the inflamed synovium. J Enzyme Inhib Med Chem 2016;31:60–3.
- a) Vullo D, Del Prete S, Osman SM, et al. Sulfonamide inhibition studies of the δ-carbonic anhydrase from the diatom Thalassiosira weissflogii. Bioorg Med Chem Lett 2014;24:275–9. b) Del Prete S, Vullo D, De Luca V, et al. Biochemical characterization of the δ-carbonic anhydrase from the marine diatom Thalassiosira weissflogii, TweCA. J Enzyme Inhib Med Chem 2014;29:906–11.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- a) Menchise V, De Simone G, Alterio V, et al. Carbonic anhydrase inhibitors: stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J Med Chem 2005;48:5721–7. b) Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors—part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents in rabbits. Eur J Med Chem 1998;33:247–54. c) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8. d) Şentürk M, Gülçin İ, Beydemir Ş, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9. e) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47. f) Dogne JM, Hanson J, Supuran C, Pratico D. Coxibs and cardiovascular side-effects: from light to shadow. Curr Pharm Des 2006;12:971–5.
- a) Krall N, Pretto F, Decurtins W, et al. A small‐molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5. b) Rehman SU, Chohan ZH, Gulnaz F, Supuran CT. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzyme Inhib Med Chem 2005;20:333–40. c) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: structure‐activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73. d) Dubois L, Peeters S, Lieuwes NG, et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 2011;99:424–31. e) Chohan ZH, Munawar A, Supuran CT. Transition metal ion complexes of Schiff-bases. Synthesis, characterization and antibacterial properties. Met Based Drugs 2001;8:137–43. f) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal β-class (Cab) and γ-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–8. g) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- a) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8. b) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3. c) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. d) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11. e) Di Fiore A, De Simone G, Alterio V, et al. The anticonvulsant sulfamide JNJ-26990990 and its S,S-dioxide analog strongly inhibit carbonic anhydrases: solution and X-ray crystallographic studies. Org Biomol Chem 2016;14:4853–8. f) Supuran CT, Scozzafava A, Mastrolorenzo A. Bacterial proteases: current therapeutic use and future prospects for the development of new antibiotics. Expert Opin Ther Pat 2001;11:221–59.
- a) Carta F, Birkmann A, Pfaff T, et al. Lead development of thiazolylsulfonamides with carbonic anhydrase inhibitory action. J Med Chem 2017;60:3154–64. b) Supuran CT, Kalinin S, Tanç M, et al. Isoform-selective inhibitory profile of 2-imidazoline-substituted benzene sulfonamides against a panel of human carbonic anhydrases. J Enzyme Inhib Med Chem 2016;31(1): 197–202. c) Pettersen EO, Ebbesen P, Gieling RG, et al. Targeting tumour hypoxia to prevent cancer metastasis. from biology, biosensing and technology to drug development: the METOXIA consortium. J Enzyme Inhib Med Chem 2015;30:689–721. d) De Vita D, Angeli A, Pandolfi F, et al. Inhibition of the α-carbonic anhydrase from Vibrio cholerae with amides and sulfonamides incorporating imidazole moieties. J Enzyme Inhib Med Chem 2017;32:798–804. e) Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed Engl 2007;46:7697–9. f) Scozzafava A, Menabuoni L, Mincione F, Supuran CT. Carbonic anhydrase inhibitors. A general approach for the preparation of water soluble sulfonamides incorporating polyamino-polycarboxylate tails and of their metal complexes possessing long lasting, topical intraocular pressure lowering properties. J Med Chem 2002;45:1466–76. g) Chohan ZH, Supuran CT, Scozzafava A. Metal binding and antibacterial activity of ciprofloxacin complexes. J Enzyme Inhib Med Chem 2005;20:303–7.
- McGinn PJ, Morel FM. Expression and regulation of carbonic anhydrases in the marine diatom Thalassiosira pseudonana and in natural phytoplankton assemblages from Great Bay, New Jersey. Physiol Plant 2008;133:78–91.
- Tachibana M, Allen AE, Kikutani S, et al. Localization of putative carbonic anhydrases in two marine diatoms, Phaeodactylum tricornutum and Thalassiosira pseudonana. Photosynth Res 2011;109:205–21.