
Abstract
In this experiment, DNA-ELISA biosensor was introduced, bearing the ability to detect specific bacteria in about 4 h. This is a more rapid system in comparison to conventional methods, like colony counting method. Moreover, this method does not require any amplification and directly detects genomic DNA of bacteria, giving a lower limit to the sensitivity of 40,000 bacteria. In this study, two specific probes capture (biotin labelled) and detector (dig labelled), were used against special regions of 16s rRNA gene of Escherichia coli ATCC 25922. The capture probe has the ability to trap the target bacterial DNA from a pool of other kinds of bacteria under specific conditions. The detector probe then was used to hybridize to the genome of trapped bacteria. The detection proceeds by adding HRP-anti dig enzyme and its substrate, ABTS to emit light. Light absorbance is measured for verifying the detection.
Introduction
Bacterial pathogens are distributed in soil, marine and intestinal tract of humans. Humans carry more than 150 kinds of bacteriaCitation1. Among these bacteria, some can have profound effect on humans and animals and may cause different infectious disease. Infectious disease account for nearly 40% of total 50 million annual estimated deathsCitation1–3. Pathogen detection is the utmost significance for health and safety reasons. Among these pathogen bacteria, Enterobacteriaceae, especially Escherichia coli, involved in infectious diseases such as urinary tract infections (UITs). Escherichia coli is the most common and thoroughly studied model bacteriumCitation4–7. The effective detection of bacteria needs method of analysis that meets a lot of challenging criteria. Time and sensitivity of analysis are the most important limitation, associated to the value of microbiological testingCitation1,Citation2. Traditional approaches for analysis of E. coli have relied on cultural techniques and many selective-differential media that have been developed. These methods usually were based on a morphological evaluation of the microorganism and require growth of single cell in to a colony. This process is time consuming because making a colony containing 106 organism will take between 18 and 24 h even in some cases up to 72 h to obtain confirmed resultsCitation1,Citation5–7. The steps of traditional methods involve, pre-enrichment, selective enrichment, biochemical screening and serological confirmationCitation1. Molecular diagnostic experiments like PCR-based methods need fewer number of primary cell but they call for equipment and expert technical personnel. Besides, amplification of cells in PCR needs lot of time before screeningCitation7–9. For this reason, diagnosis of a pathogen always requires a rapid and low cost method. The DNA hybridisation technology has opened new trends for improving detection methods with more selectivity and less time and money consumingCitation9–11. The goal of this study was to design a rapid, selective and sensitive method for pathogen detection. For this purpose, hybridisation of specific probe to the special complementary region of E. coli gene was used in place of amplification. Detection occur directly from genome. Our approach is outlined in . In brief, after hybridisation of dig labelled probe to its complementary region of 16 s rRNA of E. coli ATCC 25922, the substrates’ colour of HRP-anti dig (ABTS) turn to green so the presence of bacteria can be detected directly with faster speed.
Figure 1. Illustration of the DNA-ELISA method. 1. The bacteria were cultured in LB. 2. The detector probe hybridized to 16 s rRNA gene of the target bacteria. 3. Capture probe conjugate to the complementary sequence in the16s rRNA gene. 4. HRP enzyme added to the solution. 5. ABTS resulted in colour for detection (This figure is adopted by Joseph C. LiaCitation4).
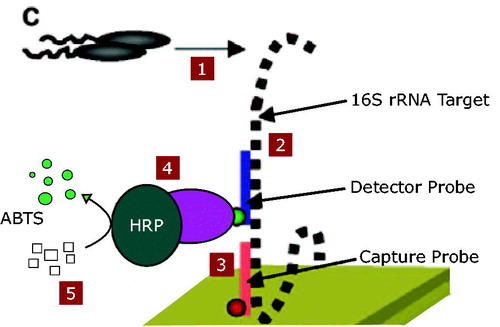
Materials and methods
Bacterial strains
Bacterial species E. coli ATCC 25922 used for the test. For cross reaction these bacteria were used: Klebsiella pneumonia ATCC 70603, Staphylococcus haemolyticus PTCC 1437, Streptococcus pyogenes PTCC 1447, Enterococcus faecalis ATCC 29212, Pseudomonas aeruginosa ATCC 27863, Proteus mirabilis ATCC 25933, Serratia marcescens ATCC 13880, E. coli PTCC1270, E. coli PTCC 1276. All the bacteria were obtained from Iranian Research Organization for Science and Technology (IROST). The bacteria were subcultured in Tryptic Soy Broth (TSB) and bacterial stocks were prepared by 70% glycerol. Bacteria were subcultured in Luria broth (LB) at 37 °C overnight, until it gets the OD (600) of 1 that is approximately 5 × 108 to 1 × 109 cells/mlCitation4.
DNA extraction
About 1–2 ml cultured bacteria were transferred in to 2-ml tube and the DNA of the bacteria extracted based on INTRON biotechnology company kit protocol (Korea). DNA concentration was adjusted to 240 ng/µl.
Probe and primer design
Two specific probes (capture and detector) for the special region of 16 s rRNA in E. coli ATCC 25,922 were selected based on the sequence reported by Joseph C. LiaCitation4. The capture probe was biotin labelled at 5′´ end to enable conjugation with the streptavidin coated in test plate (5′-GTCAATGAGCAAAGGTATTAACTTTACTCCCTTCC-3′). Detector probe contains dig at 5′ end that can attach to antidig-HRP enzyme (5′ CTGAAAGTACTTTACAACCCGAAGGCCTTCTTCAT-3′). Two 20 oligonucleotides primer (16 sf and 16 sr) were design by means of pick primer tools in NCBI for sequencing of the gene. Primers were generated for the region contains probe sequence and their performance, including self-complementary and GC percent, were evaluated by oligo program. “16 sf” Primer was located between bp 124–143. “16 sr” primer was located between bp 880–899 at the 16 s rRNA gene. The probes were located between bp 399–433 (detector) and bp 440–474 (capture). Another dig labelled 309 bp detector probe was designed by using the former 35mer detector probe and 16sf primer in PCR.
Producing PCR product for PCR-ELISA test
PCR was performed for 35 cycles. Samples were incubating at 95 °C for 15 s to denature the DNA, then at 58 °C for 30 s to annealed the primers and finally 72 °C for 60 s to extend the annealed primers. Labelling of the PCR-generated fragment was performed with Digoxigenin-11-2′-deoxy-uridine-5′-triphosphate, alkali- stable kit (USA Roche company) by adding dig in PCR samples according to the manufacture’s instruction.
Producing PCR product for developing detector probe
35-nucleotide detector probe did not result enough OD for detection so a it needs probe with more dig on it. For this purpose, PCR product was produced by using the aforementioned procedure and using the 35-nucleotide detector probe as reverse primer. The sample contains 16sf primer as forward primer, 35-nucleotide detector probe, template DNA and master mix.
PCR-ELISA test
The procedure of PCR-ELISA has been done by using the PCR-ELISA dig detection (Roche company) protocol. PCR product was incubated for 10 min in 15–20 °C in denaturation buffer contain NAOH. About 250 µl of mixture containing Capture probe and hybridisation buffer, was added to the former solution. Then the solution was added to the streptavidin coated wells and incubated for 3 h at 37 °C with shaking. After aspirating and washing the wells three to five times with washing buffer, 200 µl of HRP-anti dig enzyme was added to the wells and incubate for 30 min in 37 °C with shaking. Again wells aspirated and washed three to five times with washing buffer and 200 µl of ABTS solution was added. The absorbance of the solutions was measured in 405 nm after 30 min incubation in darkness.
DNA-ELISA test
The procedure of DNA-ELISA has been done by using the PCR-ELISA dig detection (Roche company) protocol. DNA and 309 bp detector probe were incubated for 10 min in 15–20 °C in denaturation buffer contain NAOH. About 250 µl of mixture containing Capture probe and hybridisation buffer was added to the former solution. About 200 µl of the solution including two probe, DNA and buffers was added to the streptavidin coated wells and incubated for 3 h at 37 °C with shaking. After aspirating and washing the wells three to five times with washing buffer, 200 µl of HRP-anti dig enzyme was added to the wells and incubate for 30 min in 37 °C with shaking. Again wells aspirated and washed three to five times with washing buffer and 200 µl of ABTS solution was added. The absorbance of the solutions was measured in 405 nm after 30 min incubation in darkness ().
Results
DNA-ELISA
Preparation of 300 bp, dig labelled detector probe
A 35-nucleotide detector probe with just one dig at the end was used as primer. The presence of prepared 300 bp, dig labelled probe, was proved by the appearance the band on gel agarose. The labelled PCR product was a little upper than the unlabelled.
Optimizing the capture and detector probes
To optimize the concentration of probes in the experiment, different dilution of probe as shown in for DNA-ELISA, were tested. The most reasonable which can create more colour in comparison to the concentration of probe, was detected for the experiment ( and ). For capture probe the concentration of 20 pm/µl was selected because as the concentration increases, the OD differences become lower. For detector probe the last concentration was selected because using more detector probe could unbalance the other concentration of solution. Also in this concentration we can get enough OD for detection.
Table 1. Optimization capture probe.
Table 2. Optimization detector probe.
Sensitivity
To determine the sensitivity of the tests serially diluted extracted DNA were prepared. Tenfold serial dilution of a known concentration (240 ng/µl) were used directly for DNA-ELISA test and as template for PCR-ELISA test. The detection limit of the E. coli detection was 0.024 ng/µl that is around 104 bacteria for DNA-ELISA (). The absorbance in this concentration was 0.14 that was upper than cut off valueCitation11 for the test (0.11).
Table 3. Sensitivity of the test.
Evaluation of test with other bacteria and determine the specificity
To verify the specificity of the tests nine other bacteria that is more common in UTIs was subjected to the experiment. Equal concentration of extracted DNA of nine bacteria including two E. coli tested (K. pneumonia, S. haemolyticus, S. pyogenes, E. faecalis, P. aeruginosa, P. mirabilis, S. marcescens, E. coli PTCC1270, E. coli PTCC 1276). For the DNA-ELISA test, the extracted DNA was used directly. As shown in the two E. coli were positive and has visible absorbance with green colour but other bacteria are negative and the colourless.
Table 4. Evaluation of specificity.
Assessment of repeatability of the test by inter and intra assays
The repeatability of the test was evaluated by inter- and intra-assays (). Triplicates of special concentrations in different times of a day for intra assay were tested by DNA-ELISA. The means of acquired OD for three days used for inter assay. The standard deviation and coefficient of variation of the test were calculated. Intra-assay coefficient of variations was 3.84–9.80% and intra-assay coefficient of variations were 5.26–7.14%, respectively.
Table 5. Evaluation of repeatability of the DNA-ELISA test.
PCR-ELISA
Optimizing the capture probes
To optimize the concentration of probe in PCR-ELISA different dilution of probe as shown in , were tested and the most reasonable one that can create more colour in comparison to the concentration of probe was detected for the experiment. For capture probe the last concentration was selected because using more detector probe could unbalance the other concentration of solution. Also in this concentration we can get enough ODCitation1 for detection.
Table 6. Optimization capture probe for PCR-ELISA.
Sensitivity
To determine the sensitivity of the PCR-ELISA serially diluted extracted DNA were used as template for producing PCR product. Tenfold serial dilution of a known concentration (240 ng/µl) was used for the test. The detection limit for PCR-ELISA was 0.0024 ng/µl that it is around 103 bacteria for the test (). The absorbance in this concentration was 0.5 that was upper than Cut off valueCitation11 for the test (0.11).
Table 7. Sensitivity of the PCR-ELISA.
Evaluation of test with other bacteria and determine the specificity
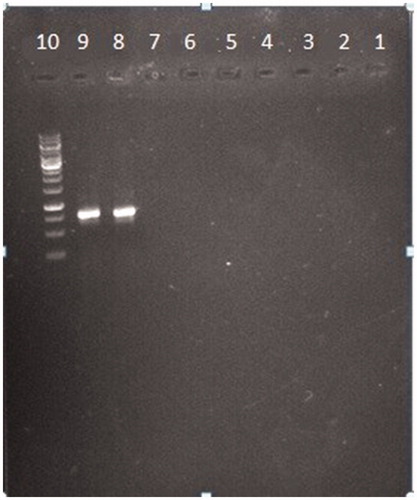
To verify the specificity of the tests nine other bacteria that is more common in UTIs were subjected to the experiment. Equal concentration of extracted DNA of nine bacteria including two E. coli were used as template for preparing PCR-product (K. pneumonia, S. haemolyticus, S. pyogenes, E. faecalis, P. aeruginosa, P. mirabilis, S. marcescens, E. coli PTCC1270, E. coli PTCC 1276). The results show that just the DNA of two E. coli have ability to appear a band on gel so, they will have absorbance in PCR-ELISA test ().
Figure 2. Evaluation of specificity. Nine common bacteria in UTI including 2 E. coli with PTCC number were subjected to the PCR. Each number is related to each of the bacteria specimen. (1) Klebsiella pneumonia. ATCC 70603. (2) Staphylococcus haemolyticus PTCC 1437. (3) Streptococcus pyogenes PTCC 1447. (4) Enterococcus faecalis ATCC 29212. (5) Pseudomonas aeruginosa ATCC 27863. (6) Proteus mirabilis ATCC 25933. (7) Serratia marcescens ATCC 13880. (8) E. coli PTCC1270. (9) E. coli PTCC 127.
Discussion
A number of other studies have been carried out to detect pathogensCitation1,Citation2,Citation5,Citation12. Detection of E. coli is important because, in most of UTIs cases, E. coli causes the diseaseCitation4. In all the traditional and new methods exist for detection of this bacterium, molecular-based methods are become widespread due to the sensitivity and specificity. Among the molecular methods, the ones which are based on hybridisation of probes, have been using more and moreCitation13–16. Also using probe obtain reasonable specificity. Safe materials like fluorescent (real time PCR) and enzymes (PCR-ELISA) can be used instead of ethidium bromide in gelCitation17–21. Methods like growing on culture and using microscope for detection are so long and takes at least 24 h. Besides, visual detection on microscope is not reliable. PCR-ELISA and real time also need expensive equipment and experts.
In current study, we introduced a novel “DNA-ELISA” method, bearing the ability to detect specific bacteria in about 4 h. The method offers simplicity, ease of use, minimal hand on time and does not need any special equipment and experts. Moreover, DNA-ELISA does not require any amplification and can directly detect genomic DNA of bacteria, giving a lower limit to the sensitivity of 40,000 bacteria. Although the detection limit for DNA-ELISA is much more than PCR-ELISA, it reduces the longevity of the detection by elimination the amplification step significantly. At present there is an ongoing effort to improve the sensitivity of detection limit.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Acknowledgements
The authors wish to thank Tarbiat Modares University for supporting the conduct of this research and also thank Karma Company which partially provide the budget of the project. There are no sources in the current document.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Ivnitski D, Abdel-Hamid I, Atanasov P, Wilkins E. Biosensors for detection of pathogenic bacteria. Biosens Bioelectron 1999;14:599–624.
- Leonard P, Hearty S, Brennan J, et al. Advances in biosensors for detection of pathogens in food and water. Enzyme Microb Technol 2003;32:3–13.
- Chen S, Cheng YF. Biosensors for bacterial detection. Int J Biosens Bioelectron 2017;2:00048.
- Liao JC, Mastali M, Gau V, et al. Use of electrochemical DNA biosensors for rapid molecular identification of uropathogens in clinical urine specimens. J Clin Microbiol 2006;44:561–70.
- Lazcka O, Del Campo FJ, Munoz FX. Pathogen detection: a perspective of traditional methods and biosensors. Biosens Bioelectron 2007;22:1205–17.
- Jothikumar N, Griffiths MW. Rapid detection of Escherichia coli O157:H7 with multiplex real-time PCR assays. Appl Environ Microbiol 2002;68:3169–71.
- Lee S, Yoo S. Optical biosensors for the detection of pathogenic microorganisms. Trends Biotechnol 2016;34:7–25.
- Rola M, Kuzmak J. The detection of bovine leukemia virus proviral DNA by PCR-ELISA. J Virol Methods 2002;99:33–40.
- Bakthavathsalam P, Rajendran VK, Mohammed JAB. A direct detection of Escherichia coli genomic DNA using gold nanoprobes. J Nanobiotechnol 2012;10:1–10.
- Shingo U, Hirotoshi E, Po-Sung C, et al. Detection of bacterial DNA by in situ hybridization in patients with decompensated liver cirrhosis. BMC Gastroenterol 2017;17:106.
- Salazar N, Caetano-Anollés G. Nucleic acid scanning-by-hybridization of enterohemorrhagic Escherichia coli isolates using oligodeoxynucleotide arrays. Nucl Acids Res 1996;24:5056–7.
- Nayak M, Kotian A, Marathe S, Chakravortty D. Detection of microorganisms using biosensors—a smarter way towards detection techniques. Biosens Bioelectron 2009;25:661–7.
- Lamture JB, LBeattie K, Burke BE, et al. Direct detection of nucleic acid hybridization on the surface of a charge coupled device. Nucl Acids Res 1994;22:2121–5.
- Rohde A, Hammerl JA, Appel B, et al. FISHing for bacteria in food-A promising tool for the reliable detection of pathogenic bacteria?. Food Microbiol 2015;46:395–407.
- Rohde A, Hammerl JA, Appel B, et al. Differential detection of pathogenic Yersinia spp. by fluorescence in situ hybridization. Food Microbiol 2017;62:39–45.
- Fortin NY, Mulchandani A, Chen W. Use of real-time polymerase chain reaction and molecular beacons for the detection of Escherichia coli O157:H7. Anal Biochem 2001;289:281–8.
- Kawasaki S, Fratamico PM, Kamisaki-Horikoshi N, et al. Development of the multiplex PCR detection kit for Salmonella spp., listeria monocytogenes, and Escherichia coli O157: H7. JARQ 2011;45:77–81.
- Laitinen R, Malinen E, Palva A. PCR-ELISA: I: application to simultaneous analysis of mixed bacterial samples composed of intestinal species. Syst Appl Microbiol 2002;25:241–8.
- Malinen E, Mättö J, Salmitie M, et al. PCR-ELISA: II: analysis of Bifidobacterium populations in human faecal samples from a consumption trial with Bifidobacterium lactis Bb-12 and a galacto-oligosaccharide preparation. Syst Appl Microbiol 2002;25:249–58.
- de Boer E, Beumer RR. Methodology for detection and typing of foodborne microorganisms. Int J Food Microbiol 1999;50:119–30.
- Singh G. Determination of cutoff score for a diagnostic test. Internet J Lab Med 2007;2:1–4.