
Abstract
In this research, we exploited derivatives of thieno[2,3-b]pyridine as dual inhibitors of the key enzymes in eicosanoid biosynthesis, cyclooxygenase (COX, subtypes 1 and 2) and 5-lipoxygensase (5-LOX). Testing these compounds in a rat paw oedema model revealed potency higher than ibuprofen. The most active compounds 7a, 7b, 8b, and 8c were screened against COX-1/2 and 5-LOX enzymes. Compound 7a was the most powerful inhibitor of 5-LOX with IC50 = 0.15 µM, while its p-chloro analogue 7b was more active against COX-2 (IC50 = 7.5 µM). The less desirable target COX-1 was inhibited more potently by 8c with IC50 = 7.7 µM. Surflex docking programme predicted that the more stable anti- conformer of compound (7a) formed a favourable complex with the active site of 5-LOX but not COX-1. This is in contrast to the binding mode of 8c, which resembles the syn-conformer of series 7 and binds favourably to COX-1.
Introduction
Eicosanoids are arachidonic acid (AA) metabolites that comprise prostaglandins (PGs) and leukotrienes (LTs), the principal inflammatory mediators in osteoarthritis (OA)Citation1,Citation2. PGs and LTs play key roles in triggering the failure of innate homeostasis of chondrocytes that contribute to pain, swelling, joint damage, and other symptoms of OACitation3,Citation4. PGs’ biosynthesis starts when cyclooxygenases (COXs) catalyse two reactions, first the bis-peroxidation of AA to PGG2 and second, the reduction of the acyclic peroxide group in PGG2 to form the key precursor PGH2. PGH2 metabolises, according to elicited biological needs, to responsive PGs under catalysis of various prostaglandin synthasesCitation5.
The “mostly” inducible subtype cyclooxygenase-2 (COX-2) is greatly upregulated during the inflammatory processes while the constitutive subtype COX-1 is “mostly” assigned to the regulation of PGs in healthy individualsCitation3. In 1990s, the approach of selective inhibition of COX-2 by coxibs was sought as the imminent strategy to control OA, taking the advantage of less side effects than the classic non-steroidal anti-inflammatory drugs (NSAIDs, )Citation6. NSAIDs (non-selective inhibitors of COX 1&2), though widely used as analgesics and anti-inflammatory drugs to date, are frequently implicated in a variety of adverse actions. Patients under NSAIDs frequently report GI inflammation, bleeding, ulcers, in addition to hepatic problems, renal toxicities, and othersCitation2. However, only few years after the first coxib approval, rofecoxib, and valdecoxib () were withdrawn from world markets subsequent to findings of links to increased cardiovascular events such as atherosclerosis and infarctionCitation7,Citation8. Other coxibs and some NSAIDs had to add warnings to their labels and limitation of uses in certain patients with risks of cardiovascular complicationsCitation9. Before withdrawals, some valuable studies placed selective COX-2 inhibitors under scrutiny as evidences accumulated about COX-1 and COX-2 overlapping functions. COX-1’s role was proved not to be limited to “housekeeping” as it was widely claimed in literatureCitation10. COX-1 was found to contribute considerably to some inflammatory responsesCitation11 leading to decreased efficacy of some COX-2 selective inhibitors unless they exert a minimum of COX-1 inhibition at the therapeutic doseCitation12. Moreover, COX-2 is constitutively expressed in certain tissues to catalyse the biosynthesis of some beneficial PGs such as PGI2, a critically needed prostaglandin for homeostasis of the cardiovascular systemCitation13. Therefore, the clinical use of COX1/2 inhibitors will remain associated with some undesirable effects regardless being a selective coxib or a non-selective NSAIDCitation14. Moreover, shutting down the COX1/2 pathway causes a shunting of AA to the other proinflammatory route, the lipoxygenase (LOX) pathway. This possibly results in decreased efficacyCitation15 as well as aggravation of GI side effect of NSAIDsCitation16. Encoding gene of 5-lipooxygensase (5-LOX), the key enzyme for biosynthesis of the inflammatory mediators LTs, is almost undetectable in healthy individualCitation17 but activated during various inflammatory eventsCitation18. Inhibitors of 5-LOX (e.g. Zileuton, ) are compassionately prescribed for controlling some inflammatory disorders such as bronchial asthmaCitation19, but it is much less efficacious in OACitation20 regardless the proven involvement of 5-LOX in arthritic progressionCitation21,Citation22.
Figure 1. Structures of reported inhibitors of arachidonic acid biosynthesis.
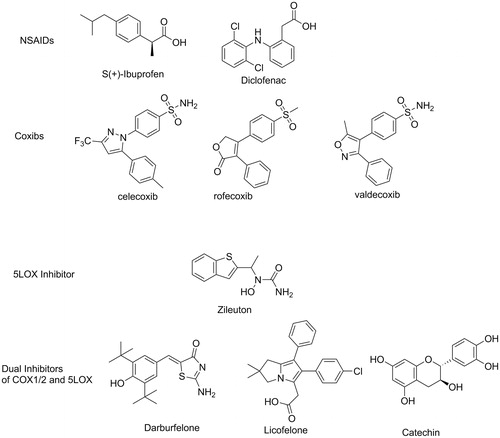
Based on undisputable findings, dual inhibitors for both COX1/2 and 5-LOX pathways should provide superior efficacies in controlling inflammation and pain in patients suffering from OA with less adverse effectsCitation23. For instance, dual COX1/2–5LOX inhibitors demonstrated excellent analgesic and anti-inflammatory activities with lower gastric irritation, bleeding and ulcerogenic side effects. In addition, dual inhibition of COX1/2 and 5-LOX provided cardioprotective activity in a noteworthy contrast to COX-2 inhibitorsCitation24. Efforts for developing clinically useful dual COX1/2–5LOX inhibitors are still active area in drug discovery, since no approval has been granted yet for the first drug in this classCitation2. Licofelone, the first clinically studied dual inhibitor has completed phase III clinical trials but it is not clear why it is not marketed yetCitation2. The potential benefits of dual COX1/2–5LOX inhibitors extend beyond OA. For instance, they demonstrated anticonvulsantCitation25 as well as anticancer activitiesCitation26,Citation27.
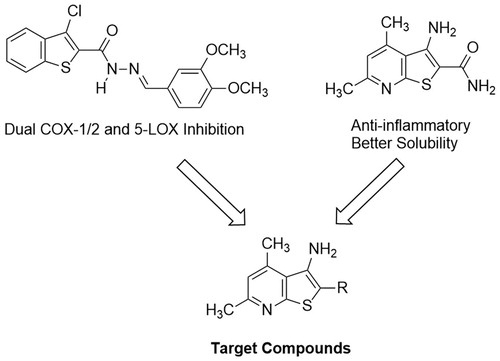
In our quest to finding novel anti-inflammatory agents, we relied on a screening model which primarily started with measuring the in vivo anti-inflammatory activitiesCitation28–30. On the other hand, we sought 3-aminothieno[2,3-b]pyridine as promising molecular entities to develop novel anti-inflammatory agents with possible dual inhibitory mechanism against COX1/2 and 5-LOX. This hetero-bicyclic scaffold has been studied for its versatile activities against inflammatory disorders, albeit for other subcellular targetsCitation31–33. Thienopyridine derivatives have also been reported for their potent anti-proliferative activitiesCitation34,Citation35. Moreover, our target thieno[2,3-b]pyridine is a close congener to benzothiophene, a nucleus recently tested for inhibition of COX1/2 and 5-LOXCitation36 ().
Figure 2. Approach to develop target compounds with dual COX1/2 and 5-LOX inhibition from previously reported compounds.
Experimental
Chemistry
General methods
All melting points were uncorrected and measured using Electro-thermal IA 9100 apparatus (Shimadzu, Japan); IR spectra were recorded as potassium bromide pellets on a Perkin-Elmer 1650 spectrophotometer (USA), Faculty of Science, Cairo University, Cairo, Egypt. 1H-NMR spectra were determined on a Varian Mercury (500 MHz) spectrometer (Varian UK) and chemical shifts were expressed as ppm against TMS as an internal reference (Faculty of Science, CairoUniversity, Cairo, Egypt). Mass spectra were recorded on 70 eV (EI Ms-QP 1000 EX, Shimadzu, Japan), Faculty of Science, CairoUniversity, Cairo, Egypt. Microanalyses were operated using Vario, Elmentar apparatus (Shimadzu, Japan), Organic Microanalysis Unit, Faculty of Science, Cairo University, Cairo, Egypt. Column Chromatography was performed on (Merck) Silica gel 60 (particle size 0.06–0.20 mm).
Synthesis
1,2-Dihydro-4,6-dimethyl-2-thioxopyridine-3-carbonitrile (1)
A mixture of 4,6-dimethyl-3-cyano-2-chloropyridine (1.66 g, 0.01 mol) and thiourea (1.52 g, 0.02 mol) was refluxed in dry ethanol (20 ml) for 6 hCitation37. Then evaporated under reduced pressure and the residue was recrystallised from methanol to give 1. Yield 68%, m.p. 183 °C (Lit. m.p. 180 °C24).
General procedure for preparation of compounds (2a–c)
A mixture of the pyridinethione 1 (1.64 g, 0.01 mol), chloroacetic acid (2.8 g, 0.03 mol), appropriate aldehyde (0.05 mol), and fused sodium acetate (2.0 g) in acetic acid/acetic anhydride (30 ml, 1:1) was heated under reflux for 8 h and left to cool. The solid formed was filtered off and washed with water and recrystallised from ethanol/H2O to afford 2a–c.
(±) 3-Benzylidene-3, 8a-dihydro-5,7-dimethyl-2-oxo-2H-thiazolo[3,2-a]pyridine-8-carbonitrile (2a)
Yield: 77%; m.p.: 105–107 °C; IR (KBr) υ (cm−1): 1725 (C=O), 2256 (C≡N). MS (EI) m/z: 294 [M+] (85%). 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.41 (s, 3H, CH3), 2.75 (s, 3H, CH3), 5.4 (s, 1H, CH), 6.5 (s, 1H, pyr-H), 6.9 (s, H, =CH), 7.1–7.6 (m, 5H, Ar-H).; Anal. Calcd. for C17H14N2OS (294.08): C, 69.36; H, 4.79; N, 9.52%. Found: C, 69.67; H, 4.42; N, 9.79%.
(±) 3-Furan-2-ylmethylene-3,8a-dihydro-5,7-dimethyl-2-oxo-2H-thiazolo[3,2-a]pyridine-8-carbonitrile (2b)
Yield: 69%; m.p.: 100–102 °C; IR (KBr) υ (cm−1): 1730 (C=O), 2220 (C≡N). MS (EI) m/z: 284 [M+] (85%). 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.12 (s, 3H, CH3), 2.3 (s, 3H, CH3), 5.5 (s, 1H, CH), 6.4 (s, 1H, pyr-H), 7.0 (s, H, =CH), 7.1–7.4 (m, 3H, Ar-H).; Anal. Calcd. for C15H12N2O2S (284.06): C, 63.36; H, 4.25; N, 9.85%. Found: C, 63.14; H, 4.59; N, 9.61%.
(±)3-(3-Phenyl-allylidene)-3,8a-dihydro-5,7-dimethyl-2-oxo-2H-thiazolo[3,2-a]pyridine-8-carbonitrile (2c)
Yield: 76%; m.p.: 80–82 °C; IR (KBr) υ (cm−1): 1580 (C = C), 1750 (C=O), 2260 (C≡N). MS (EI) m/z: 320 [M+] (85%). 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.2 (s, 3H, CH3), 2.4 (s, 3H, CH3), 5.4 (s, 1H, CH), 6.2 (s, 1H, pyr-H), 6.5 (s, H, =CH), 6.7 (s, H, =CH), 7.1 (s, H, =CH), 7.3–7.6 (m, 5H, Ar-H). Anal. Calcd. for C19H16N2OS (320.1): C, 71.22; H, 5.03; N, 8.74%. Found: C, 71.53; H, 5.28; N, 8.45%.
General procedure for preparation of compounds 3a–b
To an ethanolic sodium hydroxide solution [sodium hydroxide (0.01 mol) in ethanol (20 ml)] pyridinethione 1 (1.64 g, 0.01 mol) was added, with stirring for 30 min, and then the appropriate alkyl halide (0.02 mol) was added drop-wise with vigorous shaking. The reaction mixture was stirred for 2 h at r.t., poured onto cold water (100 ml) and neutralised with dilute HCl. The solid precipitate was filtered off and washed with water and recrystallised from methanol to afford 3a–b. The reported compound 3b was prepared in 70% yield and m.p. 93 °C (Lit. m.p. 90 °C)Citation37.
S-Methyl-4, 6-dimethyl-2-thiopyridine-3-carbonitrile (3a)
Yield: 72%; m.p.: 80–82 °C; IR (KBr) υ (cm−1): 2218 (C≡N). MS (EI) m/z: 178 [M+] (91%). 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.59 (s, 3H, CH3), 2.65 (s, 3H, CH3), 3.96 (s, 3H, S-CH3), 6.9 (s, 1H, pyr-H). Anal. Calcd. for C9H10N2S (178.25): C, 60.64; H, 5.65; N, 15.72%. Found: C, 60.41; H, 5.84; N, 15.96%.
General procedure for preparation of compounds 4a–c
An excess of aqueous 37% fomaldehyde (5–6 ml) was added to a suspension of pyridinethione 1 (1.64 g, 0.01 mol) in EtOH (15 ml). The mixture was heated with stirring to complete dissolution and then the appropriate primary amine (0.01 mol) was added in one portion. The resulting mixture was refluxed with vigorous stirring for 3 min and then stirred at 20 °C for 4 h. The precipitate formed was filtered off and crystallised from ethanol to afford 4a–c.
(±)3-Aryl-2,3,4,9a-tetrahydro-6,8-dimethylpyrido[2,1-b][1,3,5]thiadiazine-9-carbonitrile (4a)
Yield: 53%; m.p.: 180–182 °C; IR (KBr) υ (cm−1): 2230 (C≡N). MS (EI) m/z: 283 [M+] (69%). 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.30 (s, 3H, CH3), 2.58 (s, 3H, CH3), 4.5 (s, 2H, S-CH2), 4.9 (s, 2H, N-CH2), 5.2 (s, 1H, CH), 6.8 (s, 1H, pyr-H), 6.9–7.5 (m, 5H, Ar-H). Anal. Calcd. for C16H17N3S (283): C, 67.81; H, 6.05; N, 14.83%. Found: C, 67.65; H, 6.31; N, 14.62%.
(±)3-Aryl-2,3,4,9a-tetrahydro-6,8-dimethylpyrido[2,1-b][1,3,5]thiadiazine-9-carbonitrile (4b)
Yield: 68%; m.p.: 115–117 °C; IR (KBr) υ (cm−1): 2235 (C≡N). MS (EI) m/z: 297 [M+] (79%). 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.1 (s, 3H, CH3), 2.3 (s, 3H, CH3), 2.8 (s, 3H, CH3), 4.6 (s, 2H, S-CH2), 4.8 (s, 2H, N-CH2), 5.1 (s, 1H, CH), 6.2 (s, 1H, pyr-H), 7.0–7.5 (m, 4H, Ar-H). Anal. Calcd. for C17H19N3S (297): C, 68.65; H, 6.44; N, 14.13%. Found: C, 68.31; H, 6.25; N, 14.39%.
(±)3-Aryl-2,3,4,9a-tetrahydro-6,8-dimethylpyrido[2,1-b][1,3,5]thiadiazine-9-carbonitrile (4c)
Yield: 59%; m.p.: 125–127 °C; IR (KBr) υ (cm−1): 2210 (C≡N). MS (EI) m/z: 297 [M+] (72%). 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.30 (s, 3H, CH3), 2.58 (s, 3H, CH3), 4.5 (s, 2H, S-CH2), 4.7 (s, 2H, CH2), 5.2 (s, 2H, N-CH2), 5.9 (s, 1H, CH), 6.8 (s, 1H, pyr-H), 7.2–7.6 (m, 5H, Ar-H). Anal. Calcd. for C17H19N3S (297): C, 68.65; H, 6.44; N, 14.13%. Found: C, 68.88; H, 6.71; N, 14.00%.
Ethyl-3-amino-4,6-dimethylthieno[2,3-b]pyridine-2-carboxylate (5)
This compound was prepared by two methods.
Method 1: A mixture of pyridinethione 1 (1.64 g, 0.01 mol) and ethyl bromoacetate (3.34 g, 0.02 mol) in dry ethanol (50 ml) containing sodium metal (1 g) was stirred for 2 h, and then was refluxed with stirring for 4 h. The reaction mixture was cooled and poured into ice water. The solid formed was filtered off, dried, and recrystallised from methanol to afford 5.
Method 2: A mixture of ethyl 2-(3-cyano-4,6-dimethylpyridin-2-ylthio)acetate 3b (2.5 g, 0.01 mol) and dry ethanol (50 ml) containing sodium ethoxide (2%) was refluxed with stirring for 4 h. The reaction mixture was cooled and poured into ice water. The solid formed was filtered off, dried, and recrystallised from ethanol to afford 5.
Yield 70%, m.p. 138 °C (Lit. m.p. 135 °C24).
3-Amino-4,6-dimethylthieno[2,3-b]pyridine-2-carbazide (6)
A mixture of thienopyridine-2-carboxylate 5 (2.5 g, 0.01 mol) and hydrazine hydrate (0.05 mol, 99%) in absolute ethanol (15 ml); was heated under reflux for 6 h. The reaction mixture was concentrated, cooled, and the precipitate formed was filtered and crystallised from ethanol to afford 6. Yield 70%, m.p. 200 °C (Lit. m.p. 198 °C)Citation37.
General procedure for preparation of compounds 7a–c
A mixture of thienopyridine-2-carbazide (2.36 g, 0.01 mol) and the appropriate aldehyde (0.02 mol) in pyridine:ethanol (15:20 ml) was heated under reflux for 2 h. The reaction mixture was cooled and poured into ice water. The solid formed was filtered off, dried, and crystallised from the acetic acid to afford 7a–c.
3-Amino-4,6-dimethyl-thieno [2,3-b] pyridine-2-carboxylic acid benzylidene-hyrazide (7a)
Yield: 65%; m.p.: 250–252 °C; IR (KBr) υ (cm−1): 3100, 3260, 3310 (NH)(NH2), 1690 (C=O), 1230 (C=N). MS (EI) m/z: 324 [M+] (76%). 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.32 (s, 3H, CH3), 2.71 (s, 3H, CH3), 7.0 (s, 1H, pyr-H), 6.9 (s, 2H, NH2, D2O exchangeable), 7.3–7.6 (dd, 5H, Ar-H), 8.2 (s, 1H, NH, D2O exchangeable), 8.5 (s, 1H, CH). Anal. Calcd. for C17H16N4OS (324): C, 62.94; H, 4.97; N, 17.27%. Found: C, 62.97; H, 4.62; N, 17.62%.
3-Amino-4,6-dimethyl-thieno [2,3-b] pyridine-2-carboxylic acid (4-chloro-benzylidene)-hyrazide (7b)
Yield: 72%; m.p.: 280–282 °C; IR (KBr) υ (cm−1): 3150, 3290, 3310 (NH)(NH2), 1685 (C=O), 1245 (C=N). MS (EI) m/z: 358 [M+] 35Cl (66%), 360 [M + 2] 37Cl (22%), 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.45 (s, 3H, CH3), 2.62 (s, 3H, CH3), 6.9 (s, 1H, pyr-H), 7.0 (s, 2H, NH2, D2O exchangeable), 7.2–7.5 (dd, 4H, Ar-H), 8.3 (s, 1H, NH, D2O exchangeable), 8.6 (s, 1H, CH). Anal. Calcd. for C17H15ClN4OS (358): C, 56.90; H, 4.21; N, 15.61%. Found: C, 56.46; H, 4.49; N, 15.93%.
3-Amino-4, 6-dimethyl-thieno [2,3-b] pyridine-2-carboxylic acid (4-methoxy-benzylidene)-hyrazide (7c)
Yield: 74%; m.p.: 270–272 °C; IR (KBr) υ (cm−1): 3180, 3240, 3320 (NH) (NH2), 1680 (C=O), 1220 (C=N), 1150 (C–O). MS (EI) m/z: 354 [M+](58%). 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.32 (s, 3H, CH3), 2.61 (s, 3H, CH3), 3.5 (s, 3H, CH3), 6.8 (s, 1H, pyr-H), 7.0 (s, 2H, NH2, D2O exchangeable), 7.2–7.6 (dd, 4H, Ar-H), 8.0 (s, 1H, NH, D2O exchangeable), 8.6 (s, 1H, CH). Anal. Calcd. for C18H18N4O2S (354): C, 61.00; H, 5.12; N, 15.81%. Found: C, 61.33; H, 5.45; N, 15.56%.
General procedure for preparation of compounds 8a–c
To an ice-cooled solution of the corresponding thienopyridine-2-carboxylic acid aryldine-hydrazide 7a–c (0.01 mol) in AcOH (20 ml) and conc. HCl (3 ml) a cold solution of sodium nitrite (0.23 g in 5 ml of water) was added drop-wise over 5 min. The solution was stirred at room temperature for 3 h at 0–5 °C; where a solid was formed, collected by filtration, dried, and crystallised from dioxane to afford 8a–c.
7,9-Dimethyl-3-[(phenylmethylene)amino]pyrido[3′,2′:4,5]thieno[3,2-d][1,2,3]triazin-4(3H)-one (8a)
Yield: 72%; m.p.: 300–302 °C; IR (KBr) υ (cm−1): 1690 (C=O), 1250 (C=N). MS (EI) m/z: 335 [M+] (45%), 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.35 (s, 3H, CH3), 2.75 (s, 3H, CH3), 6.8 (s, 1H, pyr-H), 7.3–7.6 (m, 5H, Ar-H), 8.2 (s, 1H, CH). Anal. Calcd. for C17H13N5OS (335): C, 60.88; H, 3.91; N, 20.88%. Found: C, 60.47; H, 3.64; N, 20.57%.
7,9-Dimethyl-3-[(4-chloro-phenyl-methylene)amino]pyrido[3ˈ,2ˈ:4,5] thieno [3,2-d][1,2,3]triazin-4(3H)-one (8b)
Yield: 74%; m.p.: 255–257 °C; IR (KBr) υ (cm−1): 1685 (C=O), 1270 (C=N). MS (EI) m/z: 369 [M+] 35Cl (60%), 371 [M + 2] 37Cl (20%). 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.45 (s, 3H, CH3), 2.62 (s, 3H, CH3), 6.9 (s, 1H, pyr-H), 7.2–7.5 (dd, 4H, Ar-H), 8.3 (s, 1H, CH). Anal. Calcd. for C17H12ClN5OS (369): C, 55.21; H, 3.27; N, 18.94%. Found: C, 55.49; H, 3.51; N, 18.72%.
7,9-Dimethyl-3-[(4-methoxy-phenyl-methylene)amino]pyrido[3ˈ,2ˈ:4,5]thieno [3,2-d][1,2,3]triazin-4(3H)-one (8c)
Yield: 65%; m.p.: 240–242 °C; IR (KBr) υ (cm−1): 1680 (C=O), 1240 (C=N), 1150 (C–O). MS (EI) m/z: 365 [M+] (56%), 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.32 (s, 3H, CH3), 2.61 (s, 3H, CH3), 3.5 (s, 3H, CH3), 7.0 (s, 1H, pyr-H), 7.2–7.6 (dd, 4H, Ar-H), 8.6 (s, 1H, CH). Anal. Calcd. for C18H15N5O2S (365): C, 59.16; H, 4.14; N, 19.17%. Found: C, 59.42; H, 4.41; N, 19.54%.
General procedure for preparation of compounds 9a–b
A mixture of thienopyridine-2-carbazide 6 (2.36 g, 0.01 mol) and ethyl acetoacetate (1.3 g, 0.01 mol) or acetylacetone (1 g, 0.01 mol) in glycial acetic acid (30 ml) was heated under reflux for 5 h. The excess solvent was evaporated under reduced pressure to one-third of the solution and the reaction mixture was allowed to cool. The precipitate formed was collected and crystallised from the acetic acid to afford 9a and 9b.
3-Amino-2-[carbonyl (3-methyl-4,5-dihydro-5H-pyrazol-5-on-1-yl)]-4,6-dimethylthieno [2,3-b] pyridine (9a)
Yield: 59%; m.p.: 140–142 °C; IR (KBr) υ (cm−1): 3425, 3265 (NH2), 1686 (C=O). MS (EI) m/z: 302 [M+] (71%), 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.00 (s, 3H, CH3), 2.29 (s, 3H, CH3), 2.65 (s, 3H, CH3), 2.9 (s, 2H,CH2), 5.58 (s,2H,NH2, D2O exchangeable), 6.9 (s, 1H, pyr-H).; Anal. Calcd. for C14H14N4O2S (302.35): C, 55.61; H, 4.67; N, 18.53%. Found: C, 55.39; H, 4.90; N, 18.81%.
3-Amino-2-[carbonyl (3,5-dimethyl-pyrazol-1-yl)]-4,6-dimethylthieno[2,3-b] pyridine (9b)
Yield: 67%; m.p.: 150–152 °C; IR (KBr) υ (cm−1): 3425, 3265 (NH2), 1686 (C=O). MS (EI) m/z: 300 [M+] (62%), 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.38 (s, 3H, CH3), 2.65 (s, 3H, CH3), 2.79 (s, 6H, two CH3), 5.2 (s, 2H, NH2, D2O exchangeable), 7.1 (s, 1H, pyr-H), 8.2 (s, 1H,4H-pyrazole). Anal. Calcd. for C15H16N4OS (300.38): C, 59.98; H, 5.37; N, 18.65%. Found: C, 59.68; H, 5.68; N, 18.40%.
General procedure for preparation of compounds 10a–b
A mixture of thienopyridine-2-carbazide 6 (0.01 mol) in formic acid (15 ml) or acetic anhydride (15 ml) was heated under reflux for 6 h. The excess solvent was evaporated in vacuo (to one-third of the solution) and cooled; the solid was collected by filtration, dried, and crystallised from acetic acid to afford 10a–b.
7,9-Dimethyl-3-fomylaminopyrido [3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-one (10a)
Yield: 48%; m.p.: 220–222 °C; IR (KBr) υ (cm−1): 1715, 1760 (two C=O). MS (EI) m/z: 274 [M+] (71%), 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.4 (s, 3H, CH3), 2.8 (s, 3H, CH3), 7.1 (s, 1H, pyr-H), 7.9 (s, 1H, CH pyrimidine), 8.5 (s, 1H, H-C=O), 9.5 (s, 1H, NH). Anal. Calcd. for C12H10N4O2S (274.3): C, 52.54; H, 3.67; N, 20.43%. Found: C, 52.81; H, 3.41; N, 20.69%.
3-Diacetylamino-2,7,9-trimethylpyrido [3',2':4,5]thieno[3,2-d]pyrimidin-4(3H)-one (10b)
Yield: 65%; m.p.: 133–135 °C; IR (KBr) υ (cm−1): 1720, 1745, 1780 (three C=O). MS (EI) m/z: 344 [M+] (65%), 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 1.9 (s, 3H, CH3), 2.2 (s, 3H, CH3), 2.5 (s, 3H, CH3), 2.7 (s, 6H, 2CH3), 7.2 (s, 1H, pyr-H). Anal. Calcd. for C16H16N4O3S (344.09): C, 55.80; H, 4.68; N, 16.27%. Found: C, 56.00; H, 4.91; N, 16.50%.
Biological assay
Animals
To define the minimum total sample size and the average number for each group the a priori test with the following parameters was conducted: anticipated effect size (Cohen's d): 0.7; desired statistical power level: 0.8; probability level: 0.05. The a priori test calculation returned the following values: minimum total sample size (one-tailed hypothesis)Citation38; minimum sample size per group (one-tailed hypothesis)Citation39.
According to a priori test results, experiments were conducted on young adult male Sprague–Dawley rats (5 rats per group), weighing 140–170 g. The animals were housed at cages in a temperature controlled (25 ± 1 °C) environment and were fed ad libitum with standard laboratory chow and allowed free access to water. This investigation conforms to the ethical Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised in 1996). The animal protocol is in accordance with the Animal Ethical Care regulations in the Faculty of Pharmacy, Helwan University.
Assessment of anti-inflammatory activity
Compounds (equimolar to the reference drug) were dissolved in DMSO and administrated subcutaneously. One hour later, paw oedema was induced by subplantar injection of 0.1 ml of 1% carrageenan (Sigma-Aldrich, St. Louis, MO) into the right hind paw. Paw volume was measured using a water plethysmometer (Basile, Comerio, Italy). The difference between the right and left paw volume was measured at 1, 2, 3, and 4 h after induction of inflammation.
The control group (five rats per group) received DMSO subcutaneously and carrageenan in the subplantar region. Results were expressed as percentage inhibition of inflammation. Ibuprofen (70 mg kg−1) was used as the reference drugCitation40.
Biochemical assay
Drugs capacities to inhibit COX-1, COX-2, and 5-LOX enzymes were assessed using ELISA kits; COX-1 (human) Inhibitor Screening Assay Kit (Item № 701070), COX-2 (human) Inhibitor Screening Assay Kit (Item № 701080), and 5-LOX Inhibitor Screening Assay Kit (Item № 760700) from Cayman (Ann Arbor, MI). The used protocol was according to the manufacturer protocol guide and instructions using ELISA plate reader.
Statistics
All assays results are expressed as mean ± standard error of the mean (SEM). The comparisons between the control and the treatment groups were carried out using One-way ANOVA using a statistical package (SPSS version 17.0). A p value of <0.05 was considered significant.
Molecular modelling
All molecular modelling work was performed using SYBYL-X package (www.certara.com) installed with licence to the Faculty of Pharmacy, King Abdulaziz University on Windows 7-operated computer, equipped with Samsung SyncMaster 2233RZ 120 Hz LCD Display™ (3D ready) and Nvidia Geforce 3D Vision Glasses Kit™.
Preparation of protein and ligands
Crystal structures were downloaded as .pdb files from Protein Databank Website (www.rcsb.org). The initial biopolymers were simplified by deleting all but one monomer in the quaternary structures and then prepared for docking. The previous two steps were performed using “Biopolymer” Tools.
The Ligand Structures Library was built on ChemsketchCitation41 and saved as .sdf files. The structures were converted to 3D and optimised using SYBYL-X’s Concord embedded in “Prepare Ligands” tools.
Surflex docking
Docking was performed using Surflex programme embedded in “Dock Ligand” tools. First, the target previously prepared protein was selected and underwent final preparation for docking. Surflex docking was performed after protomol generation on ligand mode.
Manual docking
Ligand preparation
The manual docking was used in case no ligand was present to guide Surflex automatic docking procedure. This protocol was used only for docking experiments of syn-7a, anti-7a, and 8c to the active site of 5-LOX crystal structure (PDB Code 3O8Y) because it contains no ligand to guide a Surflex automatic docking. In this regards, ligands prepared above we further optimised to the global minimum conformation by energy minimisation tools until the global minimum is reached (Termination: Gradient).
Docking
We used three-step, visually-guided procedure (Place-Merge-MD) as follows.
1. Place. Prepared protein structure (as described above) and the intended ligand were imported to two different SYBYL-X screen levels. The ligand was moved until it was visually entered the active site. Careful inspection of features in the ligand and residues around to ensure maximum positive interactions with the least clashes with the active site residues.
2. Merge. When convinced that the ligand is situated in the best docking position, the ligand was merged into the protein screen. To confirm that the ligand has no serious clash with the active site residues, AAs with 5 Å sphere distance around the docked ligand were unhidden. If a clash existed, another round of moving the ligand was performed and the procedure is repeated until the least possible clash is reached. After merge, the energy of the complex it is measured and compared to uncomplexed protein. The process is repeated several times with different docking modes. The best docking complexes pose according to visual inspection of clashes and energy computation were retained and compared. The poses ranked according to their energies (first priority) and positive interactions of the ligand with the active site.
3. Molecular dynamics (MD). Molecular dynamics was performed to check the stability of the complex generated by manual docking according to the following protocol with time length 1000 fs at 300 K and 0 atm pressure. Snapshots every 5 fs were taken and inspected visually. The best snapshot in energy is considered provided that a minimum movement of the protein loops are observed.
Results
Chemical synthesis
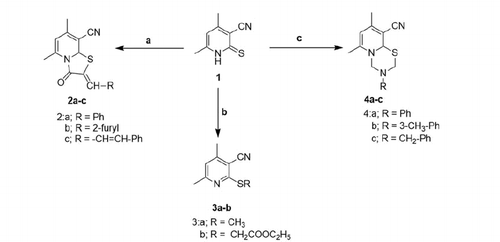
Thienopyridine synthesis has been previously reportedCitation42. Synthesis of target anti-inflammatory compounds was accomplished via the key intermediate pyridin-2-thione (compound 1), which was prepared according to the reported methodCitation37. Compound 1 was then utilised in different reactions to yield three different series of compoundsCitation2–4. Condensation of 1 with chloroacetic acid and certain aldehydes in a mixture of acetic acid and acetic anhydride gave the thiazolopyridine derivatives 2a–cCitation43. Compound 1 was also treated with different alkylating agents to afford the corresponding 2-alkylthio analogues 3a–b. Condensation of 1 with formalin or certain primary amines afforded the pyridothiadiazine derivatives 4a–c (Scheme 1).
Scheme 1. Utilisation of compound 1 to produce series 2–4 (a = ClCH2COOH, Ac2O, AcOH, NaOAc along with an aldehyde RCHO, b = RX where X = Br or I, c = RNH2 and HCHO).
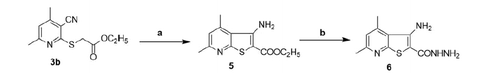
Compound 3b was subsequently converted to the thienopyridine derivatives 5 via reaction with NaOEt/EtOH. Compound 5 was treated with NH2NH2 to yield the corresponding hydrazide 6 (Scheme 2)Citation37.
Scheme 2. Conversion of 3b to thienopyridine derivatives (a = NaOEt/EtOH, b = NH2NH2).
The intermediate compound 6 was used as a diversity precursor to prepare a number of target compounds’ seriesCitation7–11. Compound 6 was reacted with certain aldehydes to give the corresponding arylidene hydrazide 7a–c which were subsequently reacted with NaNO2/HCl to afford the pyrido[3′,2′:4,5]thieno[3,2-d][1,2,3]triazin-4(3H)-one 8a–cCitation44. In addition, condensation of compound 6, with ethyl acetoacetate, acetylacetone or carbon disulphide to afford the derivatives 9a–c respectively. Finally, the reaction of 6, separately with HCOOH and acetic anhydride afforded the pyridothienodiazine analogues 10a and 10b, respectively (Scheme 3).
Scheme 3. Various reactions of compound 6 to afford the target compound series 7–10 (a = RCHO, b = NaNO2, HCl, c = ethyl acetoacetate, d = acetylacetone, e = CS2, f = HCOOH or Ac2O).
Biological screening
In vivo anti-inflammatory assay
The obtained compounds were tested for their anti-inflammatory activity using the carrageenan-induced rat paw oedema assay. Compound potency and duration of action were compared with those of the reference compound ibuprofen. The in vivo anti-inflammatory activity is shown in .
Figure 3. In vivo anti-inflammatory assay of thieno[2,3-b]pyridine derivatives. The anti-inflammatory potency is proportional to the height of the bar (inhibition %). The height of the bars (inhibition%) = (swell.drug/swell.control)100. Swell = mean difference in rat paw volume between right and left paw. RF = ibuprofen (reference drug). For more details, refer Supplementary Table S1.
![Figure 3. In vivo anti-inflammatory assay of thieno[2,3-b]pyridine derivatives. The anti-inflammatory potency is proportional to the height of the bar (inhibition %). The height of the bars (inhibition%) = (swell.drug/swell.control)100. Swell = mean difference in rat paw volume between right and left paw. RF = ibuprofen (reference drug). For more details, refer Supplementary Table S1.](/cms/asset/a7e64485-c3a5-4205-88e8-c97f9ac3dfee/ienz_a_1457657_f0003_c.jpg)
Compounds 7a, 7b, 8b, and 8c induced a satisfactory anti-inflammatory activity, exceeding that of ibuprofen at the 4 h time interval. The compounds produced 73.5, 73.2, 78.2, and 79.4% inhibition respectively, compared to 69.5% inhibition for ibuprofen. The time–activity profiles of these compounds are commensurate to that of ibuprofen, varying only in the onset of action. Compounds of series 7 showed a more rapid onset than ibuprofen, while those of series 8 showed a much slower onset (Supplementary Table S1).
In vitro inhibition assays of 5LOX, COX-2, COX-1 enzymes
The compounds belonging to series 7 and 8 were subjected to enzyme assay investigations against 5-LOX, COX-1, and COX-2. Those compounds were chosen based on the demonstrated profound anti-inflammatory activity in the above in vivo test.
Discussion
The phenotypic screening preceding isolated enzyme assays provided an opportunity to shift focus towards series of compounds that exhibited higher potential therapeutic benefit. The structure–activity relationship (SAR) of screened compounds for in vivo anti-inflammatory activities favoured thieno[2,3-b]pyridines over other scaffolds such as 2 and 4. The 5-LOX inhibition assay, however, favoured the hydrazides 7 as the most potent compound, 7a, is 35 and 87 times higher than the tricyclic derivatives 8a and 8c, respectively. A similar result was noticed in the COX-2 inhibition assay, albeit to a lower extent. In this regard, 7a inhibited COX-2 at 3.6 and 1.8 times higher than 8a and 8c, respectively.
Results, displayed in , were interesting since potency level, as well as selectivity between the two series 7 and 8, explained the potential of these compounds. The in vitro enzyme inhibition assays revealed that compounds 7a and 7b realised higher potency and selectivity towards 5-LOX and COX-2 over COX-1. They are also higher than the compounds 8b and 8c as well as ibuprofenCitation45 in their 5-LOX and COX-2 inhibition potency. Compounds 7a and 7b are also more selective than the other three compounds towards COX-2 than COX-1 with selectivity index COX-2/COX-1 = 4.6 and 4.2 for 7a and 7b, respectively. Compound 8b and 8c demonstrated approximately 2-fold more affinity towards COX-1 over COX-2.
Table 1. Enzyme inhibition assay results for COX-1, COX-2, and 5-LOX along with per cent inflammation reduction at the 4 h time interval in the rat paw oedema assay.
The lack of proportionality between in vitro and in vivo readouts can be attributed to several common factors, mainly pharmacokinetic parameters (absorption, distribution, metabolism, and excretion) that account for discriminations in drug efficaciesCitation46.
A structure-based analysis by docking selected compounds in the active site of 5-LOX, COX-2, and COX-1 was conducted to propose an understanding of the enzyme inhibition assay results. Crystal structures from the Protein Databank website (www.rcsb.org) were downloaded and employed in the study, accession codes: 3O8Y for 5-LOXCitation47, 4PH9 for COX-2Citation48 and 1EQG for COX-1Citation38. All the molecular modelling work was carried out using SYBLYL-X v.2.1 package licenced to King Abdulaziz University according to methodologies and protocols described in the “Experimental” section and detailed in Supplementary Material.
Among all crystal structures of 5-LOX, only three entries belong to H. sapiens 5-LOX (Entries: 3O8Y, 3V98, and 3V99). All of these crystal structures are mutated by replacing three lysines (K653KK635) into Glutamic acid–Aspargine–Leucine (ENL). The latter two entries had additional mutation S663D which resulted in the loss of 5-LOX activity (converted to 15-LOX-like activity)Citation49. Therefore, we settled with 3O8Y as our target for docking experiments.
The active site of 5-LOX is framed by two α-helices, the α2-helix and the arched helix, forming an elongated U-shaped hydrophobic cavity that accommodates the substrate AA, a 20-carbon polyunsaturated fatty acidCitation50. There is a catalytic iron nearby the active site but access to this iron is regulated by the side chains of Phe-177 and Tyr-181 (the FY cork), which also caps the cavity once the substrate enters the active siteCitation47. The defined conformation of this active site helped in understanding the clearly higher 5-LOX inhibitory activities of 7a and 7b (IC50 are at nanomolar level) over the cyclised analogues 8b and 8c (IC50 are at micromolar level).
To rationalise the higher activity of 7a and 7b against 5-LOX, a study of different 7a conformers was carried out. The anti-conformer is the calculated global minimum and it was found to be 3.8 kcal/mol less energy than the syn-conformer. This anti-conformer can make an intramolecular hydrogen bonding between the 3-amino and the carbonyl oxygen, more efficiently than that of the syn-conformer (Supplementary Material). It is therefore proposed that the anti-conformer of 7a attains a higher population.
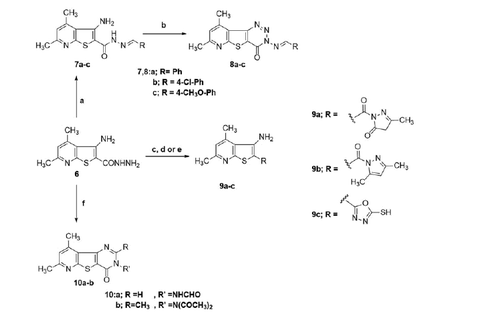
Interestingly, the pyrido[3',2':4,5]thieno[3,2-d][1,2,3]triazin-4(3H)-one derivativesCitation8 attain a global minimum conformation that well overlaps with the syn-conformation of 7a. For docking experiments into 5-LOX crystal structure, we used visually guided manual docking followed by molecular dynamics (MD), since the active site contained no ligand to guide the automatic docking process by SurflexCitation51. All three conformers (anti-7a, syn-7a and 8c) could rest inside the active site of the human 5-LOX in a hydrophobic tunnel between the α-2 helix and the arched helix considering the least steric clashes with surrounding residues (). Moreover, according to the proposed model, our compounds established several hydrophobic contacts within convenient van der Waals distances with hydrophobic residues lining the active site. The anti-conformer of the most active compound 7a made attractions with side chains of Leu-373, Leu-368, Ile-406, Leu-414, and Leu-607. In addition, anti-7a is theorised to form a crucial hydrogen bonding interaction through the hydrazide NH to Asn-407, an important residue of the arched helix that contributes to the shape of the active site. Surprisingly, the pyridine nitrogen of within the thieno[2,3-b]pyridine nucleus formed what is possibly a very important ionic interaction with Glu-376 side chain carboxylate with all three docked structures (anti-, syn-7a as well as 8a).
Figure 4. (A) Compound 7a (coloured green) docked into the active site of 5-LOX. The α2 helix is seen to the left and the arched helix is located to the right (both coloured magenta). The hydrophobic residues interacting with the ligand Leu-368, Leu-373 (of α2 helix), Ile-406, and Leu-414 (of arched helix) are shown. The FY cork residues Phe-177 and Tyr-181 are seen to the top of the image. The graphics of this image was generated using Pymol free software (https://pymol.org/2/). (B) 2D representation of the interactions of 7a with the active site of 5-LOX generated using Maestro visualiser.
Compound 8b was approximately 90 times less potent than 7a in inhibiting 5-LOX in enzyme inhibition assays. This can be explained using the structure-based analysis of an affinity model with 5-LOX active site. As mentioned above, both 8b and 8c resemble the syn-conformation of 7a. These entities were generally able to maintain the major polar and non-polar interactions with the active site residues. However, 8b approached Ile-415 and Leu-368 side chains inconveniently, thus making possible clashes with the active site in this docking mode (Supplementary Material). To be accommodated in the active site, significant shifts in the primary backbone protein loops have to be made after the MD step. Therefore, it should be challenging to predict the actual binding mode of this compound in this less flexible closed active site.
According to this model, it is noted that the 4,6-dimethylthieno[2,3-b]pyridine scaffold has a good physicochemical balance in the molecular properties as it rests in a dominantly lipophilic pocket. The two methyl groups bolstered the aromatic ring hydrophobic attractions with Leu-368 and Leu-373 side chains (residues of α2 helix, ) while the pyridine nucleus conferred a much needed polar character to our compounds.
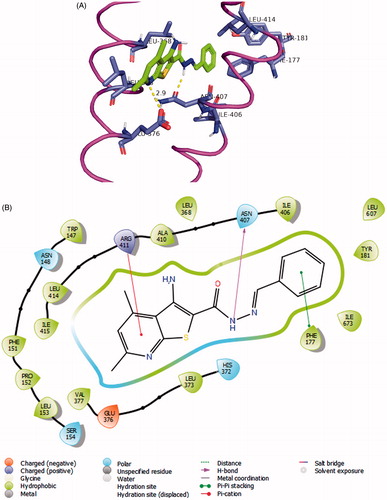
Lead compounds’ affinity towards COX-2 were investigated in a virtual screening model using Surflex docking application embedded in SYBYL-X package and the Surflex docking scores were comparedCitation51. For this purpose, we initially chose a crystal structure of COX-2 containing ibuprofen (Code: 4PH9), the reference drug for both in vivo and in vitro screenings. Our analysis suggests possible binding modes of the tricyclic analogue 8c reasonably. The active site of COX-2 is composed of four main regions according to the crystal structures; the main hydrophobic channel, the polar mouth area (mainly side chains of Arg-120 and Tyr-355), the selectivity side pocket and another smaller hydrophobic pocket near the mouth areaCitation52. Therefore, in a previous model, we reported that a pyridylurea derivative of naproxen binds possibly by inserting the naphthalene moiety properly deep in the main channel. Meanwhile, the urea group makes several hydrogen bonding with the polar area near the active site mouthCitation28. In our present work, compound 8c was predicted to insert the bulky thieno[2,3-b]pyridine moiety into the hydrophobic channel and make hydrophobic contacts with side chains of Val-344, Tyr-348, Val-349, Phe-385, Trp-387, Leu-531, and Leu-534 (). The triazine ring and imine group form a network of hydrogen bonding with the guanidine side chains of Arg-120 and the phenol group of Tyr-355 (similar to ibuprofen’s carboxylate interactions). The terminal aryl group of Schiff base was directed to the outside of the protein surface possibly because this tricyclic ligand does not enjoy much flexibility.
Figure 5. Docking of 7a and 8b to COX-2 crystal structures. (A) 2D representation of the binding modes of compounds 7a (left) and compound 8c (right) with the active site of COX-2 generated using Maestro visualiser. (B) Model of docking of 7a (violet) and 8c (Orange) on COX-2 crystal structure (Code: 3OLU). The active site pocket is coloured green as determined by Surflex’s protomol except the side pocket, which was given an orange surface. Surface of the active site’s main hydrophobic channel residues are coloured coffee-brown. Ser-530 is given a blue surface.
A limitation of this model is that it was not successful in describing the good binding of 7a, from our perspective. The top-scoring pose () of this bicyclic compound, being more flexible than 8c, was placed by Surflex as an syn-conformer while directing its thieno[2,3-b]pyridine moiety towards the hydrophobic channel. It also forced the terminal phenyl group of the Schiff base to an eclipsed torsion that penetrated the characteristic side pocket of COX-2. We had some concerns about this suggested model of 7a because it was predicted to be half the potency of 8c according to Surflex scores (). In addition, the docking conformation, though filled the side pocket, did not make any significant polar interactions (cf. 8c) with the active site polar region. This limitation highlights that this model is merely a speculative trial to explain the enzyme inhibition activities of the reported compounds.
Table 2. Surflex total scores of docked compounds into COX-2 (Code 4PH9 and 3OLU) and COX-1 (1EQG).
We suspected that the protomol formation did not help the model prediction efficiency because the process is known to be affected by the co-crystallised ligand’s size and shapeCitation51. In addition, the main channel of the active site of this crystal structure is seemingly shrunk tightly around the small isobutyl tail of ibuprofen, the co-crystallised ligand. Therefore, we decided to perform the modelling work on another COX-2 crystal structure (PDB Code: 3OLU) that contained arachidonic acid glyceryl ester as a co-crystallised ligand of larger sizeCitation53. The larger ligand gave rise to a significantly longer but narrower protomol in the main channel pocket (Supplementary Material). The docking experiments of the four compounds to the active site returned different predictions of binding modes (). The Schiff base group was inserted inside the main hydrophobic channel (reverse mode to the previous model on 4PH9). Both ligands (7a and 8c) made hydrogen bonding with Ser-530 side chain. It is worthy to mention that Ser-530 is a residue involved in the catalytic peroxidation of AA substrate, forming a hydrogen bond with the carboxylic group of diclofenac, and being acetylated by acetylsalicylic acid as proven by structural biology reportsCitation54. Compound 7a in this model existed in the previously predicted more probable anti-conformation (∼17 kcal/mol lower than that of the first docking model). The bulkier thieno[2,3-b]pyridine moiety was oriented towards the outside of the active site with shallow penetration of the selectivity pocket. Based on these assumptions, the model also helped explain the slight selectivity of 7a to COX-2 over COX-1 (Will be discussed later in further details). In our analysis, this model had improved predictions but it is not free from concerns. The much lipophilic dimethyl pyridyl ends are projecting outside the active site to the aqueous medium. In addition, the impact of the substituents on the Schiff base phenyl groups on the experimental activities are not supported by this model. Finally, the calculated scores in this model still rank 8b and 8c higher than 7a and 7b but the differences in the total Surflex scores are not as drastic as the first model (for the 4PH9 structure) (). Since the experimental differences between the ligands are not drastic either, the model might be considered qualitatively aligned with the experimental results to provide a possible explanation of the relative compounds’ activities. We expect that the scoring, as well as the binding modes, may vary if we use different crystal structure entries among the many available in the PDB website.
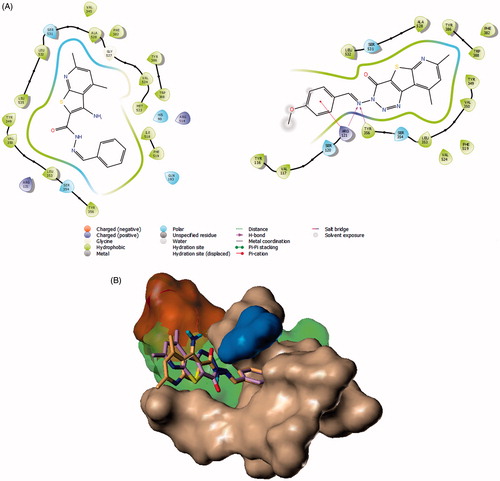
Figure 6. (A) 8c (violet) and 7a (magenta) in COX-1 active site as determined by Surflex Graphics of this image was generated using Pymol free software (https://pymol.org/2/). (B) 2D diagram showing the putative model of binding mode of compound 8c with COX-1 active site (PDB Code: 1EQG).
Modelling the COX-1 inhibition was performed on a crystal structure that also contained ibuprofen as a ligand (PDB Code: 1EQG)Citation38. It is established that there are several common features between COX-1 and COX-2 active sites, the main hydrophobic channel, the catalytic Ser-530 and the mouth having polar residues such as Arg-120. However, COX-1 is characterised by the absence of the side pocket and by a narrower main hydrophobic channelCitation55. In docking experiments, Surflex successfully granted 8c the highest scoring on COX-1, which aligns with experimental results ( and ). The dimethyl pyridine moiety reached the hydrophobic bottom of the main channel and was adjusted to form a hydrogen bond with Ser-530. The terminal triazine ring formed a bifurcate hydrogen bonding with Tyr-355. The rigid nature of 8c added convincing evidence for a high probability of its postulated binding mode. Meanwhile, Surflex placed the flexible Schiff base end of 7a to the bottom of the active site and twisted the imine side chain to fill this pocket. This way, the anti-conformer of the ligand was totally immersed inside the active site assuring that the dimethylpyridyl end is exposed to a hydrophobic microenvironment. No effective polar contacts were observed for the ligand (). We checked if the linear global energy minimum would give a better binding quality by a manual docking experiment but found serious steric clash with Val-518 (Supplementary Material).
In conclusion, this modelling work provided a hypothetical rationale of speculative binding modes of the title compounds and improved our understanding of the experimental results because it reasonably aligned these calculations with experimental results within a plausible margin of deviation.
IENZ_1457657_Supplementary_Material.pdf
Download PDF (705 KB)Acknowledgment
Authors thank the Faculty of Pharmacy, King Abdulaziz University for permission to use their SYBYL-X molecular modeling package in this research.
Disclosure statement
Authors declare no conflict of interest in this work with any potentially affected party by results of this study. The authors also declare no influence of any second party on designing of conducting the study.
Additional information
Funding
Related Research Data
References
- Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol 2015;15:511–23.
- Elliott JA, Smith HS. Handbook of acute pain management. Boca Raton (FL): CRC Press; 2016.
- Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 2001;294:1871–5.
- Harizi H, Corcuff J-B, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med 2008;14:461–9.
- Smith WL, Urade Y, Jakobsson P-J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem Rev 2011;111:5821–65.
- FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med 2001;345:433–42.
- Melnikova I. Future of COX2 inhibitors. Nat Rev Drug Discov 2005;4:453–4.
- Khanna D, Khanna PP, Furst DE. COX-2 controversy: where are we and where do we go from here? Inflammopharma-cology 2005;13:395–402.
- Bello AE, Holt RJ. Cardiovascular risk with non-steroidal anti-inflammatory drugs: clinical implications. Drug Saf 2014;37:897–902.
- Scifinder (Scifinder.cas.org) for “COX-1 Housekeeping” returned 161 references that used this term to date (6 Dec 2017).
- Prochazkova M, Dolezal T, Sliva J, Krsiak M. Different Patterns of Spinal cyclooxygenase-1 and cyclooxygenase-2 mRNA expression in inflammatory and postoperative pain. Basic Clin Pharmacol Toxicol 2006;99:173–7.
- Wallace JL, Chapman K, McKnight W. Limited anti-inflammatory efficacy of cyclo-oxygenase-2 inhibition in carrageenan-airpouch inflammation. Br J Pharmacol 1999;126:1200–4.
- Martinez-Gonzalez J, Badimon L. Mechanisms underlying the cardiovascular effects of COX-inhibition: benefits and risks. Curr Pharm Des 2007;13:2215–27.
- Suleyman H, Demircan B, Karagoz Y. Anti-inflammatory and side effects of cyclooxygenase inhibitors. Pharmacol Rep 2007;59:247.
- Marcouiller P, Pelletier J-P, Guévremont M, et al. Leukotriene and prostaglandin synthesis pathways in osteoarthritic synovial membranes: regulating factors for interleukin 1beta synthesis. J Rheumatol 2005;32:704–12.
- Rainsford K. Leukotrienes in the pathogenesis of NSAID-induced gastric and intestinal mucosal damage. Inflamm Res 1993;39:C24–26.
- Funk CD. Role of leukotrienes revealed by targeted disruption of the 5-lipoxygenase gene. Nature 1994;372:10.
- Luo M, Lee S, Brock T. Leukotriene synthesis by epithelial cells. Histol Histopathol 2003;18:587–95.
- Bouchette D, Pellegrini MV, Zileuton. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018.
- Weinblatt M, Kremer J, Coblyn J, et al. Zileuton, a 5-lipoxygenase inhibitor in rheumatoid arthritis. J Rheumatol 1992;19:1537–41.
- Samuelsson B, Dahlen S-E, Lindgren JA, et al. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science 1987;237:1171–6.
- Wittenberg RH, Willburger RE, Kleemeyer KS, Peskar BA. In vitro release of prostaglandins and leukotrienes from synovial tissue, cartilage, and bone in degenerative joint diseases. Arthrit Rheumatol 1993;36:1444–50.
- Sharma RN, Xavier FP, Vasu KK, et al. Synthesis of 4-benzyl-1, 3-thiazole derivatives as potential anti-inflammatory agents: an analogue-based drug design approach. J Enzyme Inhibit Med Chem 2009;24:890–7.
- Martel-Pelletier J, Lajeunesse D, Reboul P, Pelletier JP. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann Rheum Dis 2003;62:501–9.
- Deshmukh R, Goyal A, Sharma P. Licofelone: A dual COX/5-LOX inhibitor attenuates intracerebroventricular streptozotocin-induced cognitive deficit, oxidative stress and neuro-inflammatory cytokines in rats. Alzheimer's Dement 2013;9:P302.
- Sharma B, Pilania P, Singh P. Modeling of cyclooxygenase-2 and 5-lipooxygenase inhibitory activity of apoptosis-inducing agents potentially useful in prostate cancer chemotherapy: derivatives of diarylpyrazole. J Enzyme Inhibit Med Chem 2009;24:607–15.
- Kumar G, Patlolla JMR, Madka V, et al. Simultaneous targeting of 5-LOX-COX and ODC block NNK-induced lung adenoma progression to adenocarcinoma in A/J mice. Am J Cancer Res 2016;6:894.
- Ammar YA, Fayed EA, Bayoumi AH, et al. Design and synthesis of pyridine-amide based compounds appended naproxen moiety as anti-microbial and anti-inflammatory agents. Am J PharmTech Res 2015;5:245–73.
- Abouzid KA, Khalil NA, Ahmed EM, et al. Structure-based molecular design, synthesis, and in vivo anti-inflammatory activity of pyridazinone derivatives as nonclassic COX-2 inhibitors. Med Chem Res 2010;19:629–42.
- El-Araby M, Omar A, Hassanein HH, et al. Design, synthesis and in vivo anti-inflammatory activities of 2,4-diaryl-5-4H-imidazolone derivatives. Molecules 2012;17:12262–75.
- Zeng X-X, Zheng R-L, Zhou T, et al. Novel thienopyridine derivatives as specific anti-hepatocellular carcinoma (HCC) agents: synthesis, preliminary structure–activity relationships, and in vitro biological evaluation. Bioorg Med Chem Lett 2010;20:6282–5.
- Morwick T, Berry A, Brickwood J, et al. Evolution of the thienopyridine class of inhibitors of IkappaB kinase-beta: part I: hit-to-lead strategies. J Med Chem 2006;49:2898–908.
- Liu H, Li Y, Wang X-Y, et al. Synthesis, preliminary structure–activity relationships, and in vitro biological evaluation of 6-aryl-3-amino-thieno [2, 3-b] pyridine derivatives as potential anti-inflammatory agents. Bioorg Med Chem Lett 2013;23:2349–52.
- Zafar A, Pilkington LI, Haverkate NA, et al. Investigation into improving the aqueous solubility of the thieno[2,3-b]pyridine anti-proliferative agents. Molecules 2018;23:E145.
- Zafar A, Sari S, Leung E, et al. GPCR modulation of thieno[2,3-b]pyridine anti-proliferative agents. Molecules 2017;22:E2254.
- El-Miligy MM, Hazzaa AA, El-Messmary H, et al. New benzothiophene derivatives as dual COX-1/2 and 5-LOX inhibitors: synthesis, biological evaluation and docking study. Future Med Chem 2017;9:443–68.
- Yassin F. Synthesis, reactions and biological activity of 2-substituted 3-cyano-4, 6-dimethylpyridine derivatives. Chem Heterocyclic Comp 2009;45:35–41.
- Selinsky BS, Gupta K, Sharkey CT, Loll PJ. Structural analysis of NSAID binding by prostaglandin H2 synthase: time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry 2001;40:5172–80.
- El-Sayed M-IK, Amin HA-KA. Mechanism of endothelial cyto-protective and thrombo-resistance effects of sildenafil, vardenafil and tadalafil in male rabbit. Arch Med Sci 2015;11:190.
- Wisniewski-Rebecca ES, Rocha BA, Wiirzler LA, et al. Synergistic effects of anethole and ibuprofen in acute inflammatory response. Chem Biol Interact. 2015;242:247–53.
- ACD/Labs, ChemSketch version 12.01, Advanced Chemistry Development, Inc., Toronto, On, Canada, www.acdlabs.com, 2015.
- Pilkington LI, Haverkate NA, Van Rensburg M, et al. Synthesis of 3-Amino-2-carboxamide tetrahydropyrrolo [2, 3-b] quinolines. Synlett 2016;27:2811–4.
- Khalifa NM, Abdel-Rahman AA, El Gwaad AAA, Al-Omar MA. Synthesis and characterization of some novel substituted thiazolo [3, 2-a] pyridine and thioxopyrimido [4, 5-d] pyrimidine derivatives. Asian J Chem 2014;26:8202.
- Elneairy MA, Gad-Elkareem MA, Abdel-Fattah AM. Thieno [2, 3-b] pyridine-2-carbohydrazide in polyheterocyclic synthesis: the synthesis of pyrido [3′, 2′: 4, 5] thieno [3, 2-d] pyrimidine, pyrido [3′, 2′: 4, 5] thieno [3, 2-d][1, 2, 3] triazine, and pyrazolyl, oxadiazolylthieno [2, 3-b] pyridine derivatives. Phosphorus Sulfur Silicon 2006;181:1451–66.
- Rainsford KD. Ibuprofen: a critical bibliographic review. Boca Raton (FL): CRC Press; 2003.
- Borchardt R, Kerns E, Hageman M, et al. Optimizing the” drug-like” properties of leads in drug discovery. Berlin: Springer; 2007.
- Gilbert NC, Bartlett SG, Waight MT, et al. The structure of human 5-lipoxygenase. Science 2011;331:217–9.
- Orlando BJ, Lucido MJ, Malkowski MG. The structure of ibuprofen bound to cyclooxygenase-2. J Struct Biol 2015;189:62–6.
- Gilbert NC, Rui Z, Neau DB, et al. Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at Serine-663. FASEB J 2012;26:3222–9.
- Neau DB, Gilbert NC, Bartlett SG, et al. The 1.85 A structure of an 8R-lipoxygenase suggests a general model for lipoxygenase product specificity. Biochemistry 2009;48:7906–15.
- Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem 2003;46:499–511.
- Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996;384:644–8.
- Vecchio AJ, Malkowski MG. The structural basis of endocannabinoid oxygenation by cyclooxygenase-2. J Biol Chem 2011;286:20736–45.
- Kalgutkar AS, Crews BC, Rowlinson SW, et al. Aspirin-like molecules that covalently inactivate cyclooxygenase-2. Science 1998;280:1268–70.
- Luong C, Miller A, Barnett J, et al. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat Struct Biol 1996;3:927–33.