
Abstract
After hydrofluorination of ynesulphonamides in superacid or in the presence of hydrofluoric acid/base reagents, a series of α-fluoroenamides has been synthesised and tested for the inhibition of carbonic anhydrase (CA, EC 4.2.1.1) isoforms. This study reveals a new, highly selective family of cancer-related transmembrane human (h) CA IX/XII inhibitors. These original fluorinated ureido isosters do not inhibit the widespread cytosolic isoforms hCA I and II and selectively inhibit the transmembrane cancer-related hCA IX and XII, offering interesting new leads for future studies.
Introduction
The elevated metabolic rate of solid cancer tumors leads frequently to acidosis and hypoxiaCitation1, which can be directly related to spatial disorganisation and flow-based disruption of an abnormal microvascularisation initiated by the growing tumorCitation2. Under hypoxia stress exposition, tumor cells respond by transcription hypoxia inducible factor-1 (HIF-1α-activates), reprogramming their metabolism to overcome the reduced supply of oxygenCitation3,Citation4. The engaged nonoxygen-dependent glycolytic pathway results in increased production and export of lactic and carbonic acids to the extracellular proximal milieu, therefore decreasing extracellular pHCitation5, which induces a variation of intracellular/extracellular pH ratio (pHi/pHe ratio). This is regulated by different players including transmembrane carbonic anhydrases IX and XII (CA IX and CA XII) which are overexpressed in human cancer cellsCitation6. As a consequence, CA IX and CA XII are now recognised as especially relevant targets for cancer therapy. Sulphonamides and their bioisosteres (sulphamates, sulphamides, etc.) constitute the most investigated inhibitors of these enzymesCitation7, with useful therapeutic applicationsCitation8. They act on their deprotonated forms and bind the Zn2+ ion of the active site, disrupting the catalytic processCitation9. However, this class of inhibitors suffers from side-effects that are directly related to the undesired inhibition of the cytosolic isoform I and II, abundant in many tissues and involved in numerous physiological functionsCitation10. As a consequence, numerous efforts were dedicated over the last years to the evaluation of non-zinc binding inhibitors. This resulted for example in the discovery that coumarins, thiocoumarinesCitation11 and, more recently, sulphocoumarinsCitation12, located at the entrance of the enzyme active site, were selective inhibitors of hCA IX isozyme. Our group recently contributed to this field by exploring the activity of tertiary benzenesulphonamides derivatives: substituted N-aryl-benzenesulphamides were found to act as selective nanomolar inhibitors of hCAs IX and XIICitation13,Citation14. Despite good affinity/selectivity to hCA IX and excellent stability in plasma, a study with their 18 F-labelled analogues however showed no significant uptake in HT-29 tumors compared to normal organs/tissuesCitation15.
Considering the recent discovery of the urea derivative SLC-0111 which successfully ended Phase I clinical programmes for the treatment of patients with advanced hypoxic tumors over-expressing the isoforms hCA IX/XIICitation16,Citation17 and by the impact of ureas on pharmacokinetic properties, the evaluation of the corresponding sulphonylurea analogues must find interest. However, recent studies on the exploitation of bisarenesulphonylureas as anti-cancer agents led to unsatisfactory results in advanced clinical trialsCitation18,Citation19, due to anemia and methemioglobinemia side effects that were correlated to the in vivo oxidative cleavage of the ureas and to the generation of the corresponding aniline-derived metabolitesCitation20,Citation21. Nevertheless, sulphonylurea analogues of SLC 0111, where the sulphonyl ureido is considered as a linker, showed recently promising hCA IX and XII inhibitory propertiesCitation22. In this study, coumarinyl-substituted analogues showed even more promising profile, with nanomolar inhibition of cancer-related hCA IX and XII and low micromolar inhibition of off-targets hCA I and hCA II. Exploiting a strategy commonly used in medicinal chemistry, the use of isosters of bioactive compoundsCitation23,Citation24, we recently developed a method to design new fluoroenesulphonamides as N-sulphonylureas isostersCitation25 and demonstrated their similaritiesCitation26: these compounds, stable in solution, can be considered as good candidates to mimic unstable N-sulphonylureas. Therefore, following our seminal contribution on the use of tertiary benzene sulphonamides as selective cancer-related hCAs inhibitors, we considered that fluoroenesulphonamide group could represent an interesting novel selective chemotype and evaluated the activity of this new series against hCA I, hCA II, hCA IX and hCA XII.
Materials and methods
Chemistry
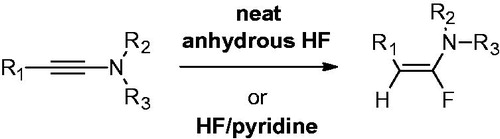
Two methods were equally used to generate the fluroenamides from their corresponding ynamides, as shown in Scheme 1.
Scheme 1. Hydrofluorination of ynamides.
General procedure A
To a solution of HF (6 ml) maintained at −50 °C or −78 °C, was added very slowly ynamide derivative (1 mmol). The mixture was magnetically stirred at the same temperature during 5 min. The reaction mixture was then neutralised with water–ice–Na2CO3, extracted with ethyl acetate (×3). The combined organic phases were dried (MgSO4) and concentrated in vacuo. Products were isolated by column chromatography on silica gel.
General procedure B
To a mixture of hydrofluoric acid and pyridine (4 ml, 70/30 w/w) maintained at the required temperature was added the starting ynamide (1 mmol). The mixture was magnetically stirred at the same temperature during the required time. The reaction mixture was then neutralised with water–ice–sodium carbonate solution, extracted with dichloromethane (3×). The combined organic phases were dried over anhydrous magnesium sulphate, filtered and concentrated in vacuo. Products were isolated by column chromatography on silica gel. The NMR spectra of the products and their detailed characterisation can be found in literatureCitation25,Citation26.
CA inhibition assay
An SX.18Mv-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the catalytic activity of various CA isozymes for CO2 hydration reactionCitation27. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 10 mM HEPES (pH 7.5) as buffer, and 0.1 M Na2SO4 (for maintaining constant ionic strength, which is not inhibitory against these CAs), following the CA-catalysed CO2 hydration reaction for a period of 10 s at 25 °C. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and activation constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial rate. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitors (10 mM) were prepared in distilled deionized water and the solution diluted to 1 nM using the assay buffer. Inhibitor and enzyme solutions were pre-incubated together for 15 min (standard assay at room temperature) prior to assay, in order to allow for the formation of the enzyme–inhibitor complex. The inhibition constant (KI), was obtained by considering the classical Michaelis–Menten equation and the Cheng-Prusoff algorithm by using non-linear least squares fitting as reported earlierCitation28–30.
Results and discussion
A series of α-fluoroenesulphonamides and imides were therefore synthesised from the corresponding ynesulphonamides and imides according to our previously reported proceduresCitation25–26. α-Fluorenesulphonamide analogue 1 was therefore tested as a carbonic anhydrase inhibitor and found to be inactive toward hCAI and II, a poor micromolar inhibitor of HCAIX but, most interestingly, a nanomolar inhibitor of HCAXII. This result was especially encouraging as it reinforces our initial hypothesis and revealed a very selective inhibitor profile for the α-fluoroenesulphonamide pharmacophore (to be compared to acetazolamide reference compound AAZ, entry 1).
Table 1. CA inhibition with acetazolamide (AAZ) as standard and compounds 1–7, against isoforms hCA I, II, IX and XII, by a stopped flow CO2 hydrase adssay [27].
To further explore substituent effect on the inhibitory activity/selectivity of this new class of hCA inhibitors, a brief structure activity relationships study was initiated. Exceptionally, all the tested fluoroenesulphonamides were found to be ineffective as offtarget hCA I and hCA II inhibitors and are selective inhibitor of the tumor associated isoforms IX and XII. Replacement of the phenyl ring on the alkene in 1 by a phenantrene or a thiophene (compounds 2 and 3) revealed a strong influence of this substituent on the efficiency and selectivity of the inhibitors. Introduction of a phenanthrene (compound 2) was indeed detrimental to the activity while the presence of a thiophene dramatically modified the inhibitory profile. 1-Thiophenyl-sustituted fluoroenesulphonamide 3 was found to be a micromolar inhibitor for hCA IX and not active for hCA XII and the presence of the heteroaromatic ring, in place of the tolyl group, shifts the inhibitor from a highly selective hCA XII inhibitor to a selective hCAIX inhibitor. These results suggest a non-zinc binding mode of action for these new chemotypes and evidence a variation of binding mode for these inhibitorsCitation8. To further verify this hypothesis, we next modified the position of the heteroatom in the thiophenyl substituent to impact eventual intra and inter molecular hydrogen bonding, analogously to what has been observed for aromatic ureas in solutionCitation31. In this case, compound 4 exhibited hCA IX nanomolar inhibition and low micromolar hCA XII inhibition. By increasing the distance between the fluoroenamide ureidoisoster moiety and the hydrophobic phenyl group, while maintaining a linear rigidity thanks to electronic conjugation between π electrons, a dual nanomolar selective inhibitor of hCAIX and hCAXII, compound 5, could be discovered. Previous report of in vivo experiments nicely demonstrated that when silencing hCA IX alone leads to a 40% reduction in xenograft tumor volume, the concomitant inhibition of both transmembrane isoforms IX and XII leads to 85% reduction of tumor growthCitation6. As a consequence, compound 5 can be considered as an interesting lead compound for further studies in this direction. To further explore the potential of fluoroenamide as a new chemotype for hCA selective inhibitors quest, fluoroenimides 6 and 7 were synthesised and tested. As for previously tested N-sulphonyl analogue 1, compound 6 was shown to be nanomolar selective inhibitor of hCAXII. This result suggests that the α-fluoroolefin core, the ureido isoster, is the essential pharmacophore for these compounds, thus confirming our initial hypothesis. Again, shifting from phenyl to alkyl chains dramatically modify the selectivity of these compounds with compound 7 being now a hCA IX selective inhibitor at the micromolar level.
On the other hand, considering the substantial interest that bacterial/fungal/protozoan CA inhibition raised ultimatelyCitation32–34, it would be of great interest to test some of these new CA inhibitors for their interaction with such enzymes belonging to other classes than the α-CAs investigated here.
Conclusions
This study reveals a new, highly selective family of cancer-related transmembrane CA inhibitors. The tested α-fluoroenamides ureidoisosters did not inhibit widespread cytosolic isoforms hCA I and II, and selectively inhibited the transmembrane cancer-related ones, hCA IX and XII. The simple modification of the C-substituent of the α-fluoroenesulphonamide and α-fluoroenimide revealed the possibility to either generate selective hCA IX, selective hCA XII or dual hCA IX and hCA XII isoform confirming the strong potential of these new pharmacophores for further studies.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- (a) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. (b) Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48. (c) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:E25.
- (a) McDonald PC, Winum JY, Supuran CT, Dedhar S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 2012;3:84–7. (b) Iessi E, Logozzi M, Mizzoni D, et al. Rethinking the combination of proton exchanger inhibitors in cancer therapy. Metabolites 2018;8:E2.
- Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases and cancer stem cells: three for the price of one. Med Res Rev 2018. doi:10.1002/med.21497
- (a) Spugnini EP, Sonveaux P, Stock C, et al. Proton channels and exchangers in cancer. Biochim Biophys Acta 2015;1848:2715–26. (b) Kusuzaki K, Matsubara T, Murata H, et al. Natural extracellular nanovesicles and photodynamic molecules: is there a future for drug delivery? J Enzyme Inhib Med Chem 2017;32:908–16. (c) Ward C, Langdon SP, Mullen P, et al. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat Rev 2013;39:171–9.
- (a) Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. J Cell Physiol 2011;226:299–308. (b) Chiche J, Ilc K, Laferrière J, et al. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor. Cancer Res 2009;69:358–68.
- (a) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun 2010;4:8371–3. (b) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II and IX with sulfonamides incorporating 1,2,4-triazine moieties. Bioorg Med Chem Lett 2004;14:5427–33.
- (a) Alterio V, Di Fiore A, D’Ambrosio K, et al. Xray crystallography of CA inhibitors and its importance in drug design. In: Supuran CT, Winum JY. eds. Drug design of zinc-enzyme inhibitors: functional, structural, and disease applications. Wiley: Hoboken (NJ); 2009:73–138. (b) Temperini C, Cecchi A, Scozzafava A, Supuran CT, Carbonic anhydrase inhibitors. Sulfonamide diuretics revisited—old leads for new applications? Org Biomol Chem 2008;6:2499–2506; (c) Di Fiore A, Maresca A, Alterio V, et al. Carbonic anhydrase inhibitors: X-ray crystallographic studies for the binding of N-substituted benzenesulfonamides to human isoform II. Chem Commun 2011;47:11636–11638.
- (a) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. (b) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. (c) Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35.
- (a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. (b) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72. (c) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (c) Casey JR, Morgan PE, Vullo D, et al. Carbonic anhydrase inhibitors. Design of selective, membrane-impermeant inhibitors targeting the human tumor-associated isozyme IX. J Med Chem 2004;47:2337–47.
- (a) Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74. (b) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- (a) Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62. (b) Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44. (c) Carta F, Maresca A, Scozzafava A, Supuran CT. Novel coumarins and 2-thioxo-coumarins as inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem 2012;20:2266–73. (d) Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73.
- (a) Tars K, Vullo D, Kazaks K, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300. (b) Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95.
- Métayer B, Mingot A, Vullo D, et al. New superacid synthesized (fluorinated) tertiary benzenesulfonamides acting as selective hCA IX inhibitors: toward a new mode of carbonic anhydrase inhibition by sulfonamides. Chem Commun 2013;49:6015–7.
- Métayer B, Mingot A, Vullo D, et al. Superacid synthesized tertiary benzenesulfonamides and benzofuzed sultams act as selective hCA IX inhibitors: toward understanding a new mode of inhibition by tertiary sulfonamides. Org Biomol Chem 2013;11:7540–9.
- Lau J, Pan J, Zhang Z, et al. Synthesis and evaluation of 18F-labeled tertiary benzenesulfonamides for imaging carbonic anhydrase IX expression in tumours with positron emission tomography. Bioorg Med Chem Lett 2014;24:3064–8.
- (a) Lomelino CL, Mahon BP, McKenna R, et al. Kinetic and X-ray crystallographic investigations on carbonic anhydrase isoforms I, II, IX and XII of a thioureido analog of SLC-0111. Bioorg Med Chem 2016;24:976–81. (b) Mastrolorenzo A, Rusconi S, Scozzafava A, et al. Inhibitors of HIV-1 protease: current state of the art 10 years after their introduction. From antiretroviral drugs to antifungal, antibacterial and antitumor agents based on aspartic protease inhibitors. Curr Med Chem 2007;14:2734–48.
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902.
- Abdel-Aziz AAM, El-Azab AS, El-Subbagh HI, et al. Design, synthesis, single-crystal and preliminary antitumor activity of novel arenesulfonylimidazolidin-2-ones. Bioorg Med Chem Lett 2012;22:2008–14.
- Howbert JJ, Grossman CS, Crowell TA, et al. Novel agents effective against solid tumors: the diarylsulfonylureas. Synthesis, activities, and analysis of quantitative structure-activity relationships. J Med Chem 1990;33:2393–407.
- Guan X, Hoffman BN, McFarland DC, et al. Glutathione and mercapturic acid conjugates of sulofenur and their activity against a human colon cancer cell line. Drug Metab Dispos 2002;30:331–5.
- (a) Fourouzesh B, Takimoto CH, Goetz A, et al. A phase I and pharmacokinetic study of ILX-295501, an oral diarylsulfonylurea, on a weekly for 3 weeks every 4-week schedule in patients with advanced solid malignancies. Clin Cancer Res 2003;9:5540–9. (b) Boyd NH, Walker K, Fried J, et al. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017;2:92928.
- Bozdag M, Ferraroni M, Carta F, et al. Structural insights on carbonic anhydrase inhibitory action, isoform selectivity, and potency of sulfonamides and coumarins incorporating arylsulfonylureido groups. J Med Chem 2014;57:9152–67.
- Lima JM, Barreiro EJ. Bioisosterism: a useful strategy for molecular modification and drug design. Curr Med Chem 2005;12:23–49.
- Angeli A, Tanini D, Peat TS, et al. Discovery of new selenoureido analogues of 4-(4-fluorophenylureido)benzenesulfonamide as carbonic anhydrase inhibitors. ACS Med Chem Lett 2017;8:963–8.
- Compain G, Jouvin K, Martin-Mingot A, et al. Stereoselective hydrofluorination of ynamides: a straightforward synthesis of novel α-fluoroenamides. Chem Commun 2012;48:5196–8.
- Métayer B, Compain G, Jouvin K, et al. Chemo- and stereoselective synthesis of fluorinated enamides from ynamides in HF/pyridine: second-generation approach to potent ureas bioisosteres. J Org Chem 2015;80:3397–410.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Menchise V, De Simone G, Alterio V, et al. Carbonic anhydrase inhibitors: stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J Med Chem 2005;4:5721–7. (b) Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors—part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents in rabbits. Eur J Med Chem 1998;33:247–54. (c) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8. (d) Şentürk M, Gülçin İ, Beydemir Ş, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9. (e) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47. (f) Dogne JM, Hanson J, Supuran C, Pratico D. Coxibs and cardiovascular side-effects: from light to shadow. Curr Pharm Des 2006;12:971–5.
- (a) Krall N, Pretto F, Decurtins W, et al. A small‐molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5. (b) Rehman SU, Chohan ZH, Gulnaz F, Supuran CT. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzyme Inhib Med Chem 2005;20:333–40. (c) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: Structure‐activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73. (d) Dubois L, Peeters S, Lieuwes NG, et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 2011;99:424–31. (e) Chohan ZH, Munawar A, Supuran CT. Transition metal ion complexes of Schiff-bases. Synthesis, characterization and antibacterial properties. Met Based Drugs 2001;8:137–43. (f) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal β-class (Cab) and γ-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–8. (g) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- (a) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8. (b) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun. 2010;46:8371–3. (c) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. (d) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11. (e) Di Fiore A, De Simone G, Alterio V, et al. The anticonvulsant sulfamide JNJ-26990990 and its S, S-dioxide analog strongly inhibit carbonic anhydrases: solution and X-ray crystallographic studies. Org Biomol Chem 2016;14:4853–8.
- Giannicchi I, Jouvelet B, Isare B, et al. Orthohalogen substituents dramatically enhance hydrogen bonding of aromatic ureas in solution. Chem Commun 2014;50:611.
- (a) Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32. (b) Supuran CT, Scozzafava A, Mastrolorenzo A. Bacterial proteases: current therapeutic use and future prospects for the development of new antibiotics. Exp Opin Ther Pat 2001;11:221–59. (c) Del Prete S, De Luca V, Vullo D, et al. A new procedure for the cloning, expression and purification of the β-carbonic anhydrase from the pathogenic yeast Malassezia globosa, an anti-dandruff drug target. J Enzyme Inhib Med Chem 2016;31:1156–61.
- (a) Supuran CT, Capasso C. New light on bacterial carbonic anhydrases phylogeny based on the analysis of signal peptide sequences. J Enzyme Inhib Med Chem 2016;31:1254–60. (b) Supuran CT, Capasso C. Carbonic anhydrase from Porphyromonas gingivalis as a drug target. Pathogens 2017;6:E30. (c) Del Prete S, De Luca V, De Simone G, et al. Cloning, expression and purification of the complete domain of the η-carbonic anhydrase from Plasmodium falciparum. J Enzyme Inhib Med Chem 2016;31:54–9.
- (a) Maresca A, Carta F, Vullo D, Supuran CT. Dithiocarbamates strongly inhibit the β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:407–11. (b) Chohan ZH, Arif M, Shafiq Z, et al. In vitro antibacterial, antifungal & cytotoxic activity of some isonicotinoylhydrazide Schiff’s bases and their cobalt (II), copper (II), nickel (II) and zinc (II) complexes. J Enzyme Inhib Med Chem 2006;21:95–103. (c) De Menezes Dda R, Calvet CM, Rodrigues GC, et al. Hydroxamic acid derivatives: a promising scaffold for rational compound optimization in Chagas disease. J Enzyme Inhib Med Chem 2016;31:964–73.