
Abstract
Dihydropyrimidinone derivatives containing piperidine moiety were synthesised in a good yield. All the compounds were confirmed by elemental analysis and spectral data. Anti-ulcer activity of novel dihydropyrimidinone-piperidine hybrids (1–18) was evaluated. Among them, four compounds (3, 8, 11 and 15) were found to be most active in 80% ethanol-induced ulcer experimental animal model. All the potent compounds were further evaluated for anti-ulcer activity by different in vivo anti-ulcer models to study the effect of compounds on anti-secretory and cytoprotective activities. All the active compounds inhibited the formation of gastric ulcers and increased the formation of gastric mucin secretion. Compound 15 was found to be the most potent compound of the series as anti-ulcer agent. Additional experimental studies on lead compound 15 will result in a new class of orally active molecule for anti-ulcer activity.
Introduction
Peptic ulcer disease (PUD) is prevalent in large population of the world. The gastric mucosal ulcer, occurring from an imbalance between the gastro protective factors (e.g. prostaglandin, mucin, bicarbonate, blood supply and nitric oxide) and the aggressive factors (e.g. pepsin and gastric acid), presents in the gastric mucosaCitation1,Citation2. The risk factors of getting PUD include Helicobacter pylori infection, frequent use of pain killer medication and stress-induced gastric mucosal lesionsCitation3. The anti-ulcer drugs act by decreasing the secretion of gastric acid and/or increasing the defence system by increasing the mucin secretion. The anti-secretory drugs include ranitidine, a histamine H2 receptor antagonist; omeprazole, irreversible proton pump inhibitor and antacids. These drugs treat PUD by reducing or neutralising the gastric acidCitation4. Drug tolerance has been reported during drug therapy of PUD by conventional drugs. Also, these drugs have serious side effects when used for a long time, which include hypergastrinemia, osteoporosis, development of carcinoids and increased risk of bacterial infection. Sucralfate is used for the treatment of gastric ulceration, but does not show good results for the ulceration caused by non-steroid anti-inflammatory drugs (NSAIDs)Citation5. NSAIDs associated ulcers can be prevented by misoprostol (analogue of prostaglandin E1), but is limited by abnormal side effectsCitation6. Therefore, there is a need for novel and potent anti-ulcer agents with improved safety profile.
Pyrimidines have played an important role in the field of medicinal chemistryCitation7. Pyrimidines are important scaffold in medicinal chemistry, because of their potential biological activities such as anti-tumour, anti-viral and anti-bacterialCitation8–10. Some of them have been used as potential anti-hypertensive agents. 4-Aryl-1,4-dihydropyridines like nifedipine was first introduced as antihypertensive in 1975. Dihydropyridines are the most effective calcium channel blockers used for various cardiovascular diseasesCitation11. Anti-ulcer activities have been reported for several calcium channel blockers including nifedipineCitation12. It is thus assumed that structural analogues of nifedipine may possess anti-ulcer potential. Dihydropyrimidines, popularly known as Biginelli’s compounds, are associated with broad spectrum of biological activitiesCitation13,Citation14. Derivatives of dihydropyrimidine have been reported to possess potent anti-ulcer and anti-secretory activityCitation15,Citation16.
Piperidine is an organic compound with the molecular formula (CH2)5NH. This heterocyclic amine consists of a six-membered ring. Piperidine is an important pharmacophore in the field of medicinal chemistry. It is reported to have various pharmacological activitiesCitation17–20. Piperidine derivatives are also reported to have anti-secretory and anti-ulcer activityCitation21,Citation22.
The literature study revealed that compounds containing these two important moieties (dihydropyrimidinone and piperidine) may have potential for the treatment of PUD. Hybrid approach, in the drug design, involves the addition two different pharmacophoric moieties to produce hybrid molecules with improved efficacy. In the present study, a series of novel dihydropyrimidinone and piperidine scaffold hybrids were synthesised, characterised by spectral data and screened for their gastric anti-ulcer activity in several in vivo ulcer models.
Experimental
Chemistry
Materials and methods
Ultraviolet light was used for the visualisation of thin layer chromatography (TLC) spots. Spectrum BX, PerkinElmer FT-IR spectrophotometer was used for performing FTIR. Gallenkamp melting point apparatus was used for performing melting points, which was uncorrected. Bruker NMR 500 MHz and 125 MHz spectrophotometer were used for 1H and 13C NMR. All the samples were processed in DMSO-d6 with tetramethylsilane as an internal standard. Molecular masses of all the compounds were measured by mass spectroscopy. CHN Elementar (Analysensysteme GmbH, Germany) was used for the elemental analysis of the compounds. The X-ray diffraction measurements were made using Bruker (2009) (Bruker AXS Inc., Madison, WI), at wavelength λ = 10,554,184 Å. Crystallographic data for compounds (III) and 13 have been deposited with Cambridge Crystallographic Data Center (CCDC) under numbers 1532826 and 1532825, respectively. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [Fax: +44-1223-336033; email: [email protected] or http://www.ccdc.cam.ac.uk].
Synthesis of 3-(dimethylamino)-1-[4-(piperidin-1-yl) phenyl]prop-2-en-1-one (III)
A mixture of 1-[4-(piperidin-1-yl) phenyl]ethan-1-one (I) (0.02 mol) and dimethylformamide-dimethylacetal (DMF-DMA) (II) (0.023 mol) was refluxed for 10 h without solvent on a heating mantle, the reaction mixture was left to cool slowly. The precipitate was obtained. Diethyl ether was added to the precipitate and filtration was performed under vacuum. The obtained product was recrystallised from absolute ethanol. Yield: 90%; m.p.: 150–152 °C; IR (KBr) cm−1: 2800 (ArC-H), 1675 (C = O), 1636 (C = O), 1618 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.5 (6H, s, 3 × –CH2, piperidine), 2.89 (3H, s, NCH3), 3.09 (4H, s, 2 × –CH2, piperidine), 3.17 (3H, s, NCH3), 5.79 (1H, d, J = 12.5 Hz, = CH), 6.91 (2H, t, J = 9.0 Hz, Ar-H), 7.65 (1H, d, J = 12.5 Hz, =CH), 7.78 (2H, d, J = 8.5 Hz, Ar-H); 13C NMR (125.76 MHz, DMSO-d6): δ = 24.4, 25.4, 48.8, 91.1, 113.9, 129.3, 129.6, 163.4, 163.5, 188.0; MS: m/z = 258.30 [M]+; analysis for C16H22N2O: C (74.38) H (5.58) N (10.84)%; found C (74.10) H (5.56) N (10.81)%.
General synthesis of 4-(substituted phenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (1–18)
A mixture of enaminone (III) (0.01 mol), differently substituted benzaldehyde (0.01 mol), urea (0.01 mol) and glacial acetic acid (10 ml), was refluxed for 3 h. The precipitates (1–18) were obtained by pouring the reaction mixture into the ice–cold water. The products were obtained by filtration under vacuum. The products were washed several times with water. The obtained products were recrystallised from glacial acetic acid.
4-Phenyl-5-[4-(piperidin-1-yl) benzoyl]-3, 4-dihydropyrimidin-2(1H)-one (1): colour: yellow; yield: 50%; m.p.: 220–222 °C; UV λmax (methanol) = 404 nm; IR (KBr) cm−1: 3273 (N–H), 2800 (ArC-H), 1675 (C = O), 1636 (C = O), 1618 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.56 (8H, s, 4 × –CH2, piperidine), 2.74 (1H, s, –CH, piperidine), 2.89 (1H, s, –CH, piperidine), 5.46 (1H, s, H-4), 6.9 (2H, d, J = 8.5 Hz, Ar-H), 7.0 (1H, s, NH, D2O exchange), 7.25–7.43 (7H, m, Ar-H), 7.78 (1H, s, =CH), 9.18 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 24.4, 25.3, 31.2, 36.2, 48.0, 48.5, 48.6, 54.1, 113.0, 113.8, 126.8, 127.2, 127.7, 128.9, 130.7, 139.3, 144.7, 152.0, 153.6, 162.7, 190.5; MS: m/z = 360.79 [M]+; analysis for C22H23N3O2: C (73.11) H (6.41) N (11.63)%; found C (73.39) H (6.43) N (11.60)%.
4-(2-Nitrophenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (2): colour: brown; yield: 60%; m.p.: 190–192 °C; UV λmax (methanol) = 426 nm; IR (KBr) cm−1: 3443 (N–H), 2852 (ArC-H), 1634 (C = O), 1595 (C = O), 1567 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.54 (8H, s, 4 × –CH2, piperidine), 3.44 (1H, s, –CH, piperidine), 3.48 (1H, s, –CH, piperidine), 6.11 (1H, s, H-4), 6.89 (2H, d, J = 9.0 Hz, Ar-H), 7.14 (1H, s, NH, D2O exchg.), 7.38–7.89 (8H, m, Ar-H), 8.10 (1H, s, =CH), 9.42 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 19.0, 24.4, 25.3, 48.5, 50.1, 56.5, 1117.7, 123.8, 124.4, 126.7, 129.1, 130.0, 130.7, 134.3, 138.8, 140.3, 148.3, 151.2, 153.6, 190.1; MS: m/z = 403.80 [M-2]+; analysis for C22H22N4O4: C (65.01) H (5.46) N (13.78)%; found C (65.26) H (5.47) N (13.73)%.
4-(4-Nitrophenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (3): colour: yellow; yield: m.p.: 180–182 °C; UV λmax (methanol) = 405 nm; IR (KBr) cm−1: 3273 (N–H), 2800 (ArC-H), 1675 (C = O), 1636 (C = O), 1618 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.55 (8H, s, 4 × –CH2, piperidine), 2.73 (1H, s, –CH, piperidine), 2.89 (1H, s, –CH, piperidine), 5.58 (1H, s, H-4), 6.89 (2H, d, J = 9.0 Hz, Ar-H), 7.09 (1H, s, NH, D2O exchange), 7.41–7.93 (8H, m, Ar-H), 8.21 (1H, s, =CH), 9.35 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 19.0, 24.4, 25.3, 48.5, 54.0, 56.5, 111.9, 113.8, 124.2, 126.9, 128.2, 130.7, 140.1, 147.1, 151.7, 151.8, 153.6, 190.2; MS: m/z = 406.00 [M]+; analysis for C22H22N4O4: C (65.01) H (5.46) N (13.78)%; found C (65.25) H (5.46) N (13.72)%.
4-(3-Nitrophenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (4): colour: yellow; yield: m.p.: 185–187 °C; UV λmax (methanol) = 404 nm; IR (KBr) cm−1: 3256 (N–H), 2800 (ArC-H), 1701 (C = O), 1685 (C = O), 1654 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.55 (8H, s, 4 × –CH2, piperidine), 2.73 (1H, s, –CH, piperidine), 2.89 (1H, s, –CH, piperidine), 5.58 (1H, s, H-4), 6.89 (2H, d, J = 9.0 Hz, Ar-H), 7.09 (1H, s, NH, D2O exchange), 7.41–7.93 (8H, m, Ar-H), 8.21 (1H, s, =CH), 9.35 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 19.0, 24.4, 25.3, 48.5, 54.0, 56.5, 111.9, 113.8, 124.2, 126.9, 128.2, 130.7, 140.1, 147.1, 151.7, 151.8, 153.6, 190.2; MS: m/z = 406.21 [M]+; analysis for C22H22N4O4: C (65.01) H (5.46) N (13.78)%; found C (65.24) H (5.45) N (13.71)%.
4-(4-Chlorophenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (5): colour: yellow; yield: 70%; m.p.: 230–232 °C; UV λmax (methanol) = 421 nm; IR (KBr) cm−1: 3261 (N–H), 2931 (ArC-H), 1654 (C = O), 1636 (C = O), 1600 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.57 (8H, s, 4 × –CH2, piperidine), 2.73 (1H, s, –CH, piperidine), 2.89 (1H, s, –CH, piperidine), 5.44 (1H, s, H-4), 6.9 (2H, d, J = 7.0 Hz, Ar-H), 7.0 (1H, s, NH, D2O exchange), 7.33–7.40 (6H, m, Ar-H), 7.81 (1H, s, =CH), 9.34 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 24.4, 25.3, 31.2, 36.2, 48.5, 53.6, 112.5, 113.8, 127.1, 128.7, 128.8, 130.7, 132.3, 139.6, 143.7, 151.9, 153.6, 190.4.; MS: m/z = 395.82 [M]+; analysis for C22H22ClN3O2: C (66.75) H (5.60) N (10.61)%; found C (66.50) H (5.61) N (10.62)%.
4-(2,4-Dichlorophenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (6): colour: yellow; yield: 75%; m.p.: 195–197 °C; UV λmax (methanol) = 406 nm; IR (KBr) cm−1: 3273 (N–H), 2800 (ArC-H), 1671 (C = O), 1630 (C = O), 1615 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.55 (8H, s, 4 × –CH2, piperidine), 3.2 (2H, s, –CH, piperidine), 5.83 (1H, s, H-4), 6.89 (2H, d, J = 8.5 Hz, Ar-H), 7.10 (1H, s, NH, D2O exchange), 7.39–7.56 (7H, m, Ar-H), 7.75 (1H, s, =CH), 9.32 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 19.0, 24.4, 25.3, 48.5, 52.4, 56.5, 111.0, 113.8, 127.6, 128.1, 129.4, 130.76, 131.36, 133.1, 133.5, 140.3, 151.2, 153.6, 190.1. MS: m/z = 430.54 [M]+; analysis for C22H21Cl2N3O2: C (61.40) H (4.92) N (9.76)%; found C (61.60) H (4.93) N (9.75)%.
4-(3,4-Dimethoxyphenyl)-5-[4-(piperidin-1-yl)benzoyl]-3,4-dihydropyrimidin-2(1H)-one (7): colour: brown; yield: 70%; m.p.: 145–147 °C; UV λmax (methanol) = 434 nm; IR (KBr) cm−1: 3478 (N–H), 2788 (ArC-H), 1634 (C = O), 1596 (C = O), 1567 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.56 (8H, s, 4 × –CH2, piperidine), 3.28 (2H, s, –CH, piperidine), 3.7 (6H, s, 2 × –OCH3), 5.42 (1H, s, H-4), 6.83–6.84 (4H, m, Ar-H), 7.0 (1H, s, NH, D2O exchange), 6.89–7.46 (8H, m, Ar-H), 7.73 (1H, s, =CH), 9.18 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 15.6, 19.0, 24.4, 25.3, 48.0, 49.0, 53.7, 55.9, 56.5, 63.3, 110.9, 112.1, 112.9, 113.9, 118.7, 127.3, 130.7, 148.5, 149.9, 152.0, 153.6, 190.6; MS: m/z = 422.18 [M + 1]+; analysis for C24H27N3O4: C (68.39) H (6.46) N (9.97)%; found C (68.57) H (6.47) N (9.99)%.
4-(2-Methoxyphenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (8): colour: yellow; yield: 50%; m.p.: 160–162 °C; UV λmax (methanol) = 429 nm; IR (KBr) cm−1: 3441 (N–H), 2931 (ArC-H), 1634 (C = O), 1595 (C = O), 1530 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.57 (8H, s, 4 × –CH2, piperidine), 2.73 (1H, s, –CH, piperidine), 2.87 (1H, s, –CH, piperidine), 3.81 (3H, s, –OCH3), 5.73 (1H, s, H-4), 6.87–7.25 (8H, m, Ar-H), 7.31 (1H, s, NH, D2O exchange), 7.45 (1H, s, =CH), 9.13 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 24.4, 25.3, 48.5, 49.6, 55.9, 112.9, 130.7, 152.2, 153.3, 190.1; MS: m/z = 391.00 [M]+; analysis C23H25N3O3: C (70.57) H (6.44) N (10.73)%; found C (70.82) H (6.43) N (10.75)%.
4-(4-Hydroxyphenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (9): colour: brown; yield: 45%; m.p.: 210–212 °C; UV λmax (methanol) = 404 nm; IR (KBr) cm−1: 3270 (N–H), 2930 (ArC-H), 1670 (C = O), 1593 (C = O), 1508 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.55 (8H, s, 4 × –CH2, piperidine), 2.73 (1H, s, –CH, piperidine), 2.88 (1H, s, –CH, piperidine), 5.37 (1H, s, H-4), 6.71–6.99 (8H, m, Ar-H), 7.14 (1H, s, NH, D2O exchange), 7.95 (1H, s, =CH), 9.20 (1H, s, –CONH, D2O exchange), 9.90 (1H, s, OH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 15.6, 24.4, 25.3, 31.2, 36.2, 48.0, 48.5, 53.6, 65.4, 113.4, 113.8, 115.5, 116.3, 127.3, 128.0, 130.7, 132.5, 135.3, 138.7, 152.1, 153.9, 157.1, 162.7, 190.6; MS: m/z = 379.61 [M + 2]+; analysis for C22H23N3O3: C (70.01) H (6.14) N (11.13)%; found C (70.25) H (6.15) N (11.11)%.
4-(3-Hydroxyphenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (10): colour: black; yield: 45%; m.p.: 190–192 °C; UV λmax (methanol) = 420 nm; IR (KBr) cm−1: 3200 (N–H), 2930 (ArC-H), 1654 (C = O), 1636 (C = O), 1600 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.55 (8H, s, 4 × –CH2, piperidine), 2.73 (1H, s, –CH, piperidine), 2.87 (1H, s, –CH, piperidine), 5.40 (1H, s, H-4), 6.7–6.9 (8H, m, Ar-H), 7.0 (1H, s, NH, D2O exchange), 7.95 (1H, s, =CH), 9.30 (1H, s, –CONH, D2O exchange), 9.70 (1H, s, OH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 15.6, 21.6, 24.4, 25.3, 25.7, 31.1, 36.2, 48.0, 48.5, 53.9, 65.4, 113.2, 113.6, 113.8, 114.7, 117.3, 127.2, 129.8, 130.7, 138.9, 146.1, 152.2, 153.6, 157.9, 162.7, 172.7, 190.5; MS: m/z = 376.94 [M]+; analysis for C22H23N3O3: C (70.01) H (6.14) N (11.13)%; found C (70.24) H (6.14) N (11.10)%.
4-(4-Dimethylamino phenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (11): colour: black; yield: 40%; m.p.: 185–187 °C; UV λmax (methanol) = 435; IR (KBr) cm−1: 3479 (N–H), 2788 (ArC-H), 1634 (C = O), 1596 (C = O), 1567 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.56 (8H, s, 4 × –CH2, piperidine), 2.81 (2H, s, –CH, piperidine), 3.0 (6H, s, -N(CH3)2, 5.30 (1H, s, H-4), 6.7–6.9 (8H, m, Ar-H), 7.0 (1H, s, NH, D2O exchange), 7.69 (1H, s, =CH), 9.67 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 15.6, 24.4, 25.3, 48.0, 48.5, 65.3, 111.5, 113.2, 130.9, 131.9, 132.8, 154.6, 190.2; MS: m/z = 405.20 [M + 1]+; analysis for C24H28N4O2: C (71.26) H (6.98) N (13.85)%; found C (71.01) H (6.96) N (13.84)%.
4-(3-Methoxyphenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (12): colour: yellow; yield: 50%; m.p.: 160–162 °C; UV λmax (methanol) = 432; IR (KBr) cm−1: 3246 (N–H), 2929 (ArC-H), 1701 (C = O), 1654 (C = O), 1600 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.57 (8H, s, 4 × –CH2, piperidine), 2.7 (1H, s, –CH, piperidine), 2.80 (1H, s, –CH, piperidine), 3.72 (3H, s, –OCH3), 5.43 (1H, s, H-4), 6.82–6.93 (6H, m, Ar-H), 7.0 (1H, s, NH, D2O exchange), 7.25–7.44 (2H, m, Ar-H), 7.78 (1H, s, =CH), 9.18 (1H, s, –CONH, D2O exchange); MS: m/z = 392.40 [M + 1]+; analysis for C23H25N3O3: C (70.57) H (6.44) N (10.73)%; found C (70.77) H (6.43) N (10.71)%.
4-(4-Ethoxyphenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (13): colour: yellow; yield: 55%; m.p.: 200–202 °C; UV λmax (methanol) = 444 nm; IR (KBr) cm−1: 3270 (N–H), 2800 (ArC-H), 1672 (C = O), 1631 (C = O), 1600 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.30 (3H, t, J = 7.0 Hz, CH3), 1.57 (8H, s, 4 × –CH2, piperidine), 2.74 (1H, s, –CH, piperidine), 2.89 (1H, s, –CH, piperidine), 3.98 (2H, q, J = 9.0 Hz, -OCH2), 5.38 (1H, s, H-4), 6.86-6.93 (4H, m, Ar-H), 6.97 (1H, s, NH, D2O exchange), 7.20 (4H, m, Ar-H), 7.69 (1H, s, =CH), 9.11 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 15.1, 24.0, 25.3, 48.0, 50.0, 65.0, 111.5, 113.2, 130.9, 131.9, 132.8, 154.6, 158.0, 162.0, 190.3; MS: m/z = 405.00 [M]+; analysis for C24H27N3O3: C (71.09) H (6.71) N (10.36)%; found C (71.34) H (6.72) N (10.34)%.
4-(2,4,5-Trimethoxyphenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (14): colour: brown; yield: 60%; m.p.:155–157 °C; UV λmax (methanol) = 449 nm; IR (KBr) cm−1: 3300 (N–H), 2800 (ArC-H), 1701 (C = O), 1686 (C = O), 1654 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.57 (8H, s, 4 × –CH2, piperidine), 2.70 (1H, s, –CH, piperidine), 2.80 (1H, s, –CH, piperidine), 3.71 (9H, s, 3 × –OCH3), 5.62 (1H, s, H-4), 6.74–6.96 (6H, m, Ar-H), 7.0 (1H, s, NH, D2O exchange), 7.50 (1H, s, =CH), 9.20 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 15.0, 19.1, 24.5, 25.3, 48.0, 48.5, 50.0, 56.2, 60.6, 61.3, 65.3, 108.1, 114.4, 113.2, 113.5, 123.5, 127.6, 129.8, 130.5, 132.6, 142.0, 151.5, 153.3, 153.5, 190.1; MS: m/z = 451.00 [M]+; analysis for C25H29N3O5: C (66.50) H (6.47) N (9.31)%; found C (66.70) H (6.48) N (9.33)%.
4-(2,3,4-Trimethoxyphenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (15): colour: brown; yield: 57%; m.p.:125–127 °C; UV λmax (methanol) = 441 nm; IR (KBr) cm−1: 3478 (N–H), 2852 (ArC-H), 1634 (C = O), 1596 (C = O), 1567 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.56 (8H, s, 4 × –CH2, piperidine), 2.70 (1H, s, –CH, piperidine), 2.80 (1H, s, –CH, piperidine), 3.70 (9H, s, 3 × –OCH3), 5.64 (1H, s, H-4), 6.75–6.97 (6H, m, Ar-H), 7.0 (1H, s, NH, D2O exchange), 7.44 (1H, s, =CH), 9.20 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 15.6, 19.0, 24.4, 25.3, 48.0, 48.5, 49.8, 56.2, 60.6, 61.3, 65.3, 108.1, 112.4, 113.2, 113.8, 123.0, 127.4, 129.9, 130.6, 132.3, 142.0, 151.5, 153.3, 153.5, 190.4; MS: m/z = 452.08 [M + 1]+; analysis for C25H29N3O5: C (66.50) H (6.47) N (9.31)%; found C (66.30) H (6.46) N (9.29)%.
4-(3,4,5-Trimethoxyphenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (16): colour: brown; yield: 60%; m.p.: 135–137 °C; UV λmax (methanol) = 441 nm; IR (KBr) cm−1: 3236 (N–H), 2933 (ArC-H), 1701 (C = O), 1650 (C = O), 1610 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.55 (8H, s, 4 × –CH2, piperidine), 2.70 (1H, s, –CH, piperidine), 2.80 (1H, s, –CH, piperidine), 3.6 (9H, s, 3 × –OCH3), 5.40 (1H, s, H-4), 6.65–6.93 (6H, m, Ar-H), 7.0 (1H, s, NH, D2O exchange), 7.5 (1H, s, =CH), 9.2 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 19.5, 24.4, 25.2, 25.3, 47.9, 48.5, 54.1, 56.2, 60.3, 65.3, 104.1, 112.4, 113.8, 127.2, 130.7, 137.2, 140.1, 152.9, 153.3, 153.6, 190.6; MS: m/z = 452.40 [M + 1]+; analysis for C25H29N3O5: C (66.50) H (6.47) N (9.31)%; found C (66.70) H (6.48) N (9.32)%.
4-(2,4,6-Trimethoxyphenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (17): colour: brown; yield: 60%; m.p.: 140–142 °C; UV λmax (methanol) = 428 nm; IR (KBr) cm−1: 3300 (N–H), 2930 (ArC-H), 1685 (C = O), 1654 (C = O), 1595 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.54 (8H, s, 4 × –CH2, piperidine), 2.7 (1H, s, –CH, piperidine), 2.80 (1H, s, –CH, piperidine), 3.70 (9H, s, 3 × –OCH3), 5.79 (1H, s, H-4), 6.90–6.93 (6H, m, Ar-H), 7.0 (1H, s, NH, D2O exchange), 7.51 (1H, s, =CH), 9.21 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 15.6, 19.0, 24.4, 25.3, 48.0, 48.5, 49.5, 56.0, 56.5, 60.6, 65.4, 112.4, 112.5, 113.8, 120.2, 124.3, 127.3, 130.7, 130.9, 137.6, 139.5, 146.5, 151.8, 152.9, 153.5, 190.3; MS: m/z = 453.92 [M + 2]+; analysis for C25H29N3O5: C (66.50) H (6.47) N (9.31)%; found C (66.35) H (6.46) N (9.30)%.
4-(2,4-Dimethoxyphenyl)-5-[4-(piperidin-1-yl) benzoyl]-3,4-dihydropyrimidin-2(1H)-one (18): colour: brown; yield: 55%; m.p.: 135–137 °C; UV λmax (methanol) = 419 nm; IR (KBr) cm−1: 3270 (N–H), 2900 (ArC-H), 1670 (C = O), 1635 (C = O), 1621 (C = C); 1H NMR (500 MHz, DMSO-d6): δ = 1.57 (8H, s, 4 × –CH2, piperidine), 2.70 (1H, s, –CH, piperidine), 2.80 (1H, s, –CH, piperidine), 3.83 (6H, s, 2 × –OCH3), 5.64 (1H, s, H-4), 6.44–6.93 (7H, m, Ar-H), 7.0 (1H, s, NH, D2O exchange), 8.0 (1H, s, =CH), 9.07 (1H, s, –CONH, D2O exchange); 13C NMR (125.76 MHz, DMSO-d6): δ = 15.6, 24.4, 25.3, 48.0, 48.5, 49.3, 55.6, 55.9, 65.4, 99.1, 104.8, 111.7, 113.2, 113.2, 124.0, 127.4, 128.6, 130.7, 132.8, 139.6, 152.2, 153.5, 158.3, 160.4, 190.4; MS: m/z = 421.67 [M]+; analysis for: C (68.39) H (6.46) N (9.97)%; found C (66.45) H (6.47) N (9.95)%.
In vivo anti-ulcer activity
Evaluation of anti-ulcer activity and gastric secretion in rats
Albino Wistar rats, weighing (150–200 g), were obtained from the animal house of College of Pharmacy, King Saud University (Riyadh, Saudi Arabia). All the animals were kept in laboratory conditions for 1 week, so that they will get acclimatised. The animals were randomly divided into groups of six rats each. Compounds (1–18) were given orally or intraperitoneally. The stomachs were removed after the rats were sacrificed and opened along the greater curvature. The animal protocol used in this study was approved by the Research Ethics Committee of College of Pharmacy, King Saud University.
Gastric lesions induced by ethanol
Albino Wistar rats, weighing (150–200 g), were divided into different groups. Animals were administered test drugs or standard drug. After 1 h, 1 ml of 80% ethanol was administered orally to each animalCitation23.
Gastric lesions induced by necrotising agents (cytoprotection)
Necrotising agent, 1 ml each (80% ethanol, 0.2 mol/l NaOH or 25% NaCl), was administered to animals. Compounds (3, 8, 11 and 15) were given half an hour prior to the administration of necrotising agents. The animals were sacrificed and examined for stomach ulcers after 1 h of the administration of necrotising agents.
Gastric lesions induced by indomethacin
Suspension of indomethacin in 1.0% of carboxymethylcellulose (CMC) in water (6 mg/ml) at a dose of (30 mg/kg) body weight was administered orally. Control rats were treated with vehicle. Compounds (3, 8, 11 and 15) were given half an hour prior to indomethacin administration at a dose of 12.5, 25 and 50 mg/kgCitation24.
Hypothermic restraint stress-induced ulcers
Thirty minutes after the oral administration of compounds (3, 8, 11, and 15), 12.5, 25 and 50 mg/kg of the rats were restrained in cages and kept inside a refrigerator for 3 hCitation25.
Pylorus-ligated rats
Pylorus ligation under ether anaesthesia was carried out. Intraperitoneal administration of compounds (3, 8, 11 and 15) was performed immediately after pylorus ligation. After 6 h, animals were sacrificedCitation26.
Determination of gastric wall mucus (GWM)
GWM was performed according to the modified procedureCitation27.
Estimation of non-protein sulfhydryls (NP-SH) MDA and total protein (TP)
Gastric mucosal non-protein sulfhydryls, MDA and TP were measured according to the reported methodCitation28.
Determination of LD50
The Karber method was used for the LD50 determination of most active compoundsCitation29.
Histopathological evaluation
Histopathological examination of gastric tissue was performed to study the anti-ulcer activity of compounds (3, 8, 11 and 15).
Results and discussions
Chemistry
As shown in Scheme 1, enaminone (III), 3-(dimethylamino)-1-[4-(piperidin-1-yl) phenyl] prop-2-en-1-one was synthesised by refluxing 1-[4-(piperidin-1-yl) phenyl] ethan-1-one (I) with DMF-DMA (II) under solvent free condition for 10 h.
Scheme 1. Synthetic route of compounds (1–18).
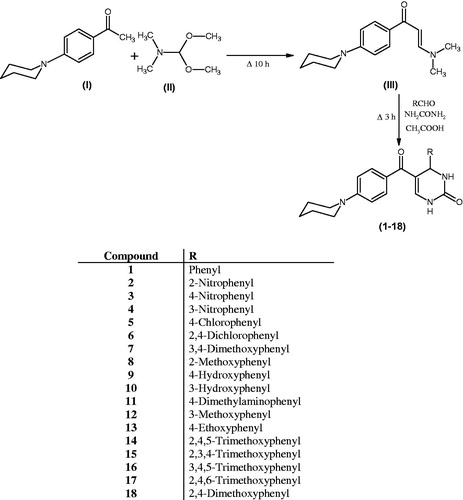
Six protons of piperidine were obtained as a singlet at δ 1.58 ppm and four piperidine protons appeared at δ 3.0 ppm. Two singlet peaks, at δ 2.89 and 3.17 ppm, were obtained due to the N,N-dimethyl protons and two doublet peaks at δ 5.79 and 7.65 ppm (J = 12.5 Hz) were obtained due to the ethylenic protons in 1H NMRCitation30. Aromatic protons were found around δ 6.91–7.78 ppm. The enaminone (III) existed in the E-configuration. A single crystal X-ray structure also confirmed the 3D structure of enaminone (III) ().
Figure 1. Single crystal X-ray structure of enaminone (III).
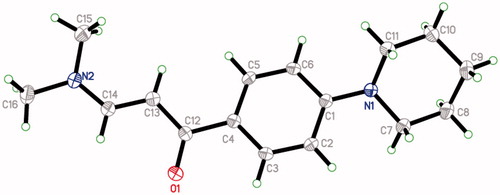
A reaction mixture of substituted benzaldehyde (0.01 mol), enaminone, 3-(dimethylamino)-1-[4-(piperidin-1-yl) phenyl] prop-2-en-1-one (III) (0.01 mol), urea (0.01 mol) and glacial acetic acid (10 ml) was refluxed for 3 h. The products were obtained by pouring the reaction mixture in cold water. The precipitates (1–18) thus formed were collected by vacuum filtration. The products were washed several times with cold water. Re-crystallisation of products was performed in glacial acetic acid. All of the compounds presented the D2O exchangeable broad singlet at δ 6.97–7.31 ppm and δ 9.07–9.67 ppm corresponding to the two NH protons. Eight protons (4 × CH2) of piperidine moiety were observed at δ 1.54–1.57 ppm. Two other piperidine protons were observed at δ 2.70–3.44 and δ 2.80–3.48 ppmCitation31. The H-4 and = CH protons of dihydropyrimidinone moiety were observed at δ 5.37–6.11 and 7.45–8.21 ppm, respectively. The presence of all carbon atoms for compounds was confirmed by 13C NMR spectra. The CH2 carbons of piperidine were obtained at around δ 24, 25, 48 and 53 ppm. The carbonyl group (C = O) peak was observed at around 190. Molecular weight of compounds was confirmed by mass spectra. All the compounds gave molecular ion peak respective to their molecular weights. The detailed spectral results of 1H NMR, 13C NMR spectra and mass spectra are given in the experimental part. The spectral and analytical data confirmed the composition of the synthesised compounds (1–18). The single crystal X-ray structure confirms the 3D structure of dihydropyrimidinone derivative 13 ().
Figure 2. Single crystal X-ray structure of compound 13.
Biological activity in vivo
In our first phase study, we screened all the synthesised compounds (1–18) at graded doses (12.5, 25 and 50 mg/kg, p.o.) in 80% ethanol induced gastric ulcer model with ranitidine (50 mg/kg, p.o.) as reference drug. The screening results are summarised in . Among the synthesised compounds, 3, 8, 11 and 15 exhibited significant protection. It gives us the impetus to further explore their anti-ulcer effects in different anti-ulcer models.
Table 1. The effect of compounds on gastric lesions induced by 80% ethanol (mean ± SE).
The animals were treated with 80% ethanol, 0.2 mol/l NaOH and 25% NaCl, which resulted in gastric lesions in the stomach in all the control animals. The ulcer index in 80% ethanol, 0.2 mol/l NaOH and 25% NaCl was 7.66 ± 0.21, 7.33 ± 0.21 and 6.83 ± 0.30, respectively, in the control animals after the 1-h administration of necrotising agents. Pre-treatment of animals with compounds 3, 8, 11 and 15 at doses of 12.5, 25, 50 mg/kg produced significant results. Compound 15 (50 mg/kg) was found to be most active as anti-ulcer agent with ulcer index in 80% ethanol, 0.2 mol/l NaOH and 25% NaCl as 2.16 ± 0.30, 1.33 ± 0.42 and 1.66 ± 0.33, p < 0.001, respectively ().
Table 2. The effect of compounds on gastric lesions induced by necrotising agents (mean ± SE).
NSAIDs are considered to be responsible for peptic ulcer in humans due to suppression of PGE2 biosynthesis and depletion of mucus. The administration of indomethacin (30 mg/kg) orally induced gastric damage of animals. The compounds 3, 8, 11 and 15 presented significant results especially compounds 3 and 15 with ulcer index of 12.66 and 14.50 respectively, which provides a proof, regarding the cytoprotective nature of these compounds. Compound 3 was found to be most active anti-ulcer agent in this test ().
Table 3. The effect of compounds on indomethacin-induced gastric mucosal lesions (mean ± SE).
Ulcer formation by hypothermic restraint stress was inhibited significantly by compounds 3 and 15 at the dose of 50 mg/kg. However, compound 15 was found to be most effective at dose of 50 mg/kg with intraluminal bleeding and gastric lesion ulcer index of 1.33 ± 0.33 and 12.33 ± 0.84, respectively. Compound 3 was observed to show similar activity as compound 15 at the same dose of 50 mg/kg ().
Table 4. The effect of compounds on hypothermic restraint stress-induced intraluminal bleeding and gastric lesion in rats (mean ± SE).
In the experiment of pylorus ligation, a large amount of gastric acid secretion were obtained (11.23 ± 0.18 ml), titratable acidity was found to be 173.88 ± 5.12 mEq/l and ulcer index was recorded as 3.33 ± 0.21 in the control group. Compounds 3 and 15 significantly reduced the gastric secretion, titratable acidity and ulcer index at the dose dependent manner. Compound 15 at the dose of 50 mg/kg was found to be most effective in reducing gastric secretion, titratable acidity and ulcer index formation 4.76 ± 0.23 ml, 73.33 ± 2.43 mEq/l and 1.00 ± 0.36 respectively as compared to the standard drug ranitidine ().
Table 5. The effect of compounds on gastric secretion, acidity and gastric lesion index in pylorus-ligated shay rats (mean ± SE).
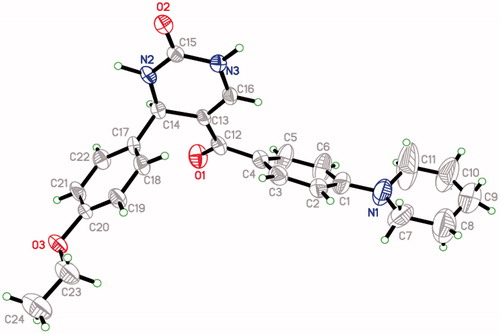
The administration of ethanol induced a significant damage to the mucosa. Treatment with 80% ethanol resulted in gastric mucosal ulceration (), ranitidine pre-treatment showed the normal gastric mucosa (), compound 3 (50 mg/kg) pre-treatment presented intact mucosa with mild ulceration (), pre-treatment with compounds 8, 11 and 15 (50 mg/kg) each showed intact normal gastric mucosa ().
Figure 3. (A) Treatment with 80% ethanol only, showing mucosal ulceration. (B) Treatment with ranitidine (50 mg/kg) showing normal mucosa. (C) Treatment with compound 3 (50 mg/kg) showing intact mucosa with mild ulceration. (D) Treatment with compound 8 (50 mg/kg) showing intact normal mucosa. (E) Treatment with compound 11 (50 mg/kg) showing intact normal mucosa. (F) Treatment with compound 15 (50 mg/kg) showing intact normal mucosa.

There is a significant reduction in the Alcian blue binding of gastric mucus (201 ± 8.32 µg/g) of tissue in animals treated with 80% ethanol as compared to control group (276.53 ± 10.19 µg/g). Pre-treatment of animals with compounds 3, 8, 11 and 15 at different doses produced dose dependent effects. Compounds 3 and 15 were found to be most effective. Compound 15 at the dose of (50 mg/kg) significantly enhances the Alician blue binding capacity of gastric mucosa (275.32 ± 5.37 µg/g), p < 0.001 ().
Table 6. The effect of compounds on the change in gastric wall mucus in stomach tissue induced by 80% ethanol (mean ± SE).
The glycogen level of the control and the pre-treated animal were also checked using the Periodic acid-Schiff (PAS). The ulcers induced by ethanol causes extensive gastric mucosal injury. Moreover, they exhibit haemorrhagic and necrotic lesions, which infiltrate into the mucosa and cause oedema and leukocyte infiltration. However, the pre-treatment with compounds 3, 8, 11 and 15 resulting in expansion of mucus gel layer that with continuous PAS-positive that lines the gastric mucosal surface (). The magenta staining colour is exhibited with the compounds 3, 8, 11 and 15 pre-treated groups. The tissue has a normal glandular pattern and mild leucocyte infiltration. On the other hand, the gastric specimen from the control did not exhibit the magenta staining colour. As shown in , the ethanol-induced ulcer exhibits pervasive injury to the gastric mucosa. The pre-treatment with ranitidine protects the gastric mucosa (). The compounds 3, 8, 11 and 15 pre-treated rats exhibited a significant decrease in ulcer index and less mucosal damage (). These results clearly indicate that compounds 3, 8, 11 and 15 have gastro-protective activity. Mucus production by gastric mucosa increased gradually in the experimental rats pre-treated with compounds 3, 8, 11 and 15. Gastric mucus plays a crucial role in gastro-protection. The pre-treatment with compounds 3, 8, 11 and 15 significantly augmented the gastro-protective activity, with enhancement of the free mucus when compared to the mucus of ulcer control animals. Thus, compounds 3, 8, 11 and 15 have gastro-protective activity against ethanol induced gastric ulcer by improving mucosal content.
Figure 4. Light micrographs showing the effect of compounds 3, 8, 11 and 15 on ethanol-induced gastric lesions of rats. (A) Treatment with ethanol (PAS); (B) pre-treatment with standard drug ranitidine (50 mg/kg) (PAS); (C) pre-treatment with compound 3 (50 mg/kg) (PAS); (D) pre-treatment with compound 8 (50 mg/kg) (PAS); (E) pre-treatment with compound 11 (50 mg/kg) (PAS); (F) pre-treatment with compound 15 (50 mg/kg) (PAS).
MDA levels in the gastric mucosa were significantly increased in ethanol only treated then in control group (7.42 ± 0.30 nmol/g; 1.14 ± 0.06 nmol/g). Compounds 15 (50 mg/kg) significantly reduced the MDA content (1.90 ± 0.06 nmol/g). Similar results were obtained for compound 3. The NP-SH level in control group was found to be 5.03 ± 0.10 nmol/g of tissue, which was significantly reduced to 3.22 ± 0.20 nmol/g of tissue following the 80% ethanol administration. Pre-treatment of animals with compounds 3, 8, 11 and 15 significantly replenished the ethanol induced depletion of NP-SH. Compounds 3 and 15 at the dose of 50 mg/kg produced highly significant results 4.56 ± 0.17 and 4.92 ± 0.30, respectively, higher than the standard drug ranitidine (4.24 ± 0.15). The level of TP in the gastric mucosa of control group was 122.55 ± 3.23 g/l, which was significantly decreased to 47.50 ± 2.08 g/l following 80% ethanol administration. Pre-treatment of animals with tested compounds significantly improved the levels of TP. Compounds 15 and 3 at the dose of 50 mg/kg produced significant results (96.60 ± 1.18 g/ml and 95.80 ± 1.51 g/ml), respectively, in comparison to the standard drug ranitidine (104.59 ± 1.59 g/ml) ().
Table 7. The effect of compounds on the levels of MDA, NP-SH and TP in stomach tissue induced by 80% ethanol (mean ± SE).
Toxicity of compounds
Karber method was used to determine the LD50 of compounds 3, 8 and 15. A 24-h observation was made for the toxicity symptoms and mortality. The dead animals were counted at the end of the study and the LD100 was calculated. The LD50 of compounds 3, 8 and 15 were found to be 125, 55.5 and 116.5 mg/kg, respectively ().
Table 8. Determination of LD50 of active compounds by Karber method.
Structure activity relationship (SAR)
The design of new compounds was based on hybrid approach. A series of compounds containing dihydropyrimidinone and piperidine were synthesised and screened for anti-ulcer activity. Structural modifications were done not only to obtain derivatives with higher activity, but also to collect data regarding SAR. We showed that the presences of pharmacophores (dihydropyrimidinone and piperidine) are both essential for the activity. Compounds 3 (R = 4-nitrophenyl substitution), 8 (R = 2-methoxyphenyl), 11 (R = N-dimethylaminophenyl) and 15 (R = 2,3,4-trimethoxyphenyl) substitutions were found to be most active compounds of the series.
Conclusion
A series of novel dihydropyrimidinone and piperidine scaffold hybrids were synthesised, characterised by spectral data and screened for their anti-ulcer activity in several in vivo ulcer models. The newly synthesised hybrids displayed significant gastro protective effect by inhibiting the formation of ulcers induced by 80% ethanol. Four compounds 3, 8, 11 and 15 were found to most potent compounds of the series. These compounds were further evaluated for anti-ulcer activity by different in vivo anti-ulcer models in animals. The anti-ulcer action of the active compounds appears to be due to both anti-secretary and gastro protective effect. The gastro protective action was mainly due to secretion of mucus. Compound 15 was found to be highly potent compounds of the series. Additional studies on lead compound 15 will result in a new orally active candidate.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Hoogerwerf WA, Paricha PJ. Pharmacotherapy of gastric acidity, peptic ulcer, and gastrointestinal reflux disease. In: Brunton LL, Lazo JS, Parker KL, eds. Goodman & Gilman’s the pharmacological basis of therapeutics. 11th ed. New York: McGraw-Hill; 2006:972.
- Robert A, Nezamis JE, Lancaster C, et al. Mild irritants prevent gastric necrosis through “adaptive cytoprotection” mediated by prostaglandins. Am J Physiol 1983;245:G113–21.
- Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet 2009;374:1449–61.
- Ahn JS, Eom CS, Jeon CY, Park SM. Acid suppressive drugs and gastric cancer: a meta-analysis of observational studies. World J Gastroenterol 2013;19:2560–8.
- Agrawal NM, Roth S, Graham DY, et al. Misoprostol compared with sucralfate in the prevention of nonsteroidal anti-inflammatory drug-induced gastric ulcer: a randomized, controlled trial. Ann Intern Med 1991;115:195–200.
- Lazzaroni M, Bianchi Porro G. Prophylaxis and treatment of non-steroidal anti-inflammatory drug-induced upper gastrointestinal side-effects. Dig Liver Dis 2001;33:S44–S58.
- Folkers K, Harwood HJ, Johnson TB. Researches on pyrimidines. cxxx. synthesis of 2-keto-1,2,3,4-tetrahydropyrimidines. J Am Chem Soc 1932;54:3751–8.
- Atwal KS, Ahmed SZ, Bird JE, et al. Dihydropyrimidine angiotensin II receptor antagonists. J Med Chem 1992;35:4751–63.
- Atwal KS, Swanson BN, Unger SE, et al. Dihydropyrimidine calcium channel blockers. 3-Carbamoyl-4-aryl-1,2,3,4-tetrahydro-6-methyl-5-pyrimidinecarboxylic acid esters as orally effective antihypertensive agents. J Med Chem 1991;34:806–11.
- Rovnyak GC, Kimball SD, Beyer BJ, et al. Calcium entry blockers and activators: conformational and structural determinants of dihydropyrimidine calcium channel modulators. J Med Chem 1995;38:119–29.
- Rana K, Kaur B, Chaudhary G. Synthesis and anti-ulcer activity of some dihydropyrimidines. Ind J Chem 2004;43B:1553–7.
- Patil A, Ganguly S, Surana S. Synthesis and antiulcer activity of 2-[5-substituted-1-H-benzo(d)imidazol-2-yl sulfinyl]methyl-3-substituted quinazoline-4-(3H) ones. J Chem Sci 2010;122:443–50.
- Beena KP, Suresh R, Rajasekaranb A. Dihydropyrimidinones-a versatile scaffold with diverse biological activity. J Pharm Sci Res 2016;8:741–6.
- Bhat MA, Al-Dhfyan A, Al-Omar MA. Targeting cancer stem cells with novel 4-(4-substitutedphenyl)-5-(3,4,5-trimethoxy/3,4-dimethoxy)-benzoyl-3,4-dihydropyrimidine-2(1H)-one/thiones. Molecules 2016;21:1746–55.
- Kodhati V, Vanga MR, Yellu NR. Synthesis and anti-bacterial and anti-ulcer evaluation of new S-mannich bases of 4,6-diaryl-3,4-dihydropyrimidin-2(1H)-thiones. J Korean Chem Soc 2013;57:234–40.
- Zhang X, Lei P, Sun T, et al. Design, synthesis, and fungicidal activity of novel thiosemicarbazide derivatives containing piperidine fragments. Molecules 2017;22:E2085.
- Forcellini E, Boutin S, Lefebvre CA, et al. Synthesis and biological evaluation of novel quinazoline-4-piperidinesulfamide derivatives as inhibitors of NPP1. Eur J Med Chem 2018;147:130–49.
- Kasturi SP, Surarapu S, Uppalanchi S, et al. Synthesis, molecular modeling and evaluation of α-glucosidase inhibition activity of 3,4-dihydroxy piperidines. Eur J Med Chem 2018;150:39–52.
- Ferro S, Deri B, Germanò MP, et al. Targeting tyrosinase: development and structural insights of novel inhibitors bearing arylpiperidine and arylpiperazine fragments. J Med Chem 2018;61:3908–17.
- Imaeda T, Ono K, Nakai K, et al. Discovery, synthesis, and structure-activity relations of 3,4-dihydro-1H-spiro(naphthalene-2,2'-piperidin)-1-ones as potassium-competitive acid blockers. Bioorg Med Chem 2017;25:3719–35.
- Scott MK, Jacoby HI, Mills JE, et al. 4-(Diphenylmethyl)-1-[(imino)methyl]piperidines as gastric antisecretory agents. J Med Chem 1983;26:535–8.
- Ivanov C, Petkov O, Petrov P. Synthesis, gastroprotective, antisecretory and anti-helicobacter effect of N-[3-(3-(1-piperidinylmethyl) phenoxy)propyl]-hydroxyacetamide 2-hydroxypropane-1,2,3-tricarboxylate bismuth (3+) complex (MX1)-MX1. J Pharm Pharmacol 1996;48:297–301.
- Yu C, Mei XT, Zheng YP, Xu DH. Gastroprotective effect of taurine zinc solid dispersios against absolute ethanol-induced gastric lesions is mediated by enhancement of antioxidant activity and endogenous PGE2 production and attenuation of NO production. Eur J Pharmacol 2014;740:329–36.
- Bhargava KP, Gupta MB, Tangri KK. Mechanism of ulcerogenic activity of indomethacin and oxyphenbutazone. Eur J Pharmacol 1973;22:191–5.
- Senay EC, Levine RL. Synergism between cold and restraint for rapid production of stress ulcers in rats. Proc Soc Exp Biol Med 1967;124:1221–31.
- Sashidhara KV, Avula SR, Mishra V, et al. Identification of quinoline-chalcone hybrids as potential antiulcer agents. Eur J Med Chem 2015;89:638–53.
- Corne SJ, Morrissey SM, Woods RJ. Proceedings: a method for the quantitative estimation of gastric barrier mucus. J Physiol 1974;242:116P–17P.
- Sedlak J, Lindsay RH. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal Biochem 1968;25:192–205.
- Karber G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch Exptl Pathol Pharmakol 1931;162:480–3.
- Bhat MA, Ahmed AF, Wen ZH, et al. Synthesis, anti-inflammatory and neuroprotective activity of pyrazole and pyrazolo[3,4-d]pyridazine bearing 3,4,5-trimethoxyphenyl. Med Chem Res 2017;26:1557–66.
- Bhat MA, Al-Rashood KA, Abdel-Aziz HA. Unexpected configuration in stereoselective synthesis of some novel (1Z)-1-(morpholin-1-yl)-N2-arylamidrazones. Lett Org Chem 2012;9:487–92.