
Abstract
A new series of NSAID thioesters were synthesized and evaluated for their in vitro antitumor effects against a panel of four human tumor cell lines, namely: HepG2, MCF-7, HCT-116 and Caco-2, using the MTT assay. Compared to the reference drugs 5-FU, afatinib and celecoxib, compounds 2b, 3b, 6a, 7a, 7b and 8a showed potent broad-spectrum antitumor activity against the selected tumour cell lines. Accordingly, these compounds were selected for mechanistic studies about COX inhibition and kinase assays. In vitro COX-1/COX-2 enzyme inhibition assay results indicated that compounds 2b, 3b, 6a, 7a, 7b, 8a and 8 b selectively inhibited the COX-2 enzyme (IC50 = ∼0.20–0.69 μM), with SI values of (>72.5–250) compared with celecoxib (IC50 = 0.16 μM, COX-2 SI: > 312.5); however, all the tested compounds did not inhibit the COX-1 enzyme (IC50 > 50 μM). On the other hand, EGFR, HER2, HER4 and cSrc kinase inhibition assays were evaluated at a 10 μM concentration. The selected candidates displayed limited activities against the various tested kinases; the compounds 2a, 3b, 6a, 7a, 7b and 8a showed no activity to weak activity (% inhibition = ∼0–10%). The molecular docking study revealed the importance of the thioester moiety for the interaction of the drugs with the amino acids in the active sites of COX-2. The aforementioned results indicated that thioester based on NSAID scaffolds derivatives may serve as new antitumor compounds.
Introduction
Malignancy is global health problem and is the leading cause of death in children until fifteen years of ageCitation1. Non-steroidal anti-inflammatory drugs (NSAIDs) such as sulindac, indomethacin and celecoxib are commonly used for treating arthritis via inhibition of the cyclooxygenase enzyme (COX)Citation2,Citation3. COX-2 levels are over-expressed in human tumours, unlike in normal cells and could develop a tumorigenic potentialCitation4. Selective enzyme inhibition and restoration of normal apoptotic responses is known as COX-2-dependent anticancer mechanismCitation4–6. On the other hand, COX-2-independent mechanisms function via apoptosis stimulation, angiogenesis arrest, or cancer cell growth inhibition by blocking signal transduction pathways for cell proliferationCitation7–10.
Drug repositioning development is a more important process for saving money and time than the production of a new drugCitation11. NSAIDs and coxib such as naproxen, ibuprofen, indomethacin, sulindac, celecoxib and their analogues () have diverse scaffolds; modifying their basic structures is relatively safe, applicable for oral use, associated with multiple therapeutic features, such as analgesic, antipyretic, anti-inflammatory and anticancer activitiesCitation12–18. For example, sulindac amides () showed a good activity against a panel of lymphoblastic leukemia cell lines in nanomolar concentrationsCitation18. Additionally, celecoxib reduced the number and size of colorectal polyps in adenomatous polyposis ()Citation19–21. Antiproliferative and apoptosis effects of celecoxib in colon, stomach, lung, prostate and pancreatic cancer cells have been observed by selective COX-2 inhibitionCitation22–25. On the other hand, a combination of drugs (NSAIDs) such as indomethacin, sulindac, tolmetin, acemetacin, zomepirac and mefenamic acid at non-toxic levels, and different chemotherapeutic drugs such as anthracyclines (doxorubicin, daunorubicin and epirubicin), in addition to VP-16, vincristine and teniposide, led to a significantly synergistic cytotoxicity of these chemotherapeutic drugs in the human COR L23R, DLKP, A549 and COR L23P lung cancer cell lines, and the human HL60/ADR leukaemia cell line3.
Figure 1. Reported NSAIDs and celecoxib as anticancer agents (A-E) and the designed compounds.
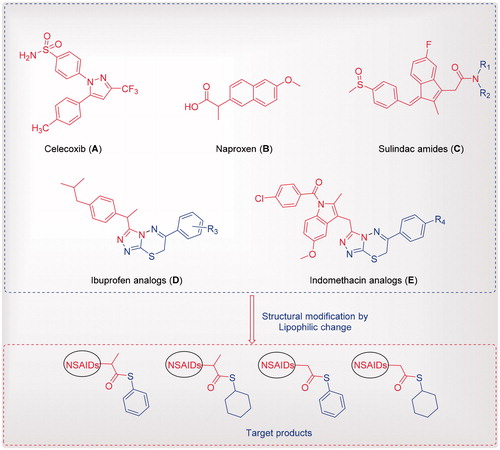
Continuing our studies as an attempt to develop effective cytotoxic agentsCitation26–42, we synthesised some NSAIDs conjugated to thioester moieties and evaluated their cytotoxic activities. Additionally, in vitro COX-1/COX-2 enzyme and kinase inhibitory assays were investigated for the most active compounds, to identify their mode of action.
A molecular docking technique was used in order to predict the binding geometry requirements of the target molecules, which is important for the antitumour activity.
Experimental
Melting points were recorded on a Barnstead 9100 Electrothermal melting apparatus. IR spectra (KBr) were recorded on an FT-IR Perkin-Elmer spectrometer (ν cm−1). 1H and 13C NMR spectra were recorded on Bruker 500 or 700 MHz spectrometers using DMSO-d6 or CDCl3 as the solvent. Microanalytical data (C, H and N) were obtained using a Perkin-Elmer 240 analyser and the proposed structures were within ±0.4% of the theoretical values. Mass spectra were recorded on a Varian TQ 320 GC/MS/MS mass spectrometer. NSAIDs thioester was obtained according to reported methodCitation43.
General method for the preparation of NSAIDs thioester
Trifluoroacetic acid (0.5 mmol) was added dropwise to a mixture of NSAIDs (0.1 mmol) and thiol (0.5 mmol) in dry acetonitrile that was heated for 10–12 h at 60 °C. The reaction mixture was cooled, quenched using ammonium chloride solution, extracted with ethylacetate, washed with brine and dried over anhydrous sodium sulphate; the solvent was then evaporated, and the product obtained was chromatographed with hexane and CHCl3.
S-phenyl-2–(4-isobutylphenyl)propanethioate (1a)Citation44
Yield, 89%; colourless oil; IR (KBr) νmax/cm−1 1700.69 (CO), 738.10, 690.48 (CS); 1H NMR (500 MHz, CDCl3): δ 7.47–7.51 (m, 5H), 7.44 (d, 2H, J = 8.0 Hz), 7.30 (d, 2H, J = 7.5 Hz), 4.13 (d, 1H, J = 7.0 Hz), 2.64 (d, 2H, J = 7.0 Hz), 2.04 (t, 1H, J = 6.5 Hz), 1.73 (d, 3H, J = 7.0 Hz), 1.08 (d, 6H, J = 6.5); 13 C NMR (125 MHz, CDCl3): δ 18.8, 22.6, 30.3, 45.2, 53.9, 127.3, 127.6, 127.9, 128.2, 129.2, 129.3, 129.6, 134.6, 136.6, 141.1, 199.1; MS; m/z (298).
S-cyclohexyl-2–(4-isobutylphenyl)propanethioate (1b)
Yield, 81%; colourless oil; 1H NMR (500 MHz, CDCl3): δ 7.27 (d, 2H, J = 8.0 H), 7.15 (d, 2H, J = 8.0 H), 3.88 (d, 1H, J = 7.0 Hz), 3.52 (s, 1H), 2.52 (d, 2H, J = 7.0 Hz), 1.98 (s, 1H), 1.90–1.94 (q, 2H, J = 7.0, 6.5 Hz), 1.74 (s, 1H), 1.69 (s, 1H), 1.62 (d, 1H, J = 12 Hz), 1.56 (d, 3H, J = 7.0 Hz), 1.35–1.45 (m, 4H), 1.29 (s, 1H), 0.96 (d, 6H, J = 6.5); 13 C NMR (125 MHz, CDCl3): δ 18.6, 22.4, 25.6, 26.0, 30.2, 32.9, 33.1, 42.4, 45.1, 54.0, 127.6, 129.3, 137.3, 140.6, 200.9; MS; m/z (304).
S-phenyl-2–(3-benzoylphenyl)propanethioate (2a)
Yield, 88%; mp: 96–98 °C; IR (KBr) νmax/cm−1 1668.97 (CO), 746.66, 694.49 (CS); 1H NMR (500 MHz, CDCl3): δ 7.73 (d, 3H, J = 8.5 Hz), 7.63 (d, 1H, J = 7.5 Hz), 7.50 (t, 2H, J = 6.0 Hz), 7.39 (dd, 3H, J = 7.5, 11.5 HZ) 7.28 (s, 5H), 3.99 (q, 1H, J = 6.5, 7.0 Hz), 1.52 (d, 3H, J = 7.0 Hz); 13 C NMR (125 MHz, CDCl3): δ 16.3, 51.5, 125.3, 126.0, 126.4, 126.9, 127.1, 127.4, 127.8, 129.6, 130.2, 132.1, 135.1, 135.7, 137.5, 194.0, 196.3; MS m/z (346).
S-cyclohexyl-2–(3-benzoylphenyl)propanethioate (2b)
Yield, 81%; mp: 69–70 °C; 1H NMR (500 MHz, CDCl3): δ 7.81 (d, 2H, J = 7.0 Hz), 7.76 (s, 1H), 7.70 (d, 1H, J = 7.5 Hz), 7.55–7.60 (m, 2H), 7.43–7.49 (m, 3H), 3.93 (q, 1H, J = 7.0 & 6.5 Hz), 3.48 (s, 1H), 1.91 (s, 1H), 1.84 (d, 1H, J = 10 Hz), 1.67 (d, 2H, J = 14.0 Hz), 1.55 (d, 4H, J = 7.0 Hz), 1.34–1.40 (m, 4H), 1.25 (s, 1H); 13 C NMR (125 MHz, CDCl3): δ 18.4, 25.5, 25.9, 32.8, 32.9, 42.6, 54.0, 128.3, 128.6, 129.1, 129.7, 130.1, 131.8, 132.5, 137.4, 137.8, 140.3, 196.3, 200.4; MS m/z (352).
S-phenyl-2–(2-fluoro-[1,1′-biphenyl]-4-yl)propanethioate (3a)
Yield, 90%; mp: 85–86 °C; IR (KBr) νmax/cm−1 1694.14 (CO), 736.75, 687.25 (CS); 1H NMR (500 MHz, CDCl3): δ 1.52 (d, 3H, J = 7 Hz), 3.94 (dd, 1H, J = 6.5 & 7.0 Hz), 7.46 (d, 2H, J = 12.5 Hz), 7.29–7.41 (m, 9H), 7.05–7.14 (m, 2H); 13 C NMR (125 MHz, CDCl3): δ 18.6, 53.4, 115.5, 115.7, 124.0, 127.5, 127.7, 128.5, 129.0, 129.2, 129.4, 130.9, 131.0, 134.4, 135.4, 140.7, 140.8, 158.7, 160.7, 198.5; MS; m/z (336).
S-cyclohexyl-2–(2-fluoro-[1,1′-biphenyl]-4-yl)propanethioate (3b)
Yield, 80%; mp: 90–92 °C; IR (KBr) νmax/cm−1 1672.76 (CO), 751.18, 690.19 (CS); 1H NMR (500 MHz, CDCl3): δ 7.45 (d, 2H, J = 7.5 Hz), 7.25–7.36 (m, 4H), 7.03–7.07 (m, 2H,), 3.79 (t, 1H, J = 7.0 Hz), 3.40 (s, 1H), 1.76–1.84 (m, 2H), 1.57–1.60 (d, 2H, J = 13 Hz), 1.45 (d, 4H, J = 7.0 H), 1.23–1.33 (m, 4H), 1.17 (s, 1H); 13 C NMR (125 MHz, CDCl3): δ 18.4, 25.5, 25.9, 32.8, 33.0, 42.7, 53.6, 115.4, 115.6, 123.8, 123.9, 127.6, 127.9, 128.0, 128.4, 128.9, 130.7, 130.8, 135.5, 141.3, 141.4, 158.7, 160.6, 200.5; MS m/z (342).
2-[(Phenylthio)carbonyl]phenyl acetate (4a)Citation45
Yield, 84%; mp: 72–73 °C; 1H NMR (500 MHz, CDCl3): δ 7.91 (dd, 1H, J = 1.0 Hz), 7.42–7.45 (m, 6H) 6.87–6.92 (m, 2H), 2.24 (s, 3H); 13 C NMR (125 MHz, CDCl3): δ 29.7, 118.36, 119.4, 126.0, 128.9, 129.4, 130.0, 135.5, 136.3, 159.7, 195.8. MS m/z (272).
2-[(Cyclohexylthio)carbonyl]phenyl acetate (4b)
Yield, 80%; mp: 55–56 °C; 1H NMR (500 MHz, CDCl3): δ 7.78 (d, 1H, J = 8.0 Hz), 7.35 (t, 1H, J = 7.0 Hz), 6.88 (d, 1H, J = 8.0 Hz), 6.79 (t, 1H, J = 7.0 Hz), 3.66 (s, 1H), 2.26 (s, 2H), 1.95 (d, 2H, J = 10 Hz), 1.69 (t, 2H, J = 4.5 Hz), 1.56 (d, 1H, J = 8.5 Hz), 1.38–1.49 (m, 4H) 1.25 (d, 2H, J = 8.5 Hz); 13 C NMR (125 MHz, CDCl3): δ 25.5, 25.9, 29.7, 33.01, 42.5, 118.1, 119.1, 120.2, 128.8, 135.6, 159.5, 197.4; MS m/z (278).
S-phenyl-(S)-2–(6-methoxynaphthalen-2-yl)propanethioate (5a)Citation44
Yield, 88%; mp: 115–117 °C; IR (KBr) νmax/cm−1 1694.16 (CO), 738.16, 683.87 (CS); 1H NMR (500 MHz, CDCl3): δ 7.88 (s, 4H), 7.59 (d, 3H, J = 7.5 Hz), 7.49 (s, 3H), 7.31(d, 1H, J = 8.5 Hz), 4.28 (d, 1H, J = 6.5 Hz), 4.05 (s, 3H), 1.80 (d, 3H, J = 6.0 Hz); 13 C NMR (125 MHz, CDCl3): δ 18.7, 54.1, 55.3 105.7, 119.1, 126.4, 126.9, 127.4, 128.0, 129.0, 129.1, 129.3, 129.4, 134.0, 134.5, 134.7, 157.8, 199.2; MS m/z (322).
S-cyclohexyl-(S)-2–(6-methoxynaphthalen-2-yl)propanethioate (5b)
Yield, 84%; mp: 105–106 °C; IR (KBr) νmax/cm−1 1679.27 (CO), 741.06, 688.41 (CS); 1H NMR (500 MHz, CDCl3): δ 7.59–7.64 (m, 3H), 7.30 (dd, 1H, J = 1.0 Hz), 7.05 (dd, 1H, J = 2.0 Hz), 7.03 (s, 1H), 3.88 (d, 1H, J = 7.0 Hz), 3.82 (s, 3H), 3.37 (s, 1H), 1.82 (d, 1H, J = 8.5 Hz), 1.72 (t, 1H, J = 5.0 & 5.5 Hz), 1.59–1.46 (m, 6H), 1.30–1.13 (m, 5H); 13 C NMR (125 MHz, CDCl3): δ 18.5, 25.5, 26.0, 32.9, 33.0, 42.5, 54.2, 55.3, 105.6, 119.0, 126.4, 126.5, 127.1, 128.9, 129.3, 133.8, 135.3, 157.7, 201.2; MS m/z (328).
S-phenyl-2–(2-((2,6-dichlorophenyl)amino)phenyl)ethanethioate (6a)
Yield, 86%; mp: 101–102 °C; IR (KBr) νmax/cm−1 1679.27 (CO), 741.06, 688.41 (CS); 1H NMR (500 MHz, CDCl3): δ 10.01 (s, 1H), 7.40–7.42 (d, 2H, J = 8.0 Hz), 7.24–7.29 (m, 4H), 7.09–7.12 (t, 3H, J = 7.5 Hz), 6.99–7.00 (d, 2H, J = 7.5 Hz), 6.30–6.32 (d, 1H, J = 7.5 Hz), 3.68 (s, 2H); 13 C NMR (125 MHz, CDCl3): δ 35.7, 109.1, 123.0, 124.3, 124.8, 125.2, 127.7, 127.9, 129.0, 130.4, 130.8, 131.0, 134.5, 135.5, 139.6, 143.3, 198.0; MS m/z (388).
S-cyclohexyl-2–(2-((2,6-dichlorophenyl)amino)phenyl)ethanethioate (6b)
Yield, 83%; mp: 88–90 °C; 1H NMR (500 MHz, CDCl3): δ 7.38–7.40 (d, 2H, J = 8.0 Hz) 7.18–7.24 (m, 2H), 7.08–7.11 (t, 1H, J = 7.5 Hz), 6.97–7.00 (t, 1H, J = 7.5 Hz), 6.29–6.31 (d, 1H, J = 8.0 Hz), 3.70 (s, 2H), 3.01 (s, 1H), 2.01–2.00 (d, 4H, J = 9.0 Hz), 1.69–1.67 (t, 2H, J = 8.5 Hz), 1.49–1.47 (t, 2H, J = 8.5 Hz), 1.37–1.36 (d, 2H, J = 8.5 Hz); 13 C NMR (125 MHz, CDCl3): δ 22.7, 29.7, 35.7, 39.2, 41.0, 123.1, 124.3, 124.8, 127.9, 129.0, 129.5, 130.4, 130.8, 131.0, 135.5, 143.3, 198.0; MS m/z (394).
S-phenyl-2–(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)ethanethioate (7a)Citation46
Yield, 86%; mp: 133–135 °C; IR (KBr) νmax/cm−1 1671.45, 1604.72 (CO), 745.04, 693.51 (CS); 1H NMR (500 MHz, CDCl3): δ 7.59 (d, 2H, J = 8.5 Hz), 7.38 (d, 2H, J = 8.5 Hz), 7.29 (s, 5H), 6.92 (d, 1H, J = 2.0 Hz), 6.82 (d, 1H, J = 9.0 Hz), 6.61 (dd, 1H, J = 2.0, 9.0 Hz), 3.87 (s, 2H), 3.76 (s, 3H), 2.36 (s, 3H); 13 C NMR (125 MHz, CDCl3): δ 13.5, 39.1, 55.7, 101.2, 111.8, 127.6, 129.1, 129.2, 129.4, 130.5, 130.9, 131.2, 133.7, 134.4, 136.8, 139.4, 156.2, 168.3, 195.1; MS m/z (449), (M + 2, 451).
S-cyclohexyl-2–(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)ethanethioate (7b)Citation46
Yield, 83%; mp: 97–98 °C; IR (KBr) νmax/cm−1 1672.24, 1600.15 (CO), 830.24, 749.96 (CS); 1H NMR (700 MHz, DMSO-d6): δ 7.76 (d, 2H, J = 5.5 Hz), 7.64 (d, 2H, J = 5.5 Hz), 7.05 (s, 1H), 6.93 (d, 1H, J = 6.5 Hz), 7.72 (d, 1H, J = 6.5 Hz), 3.98 (s, 2H), 3.75 (s, 3H), 2.55–2.51 (m, 1H), 2.23 (s, 3H), 1.79 (s, 2H), 1.59 (s, 2H), 1.50 (d, 1H, J = 8.5 Hz), 1.33–1.30 (m, 4H), 1.20 (s, 1H); 13 C NMR (125 MHz, CDCl3): δ 13.8, 25.4, 25.8, 32.8, 40.2, 42.4, 55.8, 102.2, 111.9, 112.8, 115.0, 129.5, 130.6, 130.9, 131.6, 134.4, 136.5, 138.2, 156.0, 168.3, 196.8; MS m/z (456), (M + 2, 458).
S-phenyl-2–(5-fluoro-2-methyl-1–(4-(methylsulfinyl)benzylidene)-1H-inden-3-yl)ethanethioate (8a)
Yield, 78%; mp: 66–68 °C; IR (KBr) νmax/cm−1 1700.49 (CO), 1021 (SO), 734.05, 684.77 (CS); 1H NMR (500 MHz, CDCl3): δ 7.02–7.47 (m, 13H), 3.93–3.94 (d, 2H, J = 7.0 Hz), 2.27 (s, 3H), 1.46–1.52 (dd, 3H, J = 7.0 Hz); 13 C NMR (125 MHz, CDCl3): δ 18.6, 21.3, 53.3, 115.5, 115.7, 124.0, 127.7, 128.5, 129.0, 129.3, 130.0, 130.9, 134.4, 135.4, 139.7, 140.9, 158.7, 160.7, 198.9; MS m/z (448).
S-cyclohexyl-2–(5-fluoro-2-methyl-1–(4-(methylsulfinyl)benzylidene)-1H-inden-3-yl)ethanethioate (8b)
Yield, 75%; mp: 121–122 °C; IR (KBr) νmax/cm−1 1692.84 (CO), 859.17, 808.66 (CS), (SO); 1H NMR (700 MHz, DMSO-d6): δ 7.67–7.63 (m, 5H, 7.05 (s, 1H), 6.93 (d, 1H, J = 9.1 Hz), 6.72 (d, 1H, J = 9.1 Hz), 3.98 (s, 2H), 3.75 (s, 3H), 2.23 (s, 3H), 1.799 (s, 2H), 1.59 (s, 2H), 1.50 (d, 1H, J = 11.9 Hz), 1.31 (t, 4H, J = 10.5 & 9.8 Hz), 1.20 (s, 1H); 13 C NMR (176 MHz, DMSO-d6): δ 13.8, 25.4, 25.8, 32.8, 39.3, 42.4, 55.8, 102.0, 111.9, 112.8, 115.0, 129.5, 130.6, 130.9, 131.6, 134.4, 136.5, 138.2, 156.0, 168.3, 196.8; MS m/z (454).
Biological testing
Antitumor evaluation
The evaluation of the antitumour activity was performed using tetrazolium salt MTT (3–(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay as reportedCitation47–50.
In vitro cyclooxygenase (COX) inhibition assay
The colorimetric COX (ovine) inhibitor screening assay kit (kit catalogue number 560101, Cayman Chemical, Ann Arbor, MI) was utilized according to the manufacturer’s instructions to examine the ability of the test compounds and the reference drugs to inhibit the COX-1/COX-2 isozymesCitation51,Citation52.
Kinase inhibition assay
The assay for Kinases was performed at BPS Bioscience Inc. 6044 Cornerstone Court West, Ste. E, San Diego, CA 92121, USA using Kinase-Glo Plus luminescence kinase assay kit (Promega). Luminescence signal was measured using a BioTek Synergy 2 microplate readerCitation53.
Docking methodology
All modelling experiments were conducted with MOE programs running on PC computer [MOE 2008.10 of Chemical Computing Group. Inc]Citation54. The docking protocol is summarized in supporting informationCitation51,Citation52,Citation55–57.
Results and discussion
Chemistry
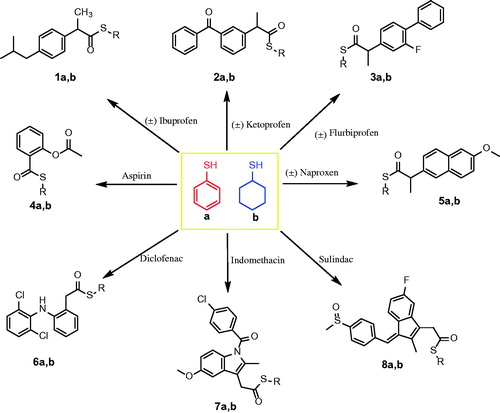
The new thioesters were synthesized by the reaction of the carboxylic acid group of NSAIDs with thiophenol and cycloxanethiol in the presence of trifluoroacetic acid (TFA)Citation43. The newly synthesized thioesters (Scheme 1) were confirmed by the presence of the carbonyl group (C=O) at 1700–1669 cm−1 and stretching of the (C–S) group at 859–683 cm−1 in the IR spectra. Additionally, the newly synthesised thioesters were confirmed by a characteristic peak at 201.21–195.15 ppm attributable to the (S–C=O) group in addition to the characteristic peaks of the cyclohexane moiety at 25.40–42.42 ppm or aromatic peaks of the thiophenol moiety in the aromatic region of the 13C NMR spectra. The 1H NMR spectra of the new thioesters showed a singlet peak because of the S–CH moiety of S-cyclohexane at 3.66–3.37 ppm, as well as the other 10 protons of the cyclohexane moiety in the aliphatic region or the aromatic peaks of the thiophenol moiety in the aromatic region.
Scheme 1. Synthesis of the designed thiocarboxylic acid esters of NSAIDs.
Biological evaluation
Antitumor evaluation using MTT assay
The synthesised thioesters were evaluated for their in vitro antitumor effects using the standard 3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) methodCitation47–50 against a panel of four human tumour cell lines: hepatocellular carcinoma cell line (HepG2), breast cancer cell line (MCF-7), colon cancer cell line (HCT-116) and colorectal cancer cell line (Caco-2). The antitumor activities of the newly synthesized compounds 1a,b–8a,b compared with those of the reference drugs, 5-FU, afatinib and celecoxib are shown in . In the present study, the active compounds revealed a characteristic selectivity potential, in addition being broad-spectrum compounds. With respect to the selectivity against the hepatocellular carcinoma cell line (HepG2), the thioesters 2b, 3b, 5b, 7a,b and 8a showed potent antitumour activity with IC50 values of 7.35–19.74 μM, while thioesters 5a, 6a and 8b showed moderate antitumour activity against HepG2 cell line, with IC50 values 22.30–36.75 μM, compared to the reference drugs 5-FU, afatinib and celecoxib (IC50 = 7.91, 5.40 and 25.60 μM, respectively). Additionally, the MCF-7 cell line showed a high sensitivity to the thioesters 2a, 3b, 6b, 7a,b and 8a, with IC50 values of 6.11–17.10 μM, whereas the thioesters 4b, 5a,b and 8b showed a moderate antitumour activity, with IC50 values 28.90–46.52 μM, compared with the reference drugs 5-FU, afatinib and celecoxib (IC50 = 5.43, 7.1 and 31.28 μM, respectively). Moreover, the colon cancer cell line (HCT-116) had a strong susceptibility to the thioesters 2a, 7a and 8a with IC50 values of 9.73–18.71 μM, but a moderate susceptibility to the thioesters 3b, 5b, 6a, 7b and 8b, with IC50 values of 23.76–46.92 μM compared to the reference drugs, 5-FU, afatinib and celecoxib (IC50 = 5.32, 6.20 and 29.54 μM, respectively). Additionally, the thioesters 2b, 6a, 7a and 8a exhibited a strong antitumour activity against colorectal cancer cell line (Caco-2), with IC50 values of 10.16–21.73 μM, whereas the thioesters 5b, 6b, 8b and 9b showed a moderate antitumour activity, with IC50 values of 26.81–43.79 μM, when compared to the reference drugs 5-FU, afatinib and celecoxib (IC50 = 6.85, 7.70 and 42.74 μM, respectively).
Table 1. In vitro antitumor activity of 5-fluorouracil, afatinib, celecoxib, and the tested compounds.
The thioesters 3b and 7b displayed a broad-spectrum antitumor activity against the HepG2 cell line (IC50 = 10.52 and 14.91 μM respectively) and MCF-7 cell line (IC50 = 13.73 and 17.10 μM respectively), while the thioester 6a showed a broad-spectrum antitumor activity against the MCF-7 cell line and Caco-2 cell line (IC50 = 6.11 and 10.16 μM, respectively). Additionally, the thioesters 2b, 7a and 8a showed strong antitumour activities against the HepG2 cell line (IC50 = 9.36, 7.86 and 7.35 μM, respectively), MCF-7 cell line (IC50 = 11.86, 9.65 and 8.62 μM respectively), HCT-116 cell line (IC50 = 18.71, 14.58 and 9.73 μM, respectively), and Caco-2 cell line (IC50 = 21.73, 18.13 and 15.44 μM, respectively). On the other hand, the thioesters 1a, b, 2b, 3a, 4a and 6b showed a weak antitumor activity with IC50 values from 48.11 to >100 μM.
In vitro COX inhibition assay
Compounds that showed promising and potent antitumor activities () were subjected to in vitro COX-1/COX-2 inhibition assays. As indicated in , seven compounds were selected for in vitro COX-1/COX-2 evaluation (). IC50 (represented in μM) (The half-maximal inhibitor concentration) values were determinedCitation56–59, and the SI values were calculatedCitation51,Citation52,Citation55 as IC50 (COX-1)/IC50 (COX-2). Interestingly, some of the tested compounds selectively inhibited COX-2 (IC50 = 0.20–0.69 μM); however, all the tested compounds did not inhibit COX-1 (IC50 > 50 μM).
Table 2. In vitro COX-1/COX-2 enzyme inhibition assay.
Compounds 2b, 3b, 7b and 8b possessed good COX-2 inhibitory activity with IC50 values of 0.66, 0.69, 0.49 and 0.60 μM, and SI values of >75.8, 72.5, 102.0 and 83.3, respectively, comparable to that of celecoxib (IC50 = 0.16 μM, COX-2 SI: > 312.5). Furthermore, compounds 6a, 7a and 8a showed a potent selective inhibition of COX-2, with IC50 values of 0.25, 0.22 and 0.20 μM, and SI values of >200, 227 and 250, respectively, compared to those of celecoxib (IC50 = 0.16 μM, COX-2 SI: > 312.5). The structure–activity relationships of the COX inhibition assays revealed the following: (i) substituted thiophenyl derivatives, such as compounds 6a, (IC50 = 0.25 μM, COX-2 SI: >200), 7a (IC50 = 0.22 μM, COX-2 SI: >227) and 8a (IC50 = 0.20 μM, COX-2 SI: >250), were more effective COX-2 inhibitors than substituted thiocyclohexyl derivatives, such as compounds 2b (IC50 = 0.66, μM, COX-2 SI: >75.8), 3b (IC50 = 0.69 μM, COX-2 SI: >72.5), 7b (IC50 = 0.49 μM, COX-2 SI: >102.0) and 8b (IC50 = 0.60 μM, COX-2 SI: >83.3); (ii) S-phenyl-2–(5-fluoro-2-methyl-1–(4-(methylsulfinyl)benzylidene)-1H-inden-3-yl)ethanethioate (8a) was more effective than S-phenyl-2–(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)ethanethioate (7a) and S-phenyl 2–(2-((2,6-dichlorophenyl)amino)phenyl)ethanethioate (6a), while the latter was less effective than compound 7a; (iii) the substituted thiocyclohexyl derivative, S-cyclohexyl 2–(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)ethanethioate (7b) was more effective than S-cyclohexyl-2–(5-fluoro-2-methyl-1–(4-(methylsulfinyl)benzylidene)-1H-inden-3-yl)ethanethioate (8b), S-cyclohexyl 2–(3-benzoylphenyl)propanethioate (2b) and S-cyclohexyl 2–(4-isobutylphenyl)propanethioate (3b).
In vitro kinase assay
Accordance to the cytotoxicity activity of the newly synthesized compounds (), six compounds were selected for further mechanistic investigations about the kinases, EGFR, HER2, HER4 and cSrc. The results of kinase inhibition assays indicated that compounds 2a, 3b, 6a, 7a, 7b and 8a showed limited activities against the kinase enzymes. As shown in , all the compounds showed no or weak activities against HER2, HER4 and cSrc, as indicated by their % inhibition when used at a concentration of 10 μM (% inhibition = ∼0–10%), comparable to the 81–100% inhibition of the reference drug staurosporine, used at a concentration of 1 μM ().
Table 3. % inhibitory effect of the compounds on kinase activities.
Docking studies
To highlight the inhibition selectivity of different core analogues towards the COX-2 enzyme, automated docking studies were carried out using the MOE 2008.10 programCitation54. The scoring functions, hydrogen bonds and hydrophobic interactions formed with the surrounding amino acids are used to predict the binding modes, the energy of interaction and orientation of the docked compounds at the active sites of the COX-2 enzyme (). The protein–ligand complex was constructed based on the X-ray structure of COX-2, with its bound inhibitor SC-558, which was available through the RCSB Protein Data Bank (PDB entry 1CX-2)Citation60. The active site of the enzyme was defined to include residues within a 10.0-Å radius around any of the inhibitor atoms. This active pocket consisted of amino acid residues such as arginine (Arg510), histidine (His90), glutamine (Gln192) or tyrosine (Tyr355), arginine (Arg120), valine (Val523) and methionine (Met535), which play fundamental roles by forming H-bonds and hydrophobic interactions (). In order to verify the reproducibility of the docking calculations, the cocrystallised ligand SC-558 was extracted from the complex and submitted for one-ligand run calculation. This reproduced 20 top scoring conformations falling within a root-mean-square deviation (rmsd) value between 0.4 Å and 2.0 Å, from the bound X-ray conformation for the COX-2 enzyme, suggesting that this method is valid enough to be used for docking studies of other compounds.
Figure 2. The 2D and 3D orientations of the docked compounds 6a (upper panel), 7a (middle panel), and 8a (lower panel) in COX-2 active pocket (H bonds and hydrophobic interactions are shown as dashed green lines or arrows).
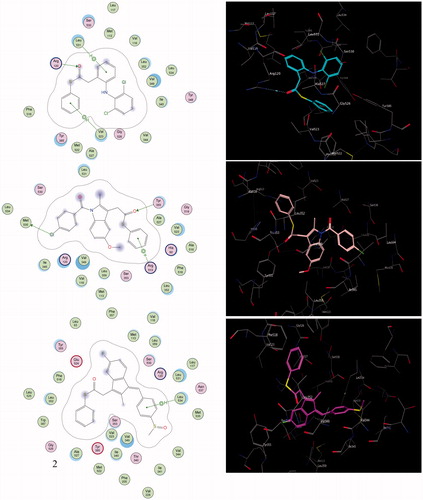
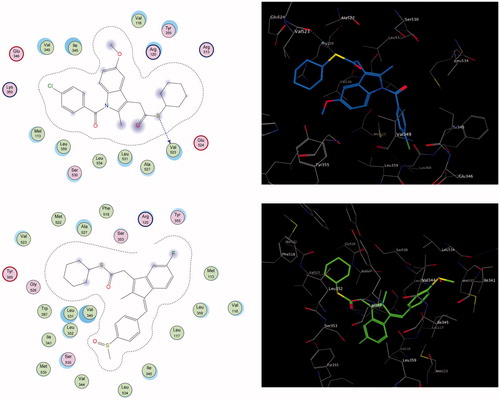
Figure 3. The 2D and 3D putative binding complexes of compound 7b (upper panel) and compound 8b (lower panel) within the binding pocket of COX-2 enzyme.
The present work is based on a comparative study to define the selectivity of most active COX-2 inhibitors, such as the thioester derivatives 6a, 7a, 7b, 8a and 8b of well-known and well-established NSAIDs, namely diclofenac, indomethacin and sulindac, by exploring their docking and complementarity to the COX-2 binding site. Generally, the results of the docking study indicated that the thioesters based on indomethacin, sulindac and diclofenac scaffolds matched perfectly with the configuration of the T-shaped merged COX-2 binding site, which easily accommodated the wide bulk SC-558 inhibitor.
Compounds 8a (IC50 = 0.20 µM), 7a (IC50 = 0.22 µM) and 6a (IC50 = 0.25 µM) were the most active analogues; they showed the highest recognition at the COX-2 binding site, which is consistent with the experimental results of the selectivity index obtained from the COX-2 assay (). Compound 8a was shown to have a unique binding configuration (, lower panel). The phenyl thioester of sulindac showed promising binding affinity and proper complementarity, because the E-conformer allows the crest-configuration to embed properly within the merged active site of the 5-flouroindenyl group, via proper hydrophobic interactions with the amino acids of the merged cleft active site. The thiocarbonyl function was impressively recognized with the polar amino acid Glu524. The two terminal phenyl groups are surrounded by hydrophobic amino acid analogs, where Leu534 showed an aromatic-aromatic interaction with the 4-methylsulfinylphenyl group. The terminal 4-methylsulfinyl fragment enhanced the strategic function that showed proper complementarity with the groove wall residues via both Val344 and Val349. According to the selectivity index and computational binding, the hydrogen-bonded compound 8a was considered the most promising selective lead.
Moreover, compound 7a was held by one hydrogen bond with Tyr355, via its carbonyl thioester, apart from the electrostatic interaction between the chloro-function and the mercapto moiety of the corresponding Met535 (, middle panel). Aromatic recognition also was observed between the aromatic phenyl thioester and the side chain of Arg513.
Additionally, the docking studies of compound 6a revealed outstanding interactions with one of the essential active-site Arg120 residues formed via proper hydrogen bonding (, upper panel). The two aminophenyl and dichlorophenyl groups augment the aromatic-aromatic interaction with a series of seven hydrophobic amino acids, Leu531, Met113, Val116, Leu352, Val349 and Ile345, arranged in a continued chain, lining the wall of the cleft. However, because of the NH-amino group being embedded inwards and away from the surrounding residues, it does not interact with the active-site amino acids, owing to the bulkiness of the two phenyl substituents. The thiophenyl function protruded towards Val523, showing notable improvement in the net lipophilic stabilisation (, upper panel).
In comparison to the aforementioned derivatives, compounds 7b and 8b showed moderately selective inhibition towards COX-2. Compound 7b revealed distinct binding wherein the cyclohexyl group was merged with the side-pocket, and the thioester function was exposed to the surrounding binding residues for interaction with the conserved amino acids Val523 via proper hydrogen bonding. Additionally, methoxy oxygen was recognized by a single conventional hydrogen bond with the conserved Arg120 (, upper panel).
Similarly, the cyclohexyl group of compound 8b was merged with the side-pocket, and the ester function was exposed to the surrounding binding residues, to be oriented ahead of the polar amino acids, Ser353 and Arg120 (, lower panel). Along the lining wall of the pocket, all the hydrophobic amino acids are oriented complementarily with the hydrophobic-facing groups indene, methylene, and the terminal phenyl. From another site, the following hydrophobic amino acids are stuffed properly and sandwiched between the cyclohexyl ring and the terminal phenyl group.
Conclusions
A new series of thioesters based on NSAID scaffolds were synthesized and evaluated for their in vitro antitumor effects against a panel of four human tumour cell lines, namely HepG2, MCF-7, HCT-116 and Caco-2 using MTT assay. The thioesters 2b, 3b, 5b, 7a,b, and 8a showed potent antitumor activity against HepG2 cell line, while thioesters 2a, 3b, 6b, 7a,b and 8a showed high sensitivity against MCF-7 cell line with IC50 values of 7.35–19.74 μM and 6.11–17.10 μM, respectively, compared with the reference drug, 5-FU, afatinib and celecoxib (IC50 = 7.91, 5.40, 25.60 μM and 5.43, 7.10, 31.28 μM, respectively). Additionally, thioesters 2a, 3b, 6b, 7a,b and 8a revealed the most antitumor activity against MCF-7 cell line, whereas HCT-116 has strong susceptibility to thioesters 2a, 7a and 8a with IC50 values of 6.11–17.10 μM and 9.73–18.71 μM, respectively, compared with the reference drugs 5-FU, afatinib and celecoxib (IC50 = 5.43, 7.10, 31.28 μM and 6.85, 7.70, 42.74 μM, respectively). Additionally, the thioesters 2b, 7a and 8a showed strong antitumor activities against HepG2 (IC50 ≅ 7.35–9.36 μM), MCF-7 (IC50 ≅ 8.62–11.86 μM), HCT-116 (IC50 ≅ 9.73–18.71 μM), and Caco-2 cell line (IC50 ≅ 15.44–21.73 μM). Thioesters 3b and 7b have broad-spectrum antitumour activity against HepG2 and MCF-7 cell lines (IC50 ≅ 10.52–17.10 μM), while thioesters 6a showed broad-spectrum antitumor activity against the MCF-7 and Caco-2 cell lines (IC50 = 6.11 and 10.16 μM). According to their cytotoxicity activities, compounds 2b, 3b, 6a, 7a, 7b, 8a and 8b were carefully chosen for mechanistic studies involving COX enzyme inhibition and kinase assays. In vitro COX-1/COX-2 enzyme inhibition assay results indicated that compounds 2b, 3b, 6a, 7a, 7b, 8a and 8b selectively inhibited the COX-2 enzyme (IC50 = ∼0.20–0.69 μM), and SI values of (>72.5–250), compared to celecoxib (IC50 = 0.16 μM, COX-2 SI: > 312.5). Although all the tested compounds did not selectively inhibit the COX-1 enzyme (IC50 > 50 μM). On the other hand, EGFR, HER2, HER4 and cSrc kinase inhibition assays were evaluated at a concentration of 10 μM. The kinase inhibition assays indicated that compounds 2a, 3b, 6a, 7a, 7b and 8a showed no activity to negligible activity (% inhibition = ∼0–10%), comparable to an 81–100% inhibition of the reference drug Staurosporine at a concentration of 1 μM.
Supplemental Material
Download Zip (2.4 MB)Acknowledgements
The authors express their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the research group project No. RGP-163. The authors thank the Deanship of Scientific Research and RSSU at King Saud University for their technical support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Sultan I, Qaddoumi I, Yaser S, et al. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol 2009;27:3391–7.
- Vane JR, Botting RM. Mechanism of action of antiinflammatory drugs. Int J Tissue React 1998;20:3–15.
- Duffy CP, Elliott CJ, O’Connor RA, et al. Enhancement of chemotherapeutic drug toxicity to human tumour cells in vitro by a subset of non-steroidal anti-inflammatory drugs (NSAIDs). Eur J Cancer 1998;34:1250–9.
- Eberhart CE, Coffey RJ, Radhika A, et al. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994;107:1183–8.
- Brown JR, DuBois RN. COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol 2005;23:2840–55.
- Husain SS, Szabo IL, Tamawski AS. NSAID inhibition of GI cancer growth: clinical implications and molecular mechanisms of action. Am J Gastroenterol 2002;97:542–53.
- Alberts DS, Hixson L, Ahnen D, et al. Do NSAIDs exert their colon cancer chemoprevention activities through the inhibition of mucosal prostaglandin synthetase?. J Cellular Biochem Suppl 1995;18–23.
- Piazza GA, Keeton AB, Tinsley HN, et al. NSAIDs: old drugs reveal new anticancer targets. Pharmaceuticals (Basel, Switzerland) 2010;3:1652–67.
- Hanif R, Pittas A, Feng Y, et al. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol 1996;52:237–45.
- Elder DJ, Halton DE, Hague A, Paraskeva C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clin Cancer Res 1997;3:1679–83.
- Gupta SC, Sung B, Prasad S, et al. Cancer drug discovery by repurposing: teaching new tricks to old dogs. Trends Pharmacol Sci 2013;34:508–17.
- Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol 2002;3:166–74.
- Piazza GA, Keeton AB, Tinsley HN, et al. A novel sulindac derivative that does not inhibit cyclooxygenases but potently inhibits colon tumor cell growth and induces apoptosis with antitumor activity. Cancer Prevent Res 2009;2:572–80.
- Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst 2002;94:252–66.
- Kim MS, Kim JE, Lim DY, et al. Naproxen induces cell-cycle arrest and apoptosis in human urinary bladder cancer cell lines and chemically induced cancers by targeting PI3K. Cancer Prevent Res 2014;7:236–45.
- Aytac PS, Durmaz I, Houston DR, et al. Novel triazolothiadiazines act as potent anticancer agents in liver cancer cells through Akt and ASK-1 proteins. Bioorg Med Chem 2016;24:858–72.
- Sever B, Altintop MD, Kus G, et al. Indomethacin based new triazolothiadiazine derivatives: Synthesis, evaluation of their anticancer effects on T98 human glioma cell line related to COX-2 inhibition and docking studies. Eur J Med Chem 2016;113:179–86.
- Mathew B, Hobrath JV, Connelly MC, et al. Diverse amide analogs of sulindac for cancer treatment and prevention. Bioorg Med Chem Lett 2017;27:4614–21.
- Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000;342:1946–52.
- Amir M, Agarwal HK. Role of COX-2 selective inhibitors for prevention and treatment of cancer. Die Pharmazie 2005;60:563–70.
- Koki AT, Masferrer JL. Celecoxib: a specific COX-2 inhibitor with anticancer properties. Cancer Contr 2002;9(2 Suppl):28–35.
- Hara A, Yoshimi N, Niwa M, et al. Apoptosis induced by NS-398, a selective cyclooxygenase-2 inhibitor, in human colorectal cancer cell lines. Jpn J Cancer Res 1997;88:600–4.
- Erickson BA, Longo WE, Panesar N, et al. The effect of selective cyclooxygenase inhibitors on intestinal epithelial cell mitogenesis. J Surg Res 1999;81:101–7.
- Liu XH, Yao S, Kirschenbaum A, Levine AC. NS398, a selective cyclooxygenase-2 inhibitor, induces apoptosis and down-regulates bcl-2 expression in LNCaP cells. Cancer Res 1998;58:4245–9.
- Hsu AL, Ching TT, Wang DS, et al. The cyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking Akt activation in human prostate cancer cells independently of Bcl-2. J Biol Chem 2000;275:11397–403.
- El-Azab AS, Al-Omar MA, Alaa A-M, et al. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: molecular docking study. Eur J Med Chem 2010;45:4188–98.
- Al-Obaid A, Abdel-Hamide S, El-Kashef H, et al. Substituted quinazolines, part 3. Synthesis, in vitro antitumor activity and molecular modeling study of certain 2-thieno-4(3H)-quinazolinone analogs. Eur J Med Chem 2009;44:2379–91.
- Al-Omary FA, Abou-Zeid LA, Nagi MN, et al. Non-classical antifolates. Part 2: synthesis, biological evaluation, and molecular modeling study of some new 2,6-substituted-quinazolin-4-ones. Bioorg Medicinal Chem 2010;18(8):2849–63.
- El-Azab AS, ElTahir KE. Design and synthesis of novel 7-aminoquinazoline derivatives: antitumor and anticonvulsant activities. Bioorg Med Chem Lett 2012;22:1879–85.
- Alanazi AM, Alaa A-M, Al-Suwaidan IA, et al. Design, synthesis and biological evaluation of some novel substituted quinazolines as antitumor agents. Eur J Med Chem 2014;79:446–54.
- Al-Suwaidan IA, Alanazi AM, Alaa A-M, et al. Design, synthesis and biological evaluation of 2-mercapto-3-phenethylquinazoline bearing anilide fragments as potential antitumor agents: molecular docking study. Bioorg Med Chem Lett 2013;23:3935–41.
- Alanazi AM, Al-Suwaidan IA, Alaa A-M, et al. Design, synthesis and biological evaluation of some novel substituted 2-mercapto-3-phenethylquinazolines as antitumor agents. Med Chem Res 2013;22:5566–77.
- Al-Suwaidan IA, Abdel-Aziz AA-M, Shawer TZ, et al. Synthesis, antitumor activity and molecular docking study of some novel 3-benzyl-4 (3H) quinazolinone analogues. J Enzyme Inhib Med Chem 2016;31:78–89.
- Alanazi AM, Abdel-Aziz AA, Shawer TZ, et al. Synthesis, antitumor and antimicrobial activity of some new 6-methyl-3-phenyl-4(3H)-quinazolinone analogues: in silico studies. J Enzyme Inhib Med Chem 2016;31:721–35.
- Mohamed MA, Ayyad RR, Shawer TZ, et al. Synthesis and antitumor evaluation of trimethoxyanilides based on 4 (3H)-quinazolinone scaffolds. Eur J Med Chem 2016;112:106–13.
- El-Azab AS, Al-Dhfyan A, Abdel-Aziz AA, et al. Synthesis, anticancer and apoptosis-inducing activities of quinazoline-isatin conjugates: epidermal growth factor receptor-tyrosine kinase assay and molecular docking studies. J Enzyme Inhib Med Chem 2017;32:935–44.
- El-Azab AS, Abdel-Aziz AA, Ghabbour HA, Al-Gendy MA. Synthesis, in vitro antitumour activity, and molecular docking study of novel 2-substituted mercapto-3-(3,4,5-trimethoxybenzyl)-4(3H)-quinazolinone analogues. J Enzyme Inhib Med Chem 2017;32:1229–39.
- Abdel-Aziz AA, El-Azab AS, El-Subbagh HI, et al. Design, synthesis, single-crystal and preliminary antitumor activity of novel arenesulfonylimidazolidin-2-ones. Bioorg Med Chem Lett 2012;22:2008–14.
- Abdel-Aziz AA, El-Azab AS, Alanazi AM, et al. Synthesis and potential antitumor activity of 7-(4-substituted piperazin-1-yl)-4-oxoquinolines based on ciprofloxacin and norfloxacin scaffolds: in silico studies. J Enzyme Inhib Med Chem 2016;31:796–809.
- Al-Suwaidan IA, Abdel-Aziz NI, El-Azab AS, et al. Antitumor evaluation and molecular docking study of substituted 2-benzylidenebutane-1,3-dione, 2-hydrazonobutane-1,3-dione and trifluoromethyl-1H-pyrazole analogues. J Enzyme Inhib Med Chem 2015;30:679–87.
- Alanazi AM, El-Azab AS, Al-Swaidan IA, et al. Single-crystal, in vitro antitumor evaluation and molecular docking of 3-substitued 5,5-diphenylimidazolidine-2,4-dione derivatives. Med Chem Res 2013;22:6129. 42.
- El-Azab AS, Alanazi AM, Abdel-Aziz NI, et al. Synthesis, molecular modeling study, preliminary antibacterial, and antitumor evaluation of N-substituted naphthalimides and their structural analogues. Med Chem Res 2013;22:2360–75.
- El-Azab AS, Abdel-Aziz AA-M. An efficient synthesis of thioesters via tfa-catalyzed reaction of carboxylic acid and thiols: remarkably facile C–S bond formation. Phosphorus, Sulfur, Silicon Related Elements 2012;187:1046–55.
- Chang C-S, Tsai S-W, Lin C-N. Enzymatic resolution of (RS)-2-arylpropionic acid thioesters by Candida rugosa lipase-catalyzed thiotransesterification or hydrolysis in organic solvents. Tetrahedron: Asymmetry 1998;9:2799–807.
- Hansen AB, Senning A. Chemical feasibility studies concerning potential prodrugs of acetylsalicylic acid. Acta Chem Scand B 1983;37:4.
- Ray RE. Thio-ester of 1 (p-chlorobenzoyl)-5-methoxy-2-methylindole-3-acetic acid. Google Patents 1978; 4,119,638, 1–14.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986;89:271–7.
- Vega-Avila E, Pugsley MK. An overview of colorimetric assay methods used to assess survival or proliferation of mammalian cells. Proc Western Pharmacol Soc 2011;54:10–4.
- Mauceri HJ, Hanna NN, Beckett MA, et al. Combined effects of angiostatin and ionizing radiation in antitumour therapy. Nature 1998;394:287–91.
- El-Sayed MA, Abdel-Aziz NI, Abdel-Aziz AA, et al. Design, synthesis, and biological evaluation of substituted hydrazone and pyrazole derivatives as selective COX-2 inhibitors: molecular docking study. Bioorg Med Chem 2011;19:3416–24.
- Alanazi AM, El-Azab AS, Al-Suwaidan IA, et al. Structure-based design of phthalimide derivatives as potential cyclooxygenase-2 (COX-2) inhibitors: anti-inflammatory and analgesic activities. Eur J Med Chem 2015;92:115–23.
- http://bpsbioscience.com/kinase/assay-kits (accessed 22 Jun 2017).
- MOE 200810 of Chemical Computing Group Inc. Available from: http://wwwchemcompcom/ (accessed 20 Jan 2018).
- El-Sayed MA, Abdel-Aziz NI, Abdel-Aziz AA, et al. Synthesis, biological evaluation and molecular modeling study of pyrazole and pyrazoline derivatives as selective COX-2 inhibitors and anti-inflammatory agents. Part 2. Bioorg Med Chem 2012;20:3306–16.
- Abdel-Aziz AA, Abou-Zeid LA, ElTahir KE, et al. Design, synthesis of 2,3-disubstitued 4(3H)-quinazolinone derivatives as anti-inflammatory and analgesic agents: COX-1/2 inhibitory activities and molecular docking studies. Bioorg Med Chem 2016;24:3818–28.
- Al-Suwaidan IA, Alanazi AM, El-Azab AS, et al. Molecular design, synthesis and biological evaluation of cyclic imides bearing benzenesulfonamide fragment as potential COX-2 inhibitors. Part 2. Bioorg Med Chem Lett 2013;23:2601–5.
- Uddin MJ, Rao PN, Knaus EE. Design and synthesis of acyclic triaryl (Z)-olefins: a novel class of cyclooxygenase-2 (COX-2) inhibitors. Bioorg Med Chem 2004;12:5929–40.
- Alaa A-M, Abou-Zeid LA, ElTahir KEH, et al. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibitory activities and molecular docking studies of substituted 2-mercapto-4 (3H)-quinazolinones. Eur J Med Chem 2016;121:410–21.
- Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996;384:644–8.