
Abstract
Carbonic anhydrases (CAs, EC 4.2.1.1) are ubiquitous metalloenzymes, grouped into seven different classes, which catalyze the reaction of CO2 hydration to bicarbonate and protons. All of the fifteen human isoforms reported to date belong to the α-class and contain zinc as a cofactor. The structure of human Zn,Cu-CA II has been solved which contains a copper ion bound at its N-terminal, coordinated to His4 and His64. In the active site a dioxygen molecule is coordinated to the zinc ion. Since dioxygen is a rather unexpected CA ligand, molecular dynamics (MD) simulations were performed which suggested a superoxide character of the zinc bound O2.
1. Introduction
One of the most abundant zinc enzymes in the blood is carbonic anhydrase (CA, EC 4.2.1.1), which catalyzes a simple but essential reaction in all life kingdoms, CO2 hydration to bicarbonate and protonsCitation1–4. This reaction, or the three chemical entities involved in it, carbon dioxide, bicarbonate and protons, are important for the pH regulation and homeostasis of the organism, CO2 and HCO3− transport in several biosynthetic processes, for the production of body fluids, bone resorption, tumorigenicity, and other physiological processes in vertebrates, whereas in some bacteria, plants and algae they are involved in photosynthetic processesCitation5–7.
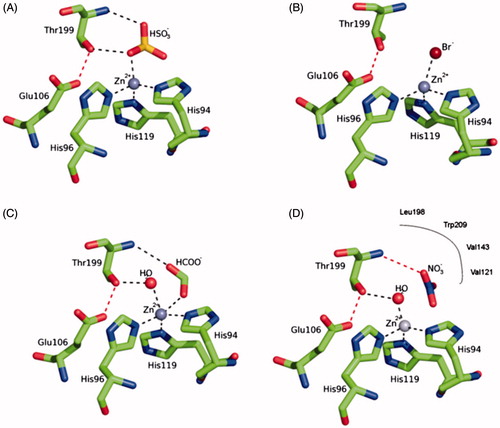
The catalytic mechanism of CAs is understood in detailCitation1–3. In all CA classes known to date (α-, β-, γ-, δ-, ζ-, η- and θ-CAs) a metal hydroxide species (L3-M2+-OH-) of the enzyme is the catalytically active species, acting as a strong nucleophile (at neutral pH) on the CO2 molecule bound in a hydrophobic pocket nearbyCitation1–6. This hydroxide species is generated from a water coordinated to the metal ion, which is found at the bottom of the active site cavity. The active center normally comprises M(II) ions in tetrahedral geometry, with three protein ligands (L) in addition to the water molecule/hydroxide ion, although Zn(II) or Co(II) were also observed in trigonal bipyramidal or octahedral coordination geometries, at least in γ-CAsCitation7. In many enzymes, generation of the hydroxide species from the metal-coordinated water one, is the rate determining step of the catalytic turnover, which for some α- and ζ-CAs achieves kcat/KM values >108 M−Citation1 × s−Citation1, making CAs among the most effective catalysts known in natureCitation1–3. The metal ion ligands are three His residues in α-, γ-, and δ-CAs or one His and two Cys residues in β- and ζ-CAsCitation1–7 .The inhibition and activation of CAs are well understood processes, with most types of inhibitors binding to the metal centerCitation8, whereas the activators bind at the entrance of the active site cavity where they participate in the proton shuttling between the metal-coordinated water molecule and the environmentCitation9. Inorganic simple anions are an important class of CA inhibitors (CAIs)Citation10. Both metal-complexing anions (such as cyanide, thiocyanate, hydrogen sulfide, etc.) as well as anions showing less affinity for metal ions in solution (such as nitrate, bisulfite, sulfate, sulfamate and sulfamidate) are known to inhibit these metalloenzymesCitation10, and for many of them detailed X-ray crystallographic studies allowed a profound understanding of the inhibition mechanismCitation10–13. As shown in , bisulfite binds to Zn(II) in a tetrahedral geometry (), bromide in a distorted tetrahedral geometry (), formate in a trigonal bipyramidal geometry () and nitrate is one of the few inhibitors non-coordinated to the zinc but binding very nearby to the catalytic metal ion ()Citation10–14.
Figure 1. Structure of hCA II complexed with (A) bisulfite (tetrahedral geometry of Zn(II)), (B) bromide (distorted tetrahedral geometry of Zn(II)), (C) formate (trigonal bipyramidal geometry of Zn(II)) and (D) nitrate (inhibitor non-coordinated to the zinc)Citation10–13. The three protein zinc ligands (His94, 96 and 119) as well as the other two amino acid residues involved in the catalytic mechanism and binding of inhibitors, Glu106 and Thr199, are also evidenced.Citation1,Citation10.
However, up until now, oxygen was never evidenced as a possible ligand of zinc in the CAs, except for one caseCitation15 which has been poorly understood and less discussed, being obtained from the apo-enzyme which has been reconstituted with diverse metal ions (e.g. Co2+). Here we report an interesting finding: when Cu(II) coordinates with His64 (an amino acid residue crucial for the catalytic cycle, as it acts as a proton shuttle between the water coordinated to the zinc and the environment)Citation16, oxygen was found bound to Zn(II) within the active site of human (h) CA II, the physiologically dominant mammalian CA isoformCitation1.
2. Material and methods
2.1. Crystallization and X-ray data collection
Crystals of native hCA II were obtained using the hanging drop vapor diffusion method. 2 µl of a solution 10 mg/ml protein solution in water were mixed with 2 µl of a solution containing 2.4 M ammonium sulfate, 50 mM Tris-HC1 pH 8.0, 2 mM HgCl2 and were equilibrated against the same solution at 296 K. Crystals grew in two weeks. The metal derivative was prepared by soaking the native crystals in 3 M ammonium sulfate, 50 mM Tris pH 8.0 and 2 mM CuSO4 for two days. Crystals were flash-frozen at 100 K using a solution obtained by adding 25% (v/v) glycerol to the mother liquor solution as cryoprotectant. X-ray data were collected at the Centro di Cristallografia Strutturale (CRIST) in Florence using an Oxford Diffraction instrument equipped with a sealed tube Enhance Ultra (Cu) and a Onyx CCD detector. Data were integrated and scaled using the program XDSCitation16. Data processing statistics are showed in .
Table 1. Summary of Data Collection and Atomic Model Refinement Statistics.Table Footnotea
2.2. Structure determination and refinement
The crystal structure of hCA II (PDB ID 4FIK) without solvent molecules and other heteroatoms was used to obtain initial phases using Refmac5Citation17. 5% of the unique reflections were selected randomly and excluded from the refinement data set for the purpose of Rfree calculations. Refinements proceeded using normal protocols of positional, isotropic atomic displacement parameters alternating with manual building of the model using COOTCitation18. Solvent molecules were introduced automatically using the program ARPCitation19. The quality of the final model was assessed with COOT and RampageCitation20. Crystal parameters and model refinement data are summarized in . Atomic coordinates were deposited in the Protein Data Bank (PDB ID 5EOI). Graphical representations were generated with ChimeraCitation21.
2.3. Computational studies
The structure of carbonic anhydrase in complex with the O2 ligand was taken from the present work (PDB ID 5EOI). Standard protonation states were used for all residues. The amber ff99SB-ILDN and GAFF forcefieldsCitation22,Citation23, were employed. The protein was solvated with TIP3P water moleculesCitation24,Citation25, in a cubic box with smallest solute-edge distance of 12 Å. Cl− counterions were added up to system charge neutrality. The system was locally optimized and equilibrated for 400 ps by classical molecular dynamics (MD) in the NVT ensemble, using the NAMD 2.9 codeCitation26. Temperature was kept at target value of 300 K by Langevin dynamics with damping parameter set at 5 ps−Citation1. The cutoff for the non-bonded interaction was set to 12 Å. Bonds were kept rigid and the integration timestep used was 2 fs. The protein region in the QM box included the whole residues Thr199, Glu106, His96, His94, His119, the Zn2+ ion and the ligand O2. During classical dynamics equilibration, this region was kept fixed. The system geometry after equilibration was used as the starting conformation for QM/MM (), which was run using the cp2k 4.1 codeCitation27. All water molecules less than 8 Å away from the Zn2+ ion were included in the QM subsystem. The orthorhombic QM box was constructed so that in the initial state the minimum solute-edge distance was larger than 8 Å. Mechanical embedding was used for the QM/MM interface. Goedecker-Teter-Hutter pseudopotentialsCitation28 and double zeta valence basis setsCitation29 with one polarization function were used for all elements. The plane wave cutoff was set to 400 Rydberg. MD simulations were performed in the NVT ensemble, using a timestep of 0.5 fs. The CSVR methodCitation30 was used to keep the temperature at the target value of 300 K. The system was initially run for 1 ps using a CSVR time constant of 0.01 ps. The time constant was then switched to 0.1 ps for further 1 ps production QM/MM run. Smooth particle mesh Ewald of order 6 and with 1 grid point per Å was used. The BLYP functionalCitation31 with DFT-D3 correctionsCitation32 was employed. The carboxylic acid of Glu106 and the catalytic Zn2+ were given a formal charge of −1 and +2, respectively. Depending on the formal charge attributed to the O2 binder, the total charge of the QM region was −1 (neutral binder) or 0 (charged binder). Average distances and standard deviations were computed using the last 500 fs of the simulations. A smaller full QM model was built by considering only the Zn2+, O2 and three imidazole binders replacing His96, His94 and His119. Geometry optimization of this model was carried out in vacuo using a cubic box with side length of 20 Å, up to a force convergence threshold of 2 · 10−4 atomic units. Calculations in the small model were performed at both BLYP and B3LYPCitation33 level of theory. The O2/Zn2+ and O2−/Zn2+ bonding distance differences between the BLYP and B3LYP approach were less than 0.05 Å. Our results for the gas phase model refer to the B3LYP calculations. All QM calculations allowed spin polarization. Orbital decomposition of electronic Kohn-Sham states was obtained by standard projection methods. Quantum theory of atom in molecule (QTAIM) analysis was performed using the BaderCitation34,Citation35 and the AngyanCitation36 approach for the charge and bond order estimations, respectively.
3. Results and discussion
The crystal structure of the Zn,Cu(II)-hCAII complex was obtained from data collected on a crystal of the native enzyme soaked in a solution containing 2 mM CuSO4. The initial |Fo - Fc| difference electron density maps showed a spherical density near His64 that was attributed to a copper ion. The Cu(II) ion was introduced at 0.75 occupancy and the B factor refined to a value of 22.3. Two protein residues His64 and His4 coordinate the Cu(II) ion, with two water molecules that complete the coordination sphere around the metal at a distance of 2.2 Å. Two other water molecules are at a distance of 2.5 and 2.8 Å from the Cu(II) ion (. Overall, the coordination geometry can be described as distorted octahedral. The distance from the copper ion to the closest (NE2) atom of His64 is 2.1 Å. The corresponding value to His4 is 2.0 Å. The side chain of His4 was modeled in two different conformations. Conversely the side chain of His64, which is actively involved in the proton shuttle and is responsible for converting the Zn-bound water molecule to hydroxide ionCitation16, was modeled as a single conformation, whereas it has been often observed to occupy two different conformations as in the first structure of a copper derivative published by Håkansson et al.Citation15 In this crystallographic studyCitation15, the X-ray structures of hCA II metal-substituted derivatives were obtained, in which diverse metal ions replaced the Zn(II) ion in the active site. In the copper derivative, two Cu(II) ions were observed, the second bound at the same N-terminal site reported in the present structure, being coordinated to His64 and His4. No solvent molecules were reported in the second copper coordination sphereCitation15. This second copper (II) binding site in hCA II has been recently characterized also thermodynamically and by spectroscopic techniquesCitation37–38.
Figure 2. Active site of Zn,Cu-hCA II. The zinc ion (gray sphere) is coordinated by His94, His96 and His119 and a O2 molecule. The Cu(II) (orange sphere) bound to His 64 and His4. An omit Fo − Fc electron density map contoured at 3.5 σ level is also shown.
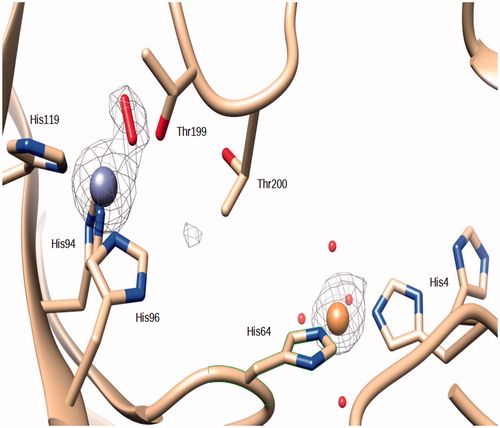
In our Zn,Cu-hCAII structure, as in many others, the zinc ion within the catalytic site is coordinate by His94, His96 and His119. An elongated density in the |Fo − Fc| electron density map was present at the position occupied by the zinc coordinated water in the wild-type enzyme (. The introduction of a water molecule resulted in residual electron density in the |Fo − Fc| map, but molecular oxygen was successfully modeled into that density (O–O distance refined to 1.2 Å, without applying any restraints, and B-factors to 25.2 and 22.3 for the the two atoms at 1.0 occupancy). O2 forms an “end-on” (ηCitation1) complex with the zinc ion. In “end-on” O2 complexes only one oxygen atom is bound to the metal and they have a bent geometry at the proximal oxygen atomCitation39. The coordination of the zinc is tetrahedral, with the oxygen of the O2 molecule at a distance of 1.9 Å. The same oxygen is at a distance of 2.6 Å from the OG1 atom of Thr199 and 2.7 Å from a water molecule. The oxygen atom that is not coordinated is at a distance of 2.8 Å from the Zn(II) ion (.
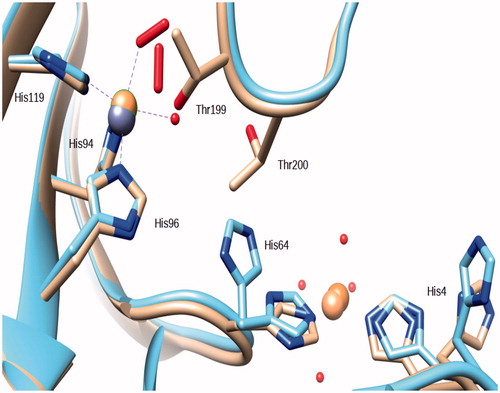
An oxygen molecule bound in the active site of hCA II has already been reported in the structure of the above mentioned copper derivativeCitation15 and also in the cobalt derivative described in the same article (PDB ID 1RZC and 1RZA). The O2 molecule was bound to the Cu(II) and Co(II) ions which replace the zinc in the active site. Contrary to the structure of the Zn,-Cu-hCA II here reported, the Cu(II) and Co(II) ions in the active site maintained also the coordinated water molecule (.
Figure 3. Superposition of the Zn,Cu-hCA II structure (this work) with the copper derivative reported in ref.Citation15. Copper ions are represented as orange spheres, zinc as a gray sphere. It should be observed that the O2 molecules occupy a different position within the coordination sphere of the two hCA II copper derivatives.
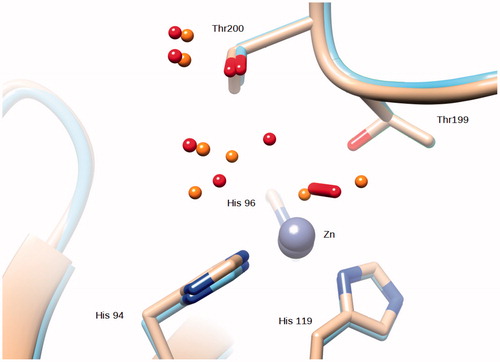
Compared to the native enzyme the O2 molecule occupies the position of the zinc-bound water and of the so-called “deep” water (.
Figure 4. Superposition of the Zn,Cu-hCA II structure (this work) with the native enzyme (PDB ID 2ILI). Zinc ions are represented as gray spheres and water molecues as red spheres (Zn,Cu-hCA II) and orange spheres (native enzyme).
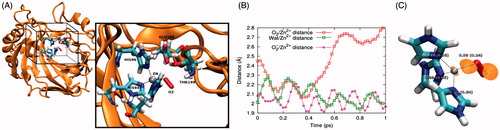
As oxygen is a rather unexpected CA ligand, and it also does not bind to the enzyme which has not been loaded with copper ions at the N-terminal region, we performed a computational study on this system (). During the molecular dynamics (MD) simulations carried out at the quantum mechanics/molecular mechanics (QM/MM) level, the binding distance between the neutral O2 and Zn2+ increased from the starting crystallographic value to above 2.80 Å (). Conversely, one water molecule reached a distance of 2.09 ± 0.06 Å from Zn2+, displacing the O2 molecule from the Zn2+ coordination shell. The resulting geometry is reminiscent of the Co2+-substituted hCA II15 but poorly represents the experimental Zn2+/O2 coordination. However, MD simulations predict O2- to stably bind Zn2+ at 2.03 ± 0.04 Å, in good agreement with the crystallographic structure. Similar binding geometries were obtained by structural optimization of the coordination shell in gas phase. In this case, the O2−/Zn2+ and O2/Zn2+ distances were 1.90 Å and 2.27 Å, respectively. The orbital composition of the HOMO in the O2−/Zn2+ complex (see also ) is predominantly O-derived (91%), but also bears a non-negligible contribution from Zn-derived orbitals (7%). This shows that the extra e- added to the system containing neutral O2 mainly increases the charge on the O2 molecule itself, turning it to a very good extent into O2−, but also contributes to the covalent character of the resulting O2−/Zn2+ bond. This was also quantified by the quantum theory of atoms in molecule (QTAIM) analysis of the small model, predicting the bond order of the O2/Zn2+ and O2−/Zn2+ systems to be 0.24 and 0.58, respectively (. Overall, computational studies suggest that negatively charged molecular oxygen binding to Zn2+ improves both covalent and electrostatic O2−/Zn2+ interactions. Furthermore, from a structural standpoint, the O2−/Zn2+ complex well agrees with the crystallographic outcome, demonstrating that a negative molecular oxygen can better fit into the experimental structure.
Figure 5. (A) Model used for MD simulations at QM/MM level. Water molecules are included in the calculation but are not shown for clarity. The QM box is reported and QM atoms are explicitly reported in the inset. (B) Time evolution of the O2/Zn2+, Wat/Zn2+ and O2−/Zn2+ distance. Here Wat represents the water molecule displacing O2 from the binding site, as described in the main text. (C) Geometry and bonding features of the complex in the small gas phase model. The numbers represent the bond order obtained by the QTAIM approach. Numbers in brackets and without brackets refer to the O2/Zn2+ and O2−/Zn2+ complex, respectively. The orange surface represents the O2−/Zn2+ complex HOMO density isosurface, computed at a density of 0.05 e−/Å3.
4. Conclusions
In native hCA II loaded with Cu(II) ions at the N-terminal region, the copper is coordinated by His4 and His64, probably creating a redox center within the active site, which leads to the transfer of one electron to an oxygen molecule which thereafter replaces the water coordinated to the zinc ion deep within the CA active site, becoming a zinc ligand. Although many details of this process are still poorly understood, our data do not preclude the fact that in biological systems copper-loaded CA may have a role in oxygen transport, apart its well-known role in bicarbonate trafficking between the metabolic sites and the excretion organs (lungs and kidneys). In fact, CA II is highly abundant in the blood with almost micromolar concentrations being reached (the hCA I + hCA II concentration in the blood is 0.2 mMCitation40 but hCA I is the predominant although catalytically less effective isoform). Future studies are thus warranted to better understand the physiological role of the present finding.
Acknowldegment
The authors are grateful to Dr. Giuseppina De Simone for .
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nature Rev Drug Disc 2008;7:168–81. b) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;4:2023–32. c) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. d) Supuran CT, Carbonic Anhydrases and Metabolism. Metabolites 2018;8:E25.
- a) Moya A, Tambutté S, Bertucci A, et al. Carbonic anhydrase in the scleractinian coral Stylophora pistillata: characterization, localization, and role in biomineralization. J Biol Chem 2008;283:25475–84. b) Nocentini A, Vullo D, Del Prete S, et al. Inhibition of the β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa with monothiocarbamates. J Enzyme Inhib Med Chem 2017;3:1064–70. c) Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum–the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;2:4389–96. c) Maresca A, Scozzafava A, Supuran CT. 7,8-disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8.
- a) Xu Y, Feng L, Jeffrey PD, et al. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008;452:56–61. b) Capasso C, Supuran CT. An overview of the alpha-, beta-and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;3:325–32. c) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;1:767–77. d) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68. e) Supuran CT, Capasso C. New light on bacterial carbonic anhydrases phylogeny based on the analysis of signal peptide sequences. J Enzyme Inhib Med Chem 2016;31:1254–60.
- a) Tripp BC, Bell 3rd CB, Cruz F, et al. A role for iron in an ancient carbonic anhydrase. J Biol Chem 2004;279:6683–6. b) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;1:61–88. c) Akocak S, Lolak N, Vullo D, et al. Synthesis and biological evaluation of histamine Schiff bases as carbonic anhydrase I, II, IV, VII, and IX activators. J Enzyme Inhib Med Chem 2017;3:1305–12. d) Angeli A, Vaiano F, Mari F, et al. Psychoactive substances belonging to the amphetamine class potently activate brain carbonic anhydrase isoforms VA, VB, VII, and XII. J Enzyme Inhib Med Chem 2017;3:1253–9. e) Licsandru E, Tanc M, Kocsis I, et al. A class of carbonic anhydrase I - selective activators. J Enzyme Inhib Med Chem 2017;32:37–46.
- a) Schlicker C, Hall RA, Vullo D, et al. Structure and inhibition of the CO2-sensing carbonic anhydrase Can2 from the pathogenic fungus Cryptococcus neoformans. J Mol Biol 2009;385:1207–20. b) Bozdag M, Carta F, Vullo D, et al. Dithiocarbamates with potent inhibitory activity against the Saccharomyces cerevisiae β-carbonic anhydrase. J Enzyme Inhib Med Chem 2016;31:132–6.
- Domsic JF, Avvaru BS, Kim CU, et al. Entrapment of carbon dioxide in the active site of Carbonic anhydrase II. J Biol Chem 2008;283:30766–71.
- Tu C, Tripp BC, Ferry JG, Silverman DN. Bicarbonate as a proton donor in catalysis by Zn(II)- and Co(II)-containing carbonic anhydrases. J Am Chem Soc 2001;123:5861–6.
- a) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;3:345–60. b) Garaj V, Puccetti L, Fasolis, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8. c) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of Action and Preliminary Pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47.
- a) Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73. b) Winum, JY, Temperini C, El Cheikh K, et al. Carbonic anhydrase inhibitors: clash with Ala65 as a means for designing inhibitors with low affinity for the ubiquitous isozyme II, exemplified by the crystal structure of the topiramate sulfamide analogue. J Med Chem 2006;49:7024–31. c) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3.
- De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- Mangani S, Håkansson K. Crystallographic studies of the binding of protonated and unprotonated inhibitors to carbonic anhydrase using hydrogen sulphide and nitrate anions. Eur J Biochem 1992;210:867–71.
- Håkansson K, Carlsson M, Svensson LA, Liljas A. Structure of native and apo carbonic anhydrase II and structure of some of its anion-ligand complexes. J Mol Biol 1992;227:1192–204.
- Håkansson K, Briand C, Zaitsev V, et al. Wild-type and E106Q mutant Carbonic anhydrase complexed with acetate. Acta Cryst D 1994;50:101–4.
- Temperini C, Scozzafava S, Supuran CT. Carbonic anhydrase inhibitors. X-ray crystal studies of the Carbonic anhydrase II-trithiocarbonate adduct-an inhibitor mimicking the sulfonamide and urea binding to the enzyme. Bioorg Med Chem Lett 2010;20:474–8.
- Håkansson K, Wehnert A, Liljas A. X-ray analysis of metal-substituted human carbonic anhydrase II derivatives. Acta Crystallogr. D Biol. Crystallogr 1994;50:93–100.
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr 2010;66:133–44.
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. D 1997;53:240–55.
- Emsley P, Lohkamp B, Scott W, Cowtan K. Features and development of Coot. Acta Cryst. D 2010;66:486–501.
- Lamzin VS, Perrakis A, Wilson KS, International tables for crystallography. Vol. F: Crystallography of biological macromolecules. Dordrecht, The Netherlands: Kluwer Academic Publishers; 2001.
- Lovell SC, Davis IW, Arendall IIIWB, et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins: Struct Funct Genet 2003;50:437–50.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12.
- Lindorff-Larsen K, Piana S, Palmo K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010;78:1950–8.
- Wang J, Wolf RM, Caldwell JW, et al. Development and testing of a general amber force field. J Comput Chem 2004;25:1157–74.
- Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983;79:926–35.
- Jorgensen WL, Madura JD. Quantum and statistical mechanical studies of liquids. Solvation and conformation of methanol in water. J Am Chem Soc 1983;105:1407–13.
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem 2005;26:1781–802.
- Hutter J, Iannuzzi M, Schiffmann F, VandeVondele J. cp2k: atomistic simulations of condensed matter systems. Wiley Interdiscip Rev Comput Mol Sci 2014;4:15–25.
- Goedecker S, Teter M, Hutter J. Separable dual-space Gaussian pseudopotentials. Phys Rev B Condens Matter 1996;54:1703–10.
- VandeVondele J, Hutter J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J Chem Phys 2007;127:114105.
- Bussi G, Donadio D, Parrinello J. Canonical sampling through velocity rescaling. J Chem Phys 2007;126:014101.
- Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev 1988;A38:3098–100.
- Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 2010;132:154104–154104.
- Becke AD. Density‐functional thermochemistry. III. The role of exact exchange. J Chem Phys 1993;98:5648–52.
- Bader RFW. A quantum theory of molecular structure and its applications. Chem Rev 1991;91:893–928.
- Yu MD, Trinkle R. Accurate and efficient algorithm for Bader charge integration. J Chem Phys 2011;134:064111
- Angyan JG, Loos M, Mayer I. Covalent bond orders and atomic valence indices in the topological theory of atoms in molecules. J Phys Chem 1994;98:5244–8.
- Nettles WL, Song H, Farquhar ER, et al. Characterization of the Copper(II) Binding sites in human carbonic anhydrase II. Inorg Chem 2015;54:5671–80.
- Song H, Weitz AC, Hendrich MP, et al. Building reactive copper center(s) in human carbonic anhydrase II. J Biol Inorg Chem 2013;18:595–8.
- Holland PL. Metal-dioxygen and metal-dinitrogen complexes: where are the electrons? Dalton Trans 2010;39:5415–25.
- Ridderstråle Y, Fierke CA, Roush ED, Wistrand PJ. Localization of a protein inhibitor of carbonic anhydrase in pig tissues. Acta Physiol Scand 2002;176:27–31.