
Abstract
Allosteric sites on proteins are targeted for designing more selective inhibitors of enzyme activity and to discover new functions. Acetylcholinesterase (AChE), which is most widely known for the hydrolysis of the neurotransmitter acetylcholine, has a peripheral allosteric subsite responsible for amyloidosis in Alzheimer’s disease through interaction with amyloid β-peptide. However, AChE plays other non-hydrolytic functions. Here, we identify and characterise using computational tools two new allosteric sites in AChE, which have allowed us to identify allosteric inhibitors by virtual screening guided by structure-based and fragment hotspot strategies. The identified compounds were also screened for in vitro inhibition of AChE and three were observed to be active. Further experimental (kinetic) and computational (molecular dynamics) studies have been performed to verify the allosteric activity. These new compounds may be valuable pharmacological tools in the study of non-cholinergic functions of AChE.
Introduction
Acetylcholinesterase (AChE) is a well-known enzyme for the hydrolysis of the neurotransmitter acetylcholine (ACh)Citation1–4, being the target of the main marketed pharmacological treatment of Alzheimer’s disease (AD). However, during the past decade it has been shown that AChE also plays other non-hydrolytic functions. Several in vitro and in vivo studies in the central nervous system suggested that developmental regulation of AChE enzyme plays a role in non-cholinergic function as morphometric processes, cell differentiation and synaptogenesis along nervous systemCitation5. It is well recognised that one of the non-cholinergic actions of AChE is neurite promotion, which is regulated by a dynamic equilibrium of different interactions sites of AChECitation6,Citation7. Zeev-Ben-Mordehai et al. published that one of the non-classical roles of AChE might be as an adhesion protein involved in synaptic development and maintenanceCitation8. Thus, pharmacological inhibitors of AChE that block the catalytic activity of the enzyme do not necessarily interfere with other biological activities of the protein. On the contrary, since damaging effects of overexpressed AChE may be related to non-catalytic activities, these drugs may actually aggravate certain conditions by elevating the levels of catalytically inactivated AChE.
To date it is well known that the active site of AChE is subdivided into several subsites: catalytic triad (CAS, Ser-Glu-His) at the base of the gorge, anionic subsite (AS), acyl-binding pocket, and peripheral anionic subsite (PAS) at the mouth of the site, being PAS an allosteric siteCitation9. Modulators binding PAS limit the catalytic efficiency by both ways, combining steric and electrostatic blockage of ligand trafficking through the gorge and changing the active site conformation. Several evidences suggest that the PAS, besides its role in allosteric regulation of AChE-catalysed hydrolysis, also mediates heterologous protein associations that contribute to cell recognition and adhesion processes during synaptogenesis, and to the nucleation of amyloid peptides during the onset of AD in humans and mammalian model systemsCitation10.
Recently, Marcelo et al. suggested another possible binding site, called site B, located outside the catalytic gorge. They showed that rosmarinic acid was able to bind this site; however, its allosteric functioning is not clearCitation11.
The “exit doors” are another very interesting structural motif in the hydrolysis mechanism of AChE. These regions are alternative routes to the gorge for product clearance, contributing to the high catalytic activity of the AChE. Computational studies together with X-ray crystallography suggest three possible regions of AChE implicated in the removal of cleavage products of the hydrolysis of ACh, known as back door (including Trp86, Gly448, Tyr449, and Ile451 (hAChE residue numbering)). Side door (including Asp74, Thr75, Leu76, Thr83, Glu84, and Asn87) and acyl loop door (including Trp236, Arg247, and Phe297)Citation12–15.
There are many challenges that need to be addressed regarding the non-classical functions of AChE, such as to identify the allosteric sites, the amino-acid residues that mediate non-classical activities and the identification of allosteric inhibitors among othersCitation16. Taking into account the relevance of this target in neurodegenerative diseases we consider that it is of great importance to identify allosteric sites as starting point to develop efficient modulators of AChE and the corresponding non-cholinergic functions.
The main goal of this work is the identification and description of new allosteric binding sites on AChE and the discovery of new allosteric inhibitors since allosterism represents one of the most common and powerful means to regulate protein function. For this purpose, we performed a search for druggable sites on the enzyme using computational approaches with the aim of identifying putative allosteric sites. Moreover, for each of these we have defined the interacting key residues by means of virtual screening of our Medicinal and Biological Chemistry (MBC) libraryCitation17, and a plausible mechanism of action is proposed.
Methods
Computational studies
Pocket search
In order to identify different cavities on the AChE enzyme, the Fpocket softwareCitation18, a highly scalable and open source pocket-detection software package, was used. Fpocket is freely available for download at http://www.sourceforge.net/projects/fpocket. Fpocket extracts the information from rigid structure and only it is based on geometric parameters. Fpocket is based on the concept of a α-sphere, which is defined as a sphere that contacts four atoms on its boundary and has no internal atom. For a protein, very small spheres are located inside the protein, large spheres at the exterior, and clefts and cavities correspond to spheres of intermediate radiiCitation19.
The use of Fpocket software involves three major steps. During the first step, the whole ensemble of α-spheres is determined from the protein structure and a pre-filtered collection of spheres is returned. The second step consists of identifying clusters of spheres close together, identifying pockets, and removing no interesting clusters. The final step is the atom properties calculation from each pocket, in order to score and rank the identified pockets.
Fourteen hAChE structures (PDB IDs: 1B41, 1F8U, 2X8B, 3LII, 4BDT, 4EY4, 4EY5, 4EY6, 4EY7, 4EY8, 4M0E, 4M0F, 4PQE, 5FPQ)Citation20–27 were downloaded from the Protein Data Bank (www.pdb.org) and subjected to pocket search using Fpocket. The PDB structures were prepared using the MaestroCitation28 Protein Preparation WizardCitation29 for removing the water molecules, ligands, and metal ions. Upon Fpocket search, the 14 structures with embedded centres of pocket α-spheres were analysed by visual inspection to identify conserved pockets.
Hotspot analysis
The Fragment Hotspot maps softwareCitation30 identifies the location and quality of binding sites on the protein by first calculating atomic hotspots and then producing Fragment Hotspot maps with simple molecular probes. These maps specifically highlight fragment-binding sites and their corresponding pharmacophores. H-Bond acceptor, donor, and apolar/aromatic interactions, reported by this software, can assist medicinal chemists search for interesting interactions in order to bind or improve the binding affinities for different ligands, and suggest modifications to the molecules. For the molecular modeller, the maps complement existing virtual screening methods because they can be visually inspected to generate docking constraints or structure-based pharmacophores. With the most important interactions highlighted, existing pharmacophore methods can be used to screen for molecules capable of making these essential interactions. The maps can also be used to generate constraints for docking and hence steer the docking towards occupying the hotspot and ensuring that the important potential interactions are satisfied.
Structure preparation of hAChE in its apo state (PDB ID: 4EY4) for the hotspot calculation was performed using Protein Preparation WizardCitation29, implemented in MaestroCitation28. Ligands and water molecules were removed, hydrogen atoms were added and protein residues were ionised at pH = 7. After the target preparation, Fragment Hotspot maps were calculated using the in-house script developed by Chris Radoux in Cambridge. The Hotspot MapsCitation30 were visualised using PyMolCitation31 software in order to identify the residues which could be involved in the interaction between ligand and protein.
Ligand preparation
The preparation of the library and the 2D-to-3D conversion was performed using the LigPrepCitation32 tool, a module of the Schrödinger software package. LigPrep allows different preparation steps of molecules such as the addition of hydrogen atoms, neutralisation of charged groups, generation of ionisation states, low-energy ring conformations, possible tautomers, followed by energy minimisation using the OPLS-2005 force fieldCitation33,Citation34. In order to carry out our studies, the compounds were prepared at physiological pH conditions, all of the compounds were desalted and finally the compounds were minimised as default. A total of 2499 protonation and tautomeric states were generated from 1830 compounds of MBC library using LigPrep.
Virtual screening
Virtual Screening of the MBC libraryCitation17 was carried out using the Glide softwareCitation35, with the Extra Precision (XP) Glide Mode, for the site 2 and site 3 of the 4EY4 structure which was previously prepared using Protein Preparation Wizard tool. This structure was selected because of its being the only apo form amongst the serie of AChE crystallographic structuresCitation22; it also has a very good resolution and excellent validation values, such as few Ramachandran outliers. The small-molecule-bound docked poses were further filtered using Maestro Pose Filter, selecting only the compounds that interact through an H-bond with the residues that are known to be critical for the ligand-target affinity in the Hotspots Maps. For site 2, the grid was centred on the site 2, ensuring that the entire cavity was included inside the box, and the molecules were ranked based on the Glide XP score. The scores for the molecules were in the range from −8.9 to 4.4 kcal/mol while rosmarinic acid was ranked with a score of −8.5 kcal/mol. His405, Glu414, and Trp532 were selected as key residues and applied as interaction filter.
For site 3, a Glide XP docking was performed ensuring the entire cavity was included in the grid. The docking score for the molecules ranges from −7.7 to 1.8 kcal/mol. Glu81, Glu452, and Arg463 were selected as key residues for the interaction filtering. Ligands that are able to interact with these residues were ranked by their XP Glide Score, and visual inspection of the fitting between the Hotspot Maps and the best ranked virtual screening results was used to select molecules for further studies.
Molecular dynamics
Molecular dynamics (MD) was performed on an Asus 1151 h170 LVX-GTX-980Ti workstation, with an Intel Core i7–6500 K Processor (12 M Cache, 3.40 GHz) and 16 GB DDR4 2133 MHz RAM. The workstation has Nvidia GeForce GTX 980Ti available for GPU computations. MD studies were performed using AMBER14Citation36 with the ff14SBCitation37 to assess the stability of the compounds and to look for the ligand’s inhibitory mechanism. Additionally, a MD trajectory for the apo-target was developed, in order to observe the behaviour of the apo-AChE enzyme and to compare it with the ligand-target trajectories. To calculate the ligands parameters for the MD simulation, RESP charges were calculated using Gaussian09Citation38, optimising the geometry for both compounds using the method HF6–31++(d,p). Once the optimisation was completed, ligands were parametrised using Antechamber moduleCitation39. Systems were solvated using TIP3P modelCitation40 for water molecules, with a cubic box, equilibrating the system charge by adding Na+ ions. Solvated systems were first minimised for 8000 steps with the initial 4500 steps using the steepest descent algorithm. The final 3500 steps used the conjugate gradient energy minimisation with constraints applied to the protein residues as mentioned above for sites 2 and 3. This was followed by two minimisation stages of 8000 steps each, with the last 3500 using the conjugate gradient decreasing the restrains to the system. The system was equilibrated to 300 K and 1 atm, using a step protocol, applying energetic restraints of 15 kcal mol−1 Å−1 from the initial step and gradually decreasing them until its disappearing. Trajectories of 25 ns were obtained in isothermal–isobaric ensembles. All bonds involving hydrogen atoms were constrained with the SHAKE algorithmCitation41. A cut-off of 10 Å was used for the Lennard–Jones interaction and the short-range electrostatic interactions. Berendsen barostatCitation42 and Langevin thermostat were used to regulate the system pressure and temperature, respectively. Trajectories of 25 ns were computed, analysed using the CpptrajCitation43 module and VMDCitation44 for visual inspection. Xmgrace softwareCitation45 was used to obtain the graphics of root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) of the MD simulations.
One of each five frames of the trajectories were saved into a new PDB format trajectory and were taken to further analysis using TRAnsient Pockets in Proteins (TRAPP) softwareCitation46, which allows the simulation, analysis, and visualisation of protein cavity dynamics for detection of transient sub-pockets using protein motion trajectories or ensembles of protein structures obtained either from experiments or from simulations. The catalytic pocket was also analysed using this software.
Biological studies
In vitro cholinesterase inhibition assays
The method was adapted from Ellman et al.Citation47 The assay solution consisted of 0.1 M phosphate buffer pH 8, 400 mM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB, Ellman’s reagent), 0.05 unit/ml AChE (Sigma Chemical Co., Madrid, Spain, Cholinesterase, acetyl human recombinant), and 800 mM acetylthiocholine iodide as the substrate of the enzymatic reaction. The compounds tested were added to the assay solution and the absorbance changes at 412 nm were recorded for 5 min with a UV/Vis Microplate and cuvette Spectrophotometer, Thermo Electron Type, Multiskan Spectrum. The reaction rates were compared, and the per cent inhibition due to the presence of test compounds was calculated. The IC50 is defined as the concentration of each compound that reduces the enzymatic activity 50% with respect to that without inhibitors. All the experiments were performed in triplicate.
Kinetic study of AChE inhibition
To investigate the mechanism of action of the compounds on AChE, a kinetic analysis was performed. The experiments were carried out using combinations of four substrate concentrations, and three inhibitor concentrations. Double-reciprocal Lineweaver–Burk plotting of the data obtained, in which each point is mean of three different experiments, were analysed.
Competitive inhibitors have the same y-intercept as uninhibited enzyme (since Vmax is unaffected by competitive inhibitors the inverse of Vmax also does not change) but there are different slopes and x-intercepts. Non-competitive inhibition produces plots with the same x-intercept as uninhibited enzyme (Km is unaffected) but different slopes and y-intercepts. Non-competitive inhibition causes different intercepts on both the y- and x-axes but the same slope. Mixed inhibitors cause intersects above or below the x-axis.
Results and discussion
Druggable site
The determination of druggable cavities in therapeutic targets is essential for structure-based drug design in order to identify binding pockets or allosteric sites and to design small-molecule ligands that bind to these with therapeutic effects. To address this goal, several computational approaches have been developed based mainly on evolutionary or structure-based algorithmsCitation48–51.
In this work, we have used a combination of methods to get a consensus prediction of protein druggable sites. We have used the free geometry-based algorithm FpocketCitation18,Citation19 (http://fpocket.sourceforge.net) together with the prediction of Fragment Hotspot mapsCitation30 to study the AChE surface with the aim to identify allosteric sites and its key residues.
The first step of this consensus protocol was performed using the Fpocket software. As Fpocket is a geometry-based pocket-detection algorithm, we performed the study using a representative set of hAChE, in both apo and different complexed forms (). Fourteen different human structures were available at the moment of the study in the Protein Data Bank (PDB) (see Material and Methods section). The software detected almost 30 cavities, with some of these appearing in all AChE structures analysed, and others, which are not conserved, in fewer crystal structures.
Table 1. Druggable binding sites.
Once all cavities were analysed and clustered, taking into account the frequency that they appear together, and the druggability prediction score given by Fpocket, only four highly reproducible cavities can be considered as putative druggable binding sites (). We observed that the four sites appear in all of the different structures, although the position of each site varies among the structures and, in the case of site 2, Fpocket detected it as two different cavities ().
Figure 1. AChE structure highlighting the best cavities found by Fpocket.
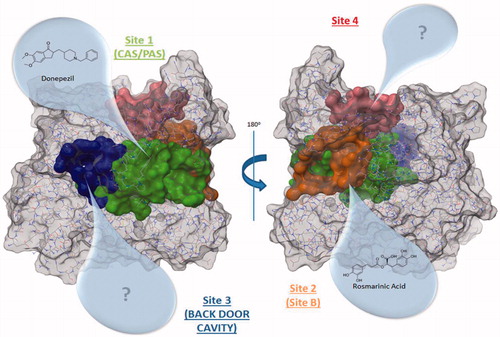
The best ranked site for all structures is the site 1 (except for 4EY4, for which is the second-best cavity) that corresponds to the well-known binding site CAS/PAS. The site 2 is ranked at the top five positions in 11 out of the 14 structures analysed. Inspection of the residues of this site () allowed us to identify Arg296 and Glu369 as possible key binding residues of the rosmarinic acid as was previously described by Marcelo et al.Citation11 Some residues are also shared with the acyl loop doorCitation14 such as Trp236, Arg247, and Phe297 ().
Figure 2. Amino acids involved in site 2 and site 3 found in hAChE (PDB ID: 4EY4). Site 2 residues are shown in orange and site 3 residues are shown in blue.
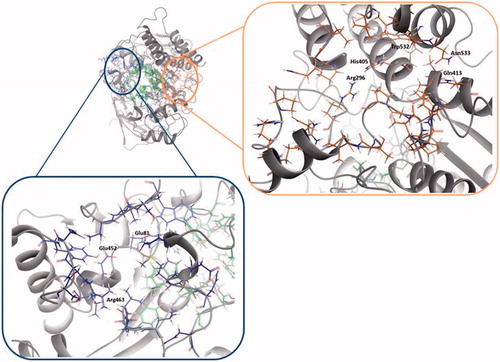
Table 2. Residues of allosteric sites, site 2 and site 3.
The site 3, in 13 out of the 14 structures analysed, is ranked in the top five positions. We identified residues Val132, Tyr449, and Glu452 as residues () that take part of the so-called back door, described as a dimple on the surface of the proteinCitation52.
The remaining site 4 corresponds to a cavity formed mostly by polar residues such as Asp333, Glu334, His381, Glu396, and Asp400. Since no molecular or biological function can be attributed to these residues, we did not pursue the study of this cavity. However, the possibility of using site 4 for the design of new allosteric modulators of AChE remains open for future work. The known side doorCitation53,Citation54 was also detected by Fpocket, although this cavity was not found in most of the structures (see Supporting Information).
Volume characterisation
Further we noticed that there was a fluctuation in the volume of the site 2 and site 3 when a ligand is bound in catalytic gorge. To analyse these fluctuations we use Fpocket to compare a series of structures crystallised in the same conditions (4EY4, 4EY5, 4EY6, 4EY7, 4EY8). We observed that the site 2 decreases its volume when a ligand is bound to the catalytic gorge except to complex 4EY6 (). These changes in the volume present a logical issue, because site 2 and CAS have some common residues, so ligands in the active gorge can interact and displace these residues as a consequence and hence modify the contiguous cavity. Similar changes might affect the active gorge when a ligand binds the site 2, explaining a possible allosteric mechanism. Moreover, it has been reported that the structural perturbations of the acyl loop occur when AChE is inhibited by a covalent inhibitor, presenting a narrowing of the gorge and a displacement of the Arg296 into the active site, which could present a steric barrier to the entry of oximes or affect oxime binding for AChE reactivationCitation55. In our case, the hypothesis that a ligand binds to site 2 and displaces the acyl loop, thus inhibiting AChE activity, will be further studied.
Figure 3. (A) Surface representation of pockets that belong to site 2 (orange) and site 3 (blue). Differences between the volume in the absence and presence in the CAS/PAS cavity of inhibitor are highlighted (4EY4 is AChE in the apo state, 4EY7 is AChE crystallised with donepezil). (B) Plots of volume measured in some structures crystallised in the same conditions for site 2 (orange) and site 3 (blue).
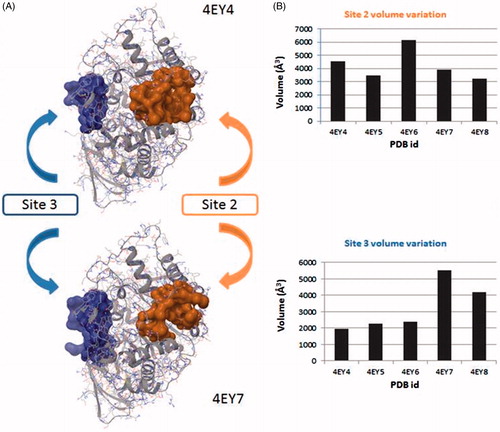
We next studied the influence of the ligand in complexes 4EY7 (with donepezil) and 4EY8 (with fasciculin-2) on the volume of the site 3. Interestingly, we found that in both cases the site 3 rises its volume approximately by two-fold as compared to the apo structure. This could be explained by the opening of the back door in both complexes, while the others remain closed.
These results, indicative of a volume fluctuation of allosteric sites, get with Fpocket are preliminary but suggesting an interrelation between catalytic site and allosteric ones.
Key residues
In order to confirm the druggability of the allosteric sites and to identify its key residues, we next performed a theoretical study of potential fragment-binding sites using Fragment Hotspot mapsCitation30 for each site detected by Fpocket. These hotspot maps provide visual guides of the fragment-binding sites and their corresponding pharmacophores. This method reports H-Bond acceptor, donor, and non-polar/aromatic interactions, helping in the search for interesting interactions in order to identify or design efficient inhibitors.
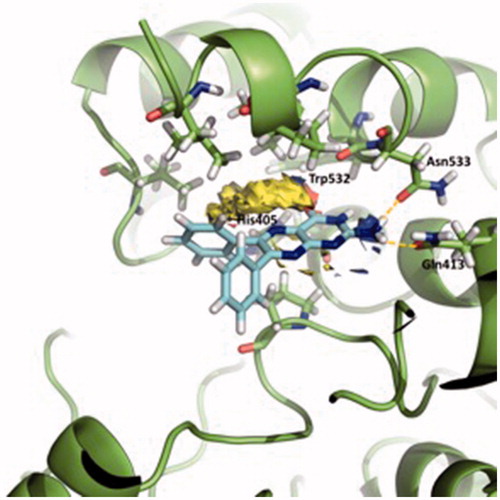
shows the Fragment Hotspot maps of the AChE apo structure (PDB ID 4EY4). The analysis of the hotspot was focussed on sites 2 and 3. We found that key residues Gln413 and Trp532 in site 2 can act as acceptors and His405 as H-Bond donor. These interactions represent the theoretical minimum binding features that will allow a fragment to bind this cavity.
Figure 4. Representation of the calculated hotspots using Fragment Hotspot maps softwareCitation30. Yellow area refers to non-polar area where the ligand should make hydrophobic interactions. Blue dots represent the area where the ligand should make a donor H-bond, and red dots where acceptor H-bond should be formed.
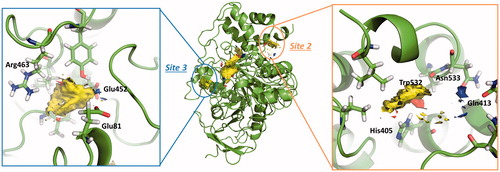
Fragment Hotspot maps for the site 3 were also analysed, demonstrating that the key residues are Glu81 and Glu452 as acceptors and Arg463 as a H-Bond donor.
In summary, considering all above results we can conclude that druggable sites (sites 2 and 3) could be possible allosteric sites and targeting them might lead to the modification of AChE activity.
Virtual screening
With the goal of searching for allosteric modulators of the sites 2 and 3, we performed a virtual screening using our in-house MBC chemical libraryCitation17. To validate the computational screening, docking of donepezil and rosmarinic acid for both sites (CAS/PAS and site 2) was also performed using Glide software (see Supporting Information, Figures S1 and S2). Upon validation, a virtual screening was performed using Glide with XP for both sites.
Site 2 virtual screening
Eleven compounds were chosen for enzymatic assay. Two of these compounds were selected based on their docking score. The rest of them were selected by an interaction-filter based on the Fragment Hotspot maps that were previously calculated. In this way, a filter of the capability of the ligands to interact through H-bond with Gln413, Trp532, and His405 was set up and the results were visually inspected. Along with the 11 compounds selected, rosmarinic acid was also sent for biological evaluation on AChE as a controlCitation11 ().
Table 3. Experimental inhibition values of the virtual screening compounds to bind site 2.
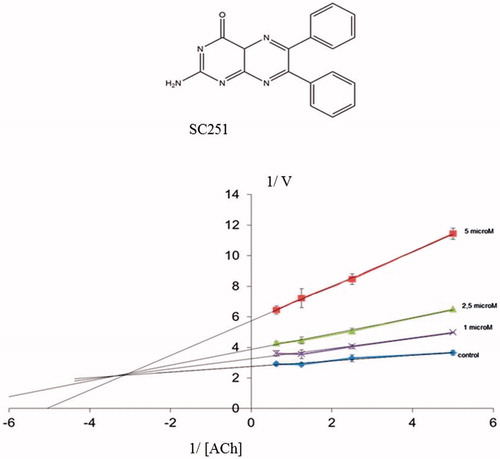
All the compounds tested showed a percentage of AChE inhibition at 10 μM greater than rosmarinic acid (the control compound in the assay), consistent with the proposed common binding site in the enzyme. Very interesting is the case of pteridine derivative SC251 with an IC50 value of 2.7 μM.
displays a three-dimensional view of the predicted binding pose of SC251 at site 2. SC251 interacts by a donor hydrogen bond with Gln413, an acceptor H-bond with His405 and a donor H-bond with Asn533, fitting properly with the Fragment Hotspot maps results.
Figure 5. Superposition of the proposed pose for SC251 at Site 2 and the hotspot calculated with Fragment Hotspot maps software.
To confirm a possible allosteric mechanism of inhibition, we perform competitive studies of SC251 with the natural substrate, acetylcholine (ACh) (). A non-competitive-mixed mechanism was observed suggesting that SC251 should bind to an allosteric site, in addition to the catalytic site.
Figure. 6. Lineweaver–Burk plots representing the reciprocal of initial enzyme velocity versus the reciprocal of ACh concentration in the absence and presence of different concentrations of SC251 (1–5 μM). Each point is the mean of three different experiments.
MD
With the aim of explaining a plausible mechanism of action for the allosteric inhibitor SC251, MD studies for AChE in the apo state and AChE-SC251 complex were carried out. MD studies provide a powerful method to follow intimate details of the conformational events in different biological systems. In this study, flexibility changes or behaviour modifications in the important sites of AChE upon inhibitor binding, at the sites 2 and 3, and the side door cavity were further analysed.
To explore the dynamic stability of the trajectories, RMSD values of the protein backbone based on the starting structure along the simulation time were calculated and plotted (Figure S4). After the initial adjustment, the longer term RMSD values are stable, ranging from 1.0 to 1.9 Å during the entire simulation time.
Along the trajectory, the ligand SC251 fluctuates around the predicted docking poses (Figure S5). Accordingly, during the simulation, the ligand maintains some of the strong interaction as the H-bond donor with the Asn533 (during the 85% of the simulation) and the H-bond acceptor with the His405 (during the 50% of the simulation). However, the H-bond donor with Gln413 is lost in the majority of the simulation, and appears only 2% of the simulation time as it can be observed in Figure S6 (in order to clarify the final bind mode of the complex, a picture is shown in Figure S7).
The RMSF of the apo and bound state were compared in order to see any differences in the fluctuations of the residues (Figure S8). The most interesting change corresponds to the residues 70–78, that belong to the omega loop, that are also implicated in the side door. We can identify rigidity of these residues when SC251 binds the site 2, as demonstrated by the decrease in fluctuations from approximately 3 Å to barely 1 Å, which might suggest the impossibility of opening the side door while the ligand remains bound to the site 2.
To further analyse the conformational changes between the trajectories, we used TRAPP softwareCitation46. TRAPP is a tool that allows the analysis of the evolution of the spatial and physicochemical properties of a specified pocket in a protein during a MD simulation. We selected one out of five frames of the simulation as an input for TRAPP. We first analysed the CAS/PAS cavity of the apo AChE trajectory, which reveals that our molecular dynamic study also shows the presence of the side and back doors in at least the 50% of the trajectory, as other previous studies have already publishedCitation56 (). The acyl loop door is less flexible therefore the gate opening is more difficult to occur, as other studies have also previously reportedCitation56, thus we only can see its opening in 25% of the total snapshots of the simulation.
Figure 7. (A) TRAPP analysis for the apo AChE trajectory. (B) TRAPP analysis for the AChE-SC251 trajectory. Blue areas represent disappearing areas at the 50% of the snapshots, red areas represent appearing areas at the 50% of the snapshots. Green loop corresponds to the side door, orange loop to the acyl-loop and the blue residues to the back door.
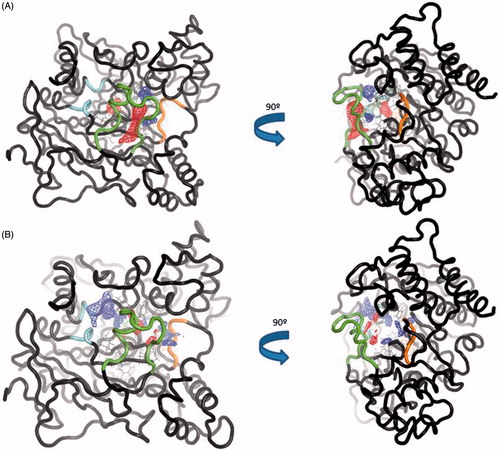
Analysing the CAS/PAS cavity of the complex AChE-SC251 trajectory, some interesting differences with the apo form are highlighted. As we commented above, while the omega loop of the apo state of AChE is very flexible and allows the opening of the side door, in the AChE-SC251 trajectory this loop is much more rigid and as a result, the side door opens only in <50% of the snapshots of the trajectory. A red area appears in the back door region of the apo AChE trajectory, indicating a transient pocket when the back door is opening (). However, for the AChE-SC251 trajectory we can see instead a big disappearing area (in blue, ), suggesting the impossibility for this door to open. Another difference is also noticed near the acyl loop, while the loop remains rigid in the apo trajectory, for the AChE-SC251 trajectory a disappearing area is situated near it, suggesting a slight displacement of these residues into the CAS/PAS cavity.
In general terms, we observe a stiffness of the doors. All of these data are in agreement with the allosteric theory that proposes that the binding of a ligand can reduce the entropy of the system in such a way that the conformation of the protein is fixed, restricting its movement and thus modifying the target behaviourCitation57.
Site 3 virtual screening
For the site 3, no ligands can be used as a control for docking calculations since no previous studies have been carried out. Thus, the same docking conditions as for the virtual screening in the site 2 were used, following Fragment Hotspot maps leads.
In this case, 14 compounds were selected for biological evaluation using the hotspots calculated as a filter for the screening. Four different filters were set, taking into account the ability of the ligands to make an H-bond interaction with Arg463, Glu452, and Glu81.
As given in , two compounds, VP2.33 and SC035, showed an IC50 of ∼50 μM. A competition study for both was performed showing a non-competitive inhibition. At larger concentrations of substrate, the inhibition percentage slightly decreases, as shown in graphic of .
Figure 8. Lineweaver–Burk plots representing the reciprocal of initial enzyme velocity versus the reciprocal of ACh concentration in the absence and presence of different concentrations of VP2.33 and SC035. Each point is the mean of three different experiments.
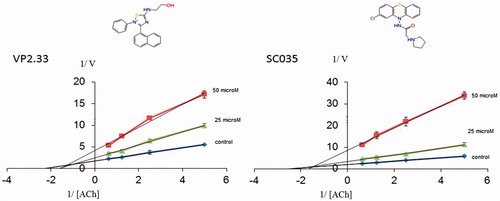
Table 4. Experimental inhibition value of the identified compounds to bind site 3 and binding interactions with AChE.
Clearly, both compounds bind to AChE at an allosteric site. To further validate the allosteric behaviour of these compounds we measured their inhibitory capacity in the presence of the pure competitive inhibitor JTE-907Citation58. If our hypothesis is right, the binding of VP2.33 and SC035 to the allosteric site 3 in presence of JTE-907 should show a cooperative behaviour in terms of inhibition of the AChE.
shows the inhibitory value of each compound and their sum. In the case of VP2.33 the data indicate an effect of cooperative activities. These data support a model where both compounds collaboratively participate in the inhibition of AChE by targeting different sites in the enzyme.
Table 5. Results of the inhibition of AChE with JTE-907 and VP2.33.
However, when the experiment was performed with SC035, there is no addition of activities (Table S1) and therefore the results are not conclusive for this compound.
Since the interactions of SC035 and VP2.33 are different (), both compounds could be inhibiting the enzymatic activity but with different mechanisms of action, explaining the difference when the sum of activities is measured.
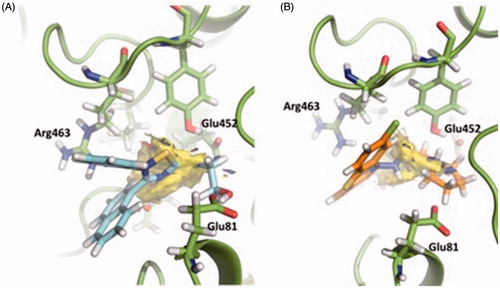
Figure 9. Superimposition of the proposed pose for VP2.33 (blue) (A) and SC035 (orange) (B) at Site 3 and the hotspot calculated with Fragment Hotspot maps software.
Molecular dynamics
Further MD studies have been carried out to study the behaviour of the complex AChE-VP2.33 in order to propose a possible mechanism of action of this compound.
The RMSD values of the protein backbone based on the starting structure along the simulation time were calculated and plotted (Figure S11). After the initial adjustment, the longer term RMSD values are stable, ranging from 1.2 to 1.8 Å during the entire simulation process.
Along the simulation of the AChE-VP2.33 complex, we found that the compound pose remains stable along the trajectory (Figure S12), maintaining the H-bond donor interactions with the Glu452 (during the 95% of the simulation) and H-bond donor and acceptor interactions with the Thr436 (during 30% of the simulation) (Figures S13–S14).
The RMSF of the apo and bound forms were computed and compared in order to see any differences in the fluctuations of the residues (Figure S15). One of the differences lie on the omega loop residues, in which a decrease in the loop flexibility occurs when VP2.33 binds the site 3. Other two regions that also lose flexibility are residues 251–260 and residues 330–347 that comprise loops exposed to the solvent. This rigidity process, when VP2.33 binds AChE, suggests the stabilisation of the complex, reducing its Gibbs free energy and entropy valuesCitation59.
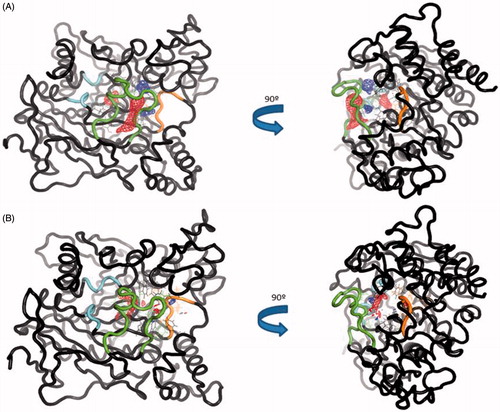
To verify these changes, we studied different areas of AChE along the MD trajectory using TRAPP software. The normal behaviour of the omega loop that corresponds to the opening of the side door is modified when the ligand is bound to the target. In the apo form a red area appears (new transient pocket) pointing out the opening of the side door, meanwhile this new transient pocket does not appear in the complex AChE-VP2.33 (). This fact could suggest that compound VP2.33 could be able to modify the behaviour of the omega loop residues, as shown in the RMSF data in Figure S4.
Figure 10. (A) TRAPP analysis for the apo AChE trajectory. (B) TRAPP analysis for the AChE-VP2.33 trajectory. Blue areas represent disappearing areas at the 50% of the snapshots, red areas represent appearing areas at the 50% of the snapshots. Green loop corresponds to the side door, orange loop to the acyl-loop and the blue residues to the back door.
Regarding the back door, significant differences are not appreciated in the movement of the residues involved in the opening of this door between the two trajectories. This suggests that the compound VP2.33 might block the clearance of ACh, without modifying the movement of the residues involved in the back door.
In summary, we do not find any significative changes in acyl loop neither back door. Therefore, we can suggest an innovative allosteric mechanism of VP2.33 due to the prevention of side door opening. The residues of this alternative channel present less movement when VP2.33 is bound to site 3 than the apo trajectory correlating with a lower efficiency of the clearance of the degradation products of ACh.
Conclusions
Our main goal in this work was to extend knowledge of the druggable sites of AChE. Since allosterism represents one of the most common and powerful means to regulate protein function, we aimed to study the AChE surface to identify allosteric sites. A combination of the free geometry-based algorithm Fpocket with the Fragment Hotspot maps have allowed the identification of new allosteric binding sites (sites 2 and 3). We carried out a virtual screening study of our in-house library to identify allosteric inhibitors of sites 2 and 3. We validated the predicted hits with experimental studies (in vitro and kinetic studies). These studies have culminated with the identification of allosteric compounds. SC251 has been identified as an allosteric inhibitor of site 2 showing a non-competitive-mixed inhibition. Further MD allowed us to propose a possible action mechanism of this compound. In relation to site 3, VP2.33 and SC035 have been identified. Both compounds show a non-competitive inhibition. Further experimental studies (cooperative activities) have allowed us to validate VP2.33 as an allosteric inhibitor of site 3. These new allosteric modulators are potentially useful pharmacological tools for study of non-hydrolytic functions of cholinergic system.
Supplemental Material
Download PDF (761.5 KB)Acknowledgments
We thank to Ministerio de Educación, Cultura y Deporte (Grant of V.S.P, FPU15/01465 and Grant of N.E.C., Programa “Salvador de Madariaga” PR2015–00476). T.L.B. & S.M. thank the MRC Newton Fund and C.R. thanks the BBSRC and UCB for a CASE studentship.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Nachmansohn D, Wilson IB. The enzymic hydrolysis and synthesis of acetylcholine. Adv Enzymol Relat Subj Biochem 1951;12:259–339.
- Rosenberry TL. Catalysis by acetylcholinesterase: evidence that the rate-limiting step for acylation with certain substrates precedes general acid-base catalysis. Proc Natl Acad Sci USA 1975;72:3834–8.
- Rosenberry TL. Acetylcholinesterase. Adv Enzymol Relat Areas Mol Biol 1975;43:103–218.
- Nijholt I, Farchi N, Kye M, et al. Stress-induced alternative splicing of acetylcholinesterase results in enhanced fear memory and long-term potentiation. Mol Psychiatry 2004;9:174–83.
- Silman I, Sussman JL. Acetylcholinesterase: ‘classical’ and ‘non-classical’ functions and pharmacology. Curr Opin Pharmacol 2005;5:293–302.
- Halliday AC, Greenfield SA. From protein to peptides: a spectrum of non-hydrolytic functions of acetylcholinesterase. Protein Pept Lett 2012;19:165–72.
- Johnson G, Swart C, Moore SW. Interaction of acetylcholinesterase with the g4 domain of the laminin alpha1-chain. Biochem J 2008;411:507–14.
- Zeev-Ben-Mordehai T, Rydberg EH, Solomon A, et al. The intracellular domain of the drosophila cholinesterase-like neural adhesion protein, gliotactin, is natively unfolded. Proteins 2003;53:758–67.
- Johnson G, Moore SW. The peripheral anionic site of acetylcholinesterase: structure, functions and potential role in rational drug design. Curr Pharm Des 2006;12:217–25.
- Bourne Y, Renault L, Marchot P. Crystal structure of snake venom acetylcholinesterase in complex with inhibitory antibody fragment fab410 bound at the peripheral site: evidence for open and closed states of a back door channel. J Biol Chem 2015;290:1522–35.
- Marcelo F, Dias C, Martins A, et al. Molecular recognition of rosmarinic acid from salvia sclareoides extracts by acetylcholinesterase: a new binding site detected by NMR spectroscopy. Chemistry 2013;19:6641–9.
- Sanson B, Colletier JP, Xu Y, et al. Backdoor opening mechanism in acetylcholinesterase based on x-ray crystallography and molecular dynamics simulations. Protein Sci 2011;20:1114–18.
- Bennion BJ, Essiz SG, Lau EY, et al. A wrench in the works of human acetylcholinesterase: soman induced conformational changes revealed by molecular dynamics simulations. PLoS One 2015;10:e0121092.
- Colletier JP, Royant A, Specht A, et al. Use of a ‘caged’ analogue to study the traffic of choline within acetylcholinesterase by kinetic crystallography. Acta Crystallogr D Biol Crystallogr 2007;63:1115–28.
- Xu Y, Cheng S, Sussman JL, et al. Computational studies on acetylcholinesterases. Molecules 2017;22:1–20.
- Soreq H, Seidman S. Acetylcholinesterase-new roles for an old actor. Nat Rev Neurosci 2001;2:294–302.
- Sebastian-Perez V, Roca C, Awale M, et al. Medicinal and Biological Chemistry (MBC) library: an efficient source of new hits. J Chem Inf Model 2017;57:2143–51.
- Schmidtke P, Le Guilloux V, Maupetit J, Tuffery P. Fpocket: online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res 2010;38:W582–9.
- Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics 2009;10:168.
- Allgardsson A, Berg L, Akfur C, et al. Structure of a prereaction complex between the nerve agent sarin, its biological target acetylcholinesterase, and the antidote hi-6. Proc Natl Acad Sci USA 2016;113:5514–19.
- Carletti E, Colletier JP, Dupeux F, et al. Structural evidence that human acetylcholinesterase inhibited by tabun ages through o-dealkylation. J Med Chem 2010;53:4002–8.
- Cheung J, Rudolph MJ, Burshteyn F, et al. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J Med Chem 2012;55:10282–6.
- Cheung J, Gary EN, Shiomi K, Rosenberry TL. Structures of human acetylcholinesterase bound to dihydrotanshinone I and territrem B show peripheral site flexibility. ACS Med Chem Lett 2013;4:1091–6.
- Dvir H, Silman I, Harel M, et al. Acetylcholinesterase: from 3d structure to function. Chem Biol Interact 2010;187:10–22.
- Kryger G, Harel M, Giles K, et al. Structures of recombinant native and e202q mutant human acetylcholinesterase complexed with the snake-venom toxin fasciculin-ii. Acta Crystallogr D Biol Crystallogr 2000;56:1385–94.
- Millard CB, Kryger G, Ordentlich A, et al. Crystal structures of aged phosphonylated acetylcholinesterase: nerve agent reaction products at the atomic level. Biochemistry 1999;38:7032–9.
- Nachon F, Carletti E, Ronco C, et al. Crystal structures of human cholinesterases in complex with huprine w and tacrine: elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem J 2013;453:393–9.
- Schrödinger Release 2017-1: Maestro S, LLC, New York, NY, 2017. Maestro, schrödinger. In: Maestro, schrödinger; 2017.
- Schrödinger Release 2017-1: Schrödinger Suite 2017-1 Protein Preparation Wizard; Epik S, LLC, New York, NY, 2016; Impact, Schrödinger, LLC, New York, NY, 2016; Prime, Schrödinger, LLC, New York, NY, 2017. In: 2017.
- Radoux CJ, Olsson TS, Pitt WR, et al. Identifying interactions that determine fragment binding at protein hotspots. J Med Chem 2016;59:4314–25.
- The PyMOL Molecular Graphics System VS, LLC.
- Schrödinger Release 2017-1: LigPrep L, New York, NY, 2017. Maestro, Schrödinger.
- Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the opls all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 1996;118:11225–36.
- Banks JL, Beard HS, Cao Y, et al. Integrated modeling program, applied chemical theory (impact). J Comput Chem 2005;26:1752–80.
- Schrödinger Release 2017-1: Glide S, LLC, New York, NY, 2017. Glide, schrödinger. In: Glide, schrödinger; 2017.
- Case DA, Babin V, Berryman JT, et al. Amber14. In: Amber14; University of California, San Francisco; 2014.
- Maier JA, Martinez C, Kasavajhala K, et al. Ff14sb: improving the accuracy of protein side chain and backbone parameters from ff99sb. J Chem Theory Comput 2015;11:3696–713.
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, revision a.02. Wallingford, CT: Gaussian, Inc.; 2016.
- Wang J, Wang W, AKP, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 2006;25:247–60.
- Jorgensen WL, Chandrasekhar J, Buckner JK, Madura JD. Computer simulations of organic reactions in solution. Ann N Y Acad Sci 1986;482:198–209.
- Lippert RA, Bowers KJ, Dror RO, et al. A common, avoidable source of error in molecular dynamics integrators. J Chem Phys 2007;126:046101.
- Berendsen HJC, Postma JPM, van Gunsteren WF, et al. Molecular dynamics with coupling to an external bath. J Chem Phys 1984;81:3684.
- Roe DR, Cheatham TE III., Ptraj and cpptraj: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 2013;9:3084–95.
- Humphrey W, Dalke A, Schulten K. Vmd: visual molecular dynamics. J Mol Graph 1996;14:33–8, 27–38.
- XMGRACE V. In: Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, Beaverton, OR; 2005.
- Stank A, Kokh DB, Horn M, et al. Trapp webserver: predicting protein binding site flexibility and detecting transient binding pockets. Nucleic Acids Res 2017;45:w325–30.
- Ellman GL, Courtney KD, Andres V Jr, Feather SRM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95.
- Broomhead NK, Soliman ME. Can we rely on computational predictions to correctly identify ligand binding sites on novel protein drug targets? Assessment of binding site prediction methods and a protocol for validation of predicted binding sites. Cell Biochem Biophys 2017;75:15–23.
- Seco J, Luque FJ, Barril X. Binding site detection and druggability index from first principles. J Med Chem 2009;52:2363–71.
- Barril X. Druggability predictions: methods, limitations, and applications. WIREs Comput Mol Sci 2013;3:327–38.
- Schmidtke P, Souaille C, Estienne F, et al. Large-scale comparison of four binding site detection algorithms. J Chem Inf Model 2010;50:2191–200.
- Alisaraie L, Fels G. Molecular docking study on the “back door” hypothesis for product clearance in acetylcholinesterase. J Mol Model 2006;12:348–54.
- Tai K, Shen T, Henchman RH, et al. Mechanism of acetylcholinesterase inhibition by fasciculin: a 5-ns molecular dynamics simulation. J Am Chem Soc 2002;124:6153–61.
- Bui JM, Tai K, McCammon JA. Acetylcholinesterase: enhanced fluctuations and alternative routes to the active site in the complex with fasciculin-2. J Am Chem Soc 2004;126:7198–205.
- Franklin MC, Rudolph MJ, Ginter C, et al. Structures of paraoxon-inhibited human acetylcholinesterase reveal perturbations of the acyl loop and the dimer interface. Proteins 2016;84:1246–56.
- Fang L, Pan Y, Muzyka JL, Zhan CG. Active site gating and substrate specificity of butyrylcholinesterase and acetylcholinesterase: insights from molecular dynamics simulations. J Phys Chem B 2011;115:8797–805.
- Laskowski RA, Gerick F, Thornton JM. The structural basis of allosteric regulation in proteins. FEBS Lett 2009;583:1692–8.
- Gonzalez-Naranjo P, Campillo NE, Perez C, Paez JA. Multitarget cannabinoids as novel strategy for Alzheimer disease. Curr Alzheimer Res 2013;10:229–39.
- Tsai CJ, del Sol A, Nussinov R. Allostery: absence of a change in shape does not imply that allostery is not at play. J Mol Biol 2008;378:1–11.