
Abstract
Mitochondria play important roles in the development of diabetic kidney disease (DKD). The SS peptide is a tetrapeptide that is located and accumulated in the inner mitochondrial membrane; it reduces reactive oxygen species (ROS) and prevents mitochondrial dysfunction. Podocytes are key cellular components in DKD progression. However, whether the SS peptide can exert renal protection through podocytes and the mechanism involved are unknown. In the present study, we explored the mechanisms of the SS peptide on podocyte injury in vivo and in vitro. Compared with the control group, the glomerular podocyte number and expression of WT1 were significantly reduced and TUNEL-positive podocytes were significantly increased in renal tissues in the diabetic rat. These effects were further exacerbated by hypochlorite-modified albumin (HOCl-alb) challenge but prevented by SS-31. In vitro, SS-31 blocked apoptosis in podocyte cell line induced by HOCl-alb. SS-31 prevented oxidative stress and mitochondria-dependent apoptosis signalling by HOCl-alb in vivo and in vitro, as evidenced by the release of cytochrome c (cyt c), binding of apoptosis activated factor-1 (Apaf-1) and caspase-9, and activation of caspases. These data suggest that SS-31 may prevent podocyte apoptosis, exerting renal protection in diabetes mellitus, probably through an apoptosis-related signalling pathway involving oxidative stress and culminating in mitochondria.
Introduction
Diabetic kidney disease (DKD), the most common chronic complication of diabetes mellitus (DM) occurs in ∼20–40% of patients with DM and is characterised by progressive lesions of glomerular filtration and the development of Kimmelstiel–Wilson nodules, leading to end-stage renal failure (ESRD)Citation1. DKD is also closely associated with the morbidity and mortality of cardiovascular disease and has an extremely adverse impact on patients’ quality of life and survivalCitation2. Unfortunately, the mechanism of DKD is still not clear, resulting in a poor clinical outcome.
Following the discovery of the association of the reduction of the podocyte number, proteinuria and glomerular filtration membrane impairment in Pima Indians with type 2 DM in 1997 by Pagtalunan et al.Citation3, the relationship between podocyte loss and DKD was also confirmed by multiple clinical studiesCitation4. Podocytes are terminally differentiated epithelial cells that are important components of the glomerular filtration membrane. Podocyte number reduction mainly occurs through apoptosis and detachment, resulting in irreversible damage to the glomerular filtration functionCitation5. This suggests that podocytes act as a key cellular basis in the development of DKD and a new effective target in DKD therapy.
In the past decade, excessive reactive oxygen species (ROS) production and oxidative stress in the matrix were considered to be important pathophysiological mechanisms in DKDCitation6. Hypochlorite-modified albumin (HOCl-alb) is an active compound that is formed during the reaction between proteins (mainly albumin) and hypochlorite (HOCl) that originates through the action of the myeloperoxidase (MPO) of neutrophils, a process that is concurrent with oxidative stressCitation7. HOCl-alb was found to be much higher in concentration and to act as a mediator of oxidative stress and inflammation in patients with uraemia or DMCitation8.
Sources of endogenous ROS include mitochondria, NADPH oxidases, nitric oxide synthase, lipoxygenases, and xanthine oxidase, of which the estimated concentrations of ROS within mitochondria are 5- to 10-fold higher than other cytosolic and nuclear compartmentsCitation9. Mitochondria are not only the main source of ROS but also the organelle that is the most sensitive to oxidative stressCitation10. During DM, excessive intake of glucose by sensitive cells resulted in increased pyruvate in mitochondria and formation of superoxide caused by abnormal electron transport chain activity leading to an increased one-electron reduction of oxygen molecules and weakened mitochondrial antioxidant defences. Overproduction of mitochondrial ROS (mtROS) in turn leads to mitochondrial damage and functional deterioration, activates signalling pathways, promotes cell deathCitation11, and regulates DM complicationsCitation12, thus playing key roles in the development of DKD.
General antioxidants, such as vitamin E, β-carotene, and vitamin A, have been shown to have no obvious benefits in reducing DM complicationsCitation13,Citation14, inferring that these compounds might be distributed in the cytoplasm or extracellularly, could not enter the mitochondria, and had difficulty in exerting antioxidant effects. A new type of cell-permeable tetrapeptide was initially designed and synthesised by Szeto and Schiller and named the SS peptides. Among these peptides, SS-31 was reported to target and accumulate in the inner membrane of mitochondria and scavenge mtROS, thereby preventing mitochondrial depolarisation, mitochondrial permeability transition, and cytochrome c (cyt c) release from mitochondria to cytoplasmCitation15. The SS peptide protects N2A neuroblastoma cells, Caco-2 colon cancer cellsCitation16, HLEB-3 human lens epithelial cellsCitation17, NRK52E rat renal tubular cellsCitation18, and mouse mesangial cellsCitation19 from death. However, the roles and mechanisms of the SS peptide in podocytes under the circumstances of DM and oxidative stress remain largely unknown.
Here, we hypothesised that the SS peptide might attenuate podocyte apoptosis enhanced by HOCl-alb in DM through the mitochondrial pathway. We used streptozotocin-induced DM rats and MPC5 mouse podocytes as models to examine our hypothesis in vivo and in vitro.
Materials and methods
HOCl-RSA/MSA preparation and content determination
Hypochlorite modified rat serum albumin (HOCl-RSA) was prepared in vitro per our previous methodCitation20 by incubation of fatty acid-free RSA (100 g/l; Sigma-Aldrich, St. Louis, MO) with an equivalent volume of hypochlorous acid (HOCl, 200 mmol/l) or phosphate-buffered saline (PBS) for 30 min at room temperature. To prepare hypochlorite modified mouse serum albumin (HOCl-MSA), fatty acid-free MSA instead of RSA was incubated with HOCl or PBS. Prepared samples were dialysed in PBS at 4 °C overnight to remove free HOCl and then passed through a Detoxi-Gel (Thermo, Waltham, MA) to remove endotoxin contaminants.
The concentrations of endotoxin in the preparation were measured with the Limulus Amoebocyte Lysate kit (Sigma-Aldrich) and found to be <0.025 EU/ml. The content of HOCl-alb in the preparations was measured by monitoring the absorbance at 340 nm using a microplate reader under acetic acid conditions and calibrated with chloramine-T equivalents. The HOCl-alb contents in the HOCl-RSA/HOCl-MSA and unmodified RSA/MSA preparations were 6.35 ± 0.40/7.87 ± 0.39 μmol/g and 0.14 ± 0.050/0.22 ± 0.016 μmol/g protein, respectively.
Preparation and location determination of SS-31
SS-31 (D-Arg-Dmt-Lys-Phe-NH2, where Dmt refers to 2′,6′-dimethyltyrosine) was prepared by solid-phase synthesis by GL Biochem, Inc. (Shanghai, China).
Tissue mitochondrial uptake of SS-31 was determined as previously reportedCitation15,Citation20. In brief, SS-31 was labelled with [3H] and mixed with 5 g/l rat kidney mitochondria and 1 μM SS-31 in a binding buffer at room temperature. The uptake of [3H]SS-31 was expressed as the radioactivity in the mitochondrial suspension as determined by a liquid scintillation counter (Tri-Carb 3110TR; PerkinElmer, Waltham, MA). To determine the distribution of SS-31 within mitochondria, the above mixture was subjected to three freeze-thaw cycles. The supernatant (fraction of matrix) and precipitant (fraction of inner and outer membranes) were collected. The precipitant was treated with 0.2% digitonin to disrupt the outer membrane and isolate the inner membrane. The radioactivity in the fractions containing the matrix, outer membrane, and inner membrane was measured and compared with the total radioactivity in the mitochondria. Approximately 50% of [3H]SS-31 was retained in the inner mitochondrial membrane (IMM), 30% in the matrix, and 20% in the outer mitochondrial membrane (OMM).
To determine the cellular mitochondrial uptake of SS-31, podocytes (2 × 107) were incubated with [3H]SS-31 at 37 °C for 60 min15 and collected. Isolation of mitochondria and cytosol was carried out using a Cell Mitochondria Isolation Kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions. The radioactivity was measured in the mitochondrial and in cytosolic fractions and corrected with protein concentration. The ratio of [3H]SS-31 retained in mitochondrial and cytosolic fractions was 35:1.
Cell culture
Murine podocytes were generously provided by Professor Peter Mundel (Sinai School of Medicine, New York, NY) and cultured as described previouslyCitation21. Undifferentiated podocytes were grown in RPMI 1640 containing 10% foetal bovine serum, penicillin (100 U/ml), streptomycin (100 μg/ml), sodium pyruvate (1 mmol/l), HEPES buffer (10 mmol/l), and sodium bicarbonate (0.075%). To passage the cells, Interferon-γ (50 U/ml; Sigma-Aldrich) was added to the medium and podocytes were grown under “growth-permissive” conditions at 33 °C and 5% CO2. To acquire a differentiated phenotype, cells were grown in RPMI 1640 containing 10% foetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) under “restrictive conditions” at 37 °C for more than 12–14 days. Experiments were carried out using passages 10 –18, differentiated and conditionally immortalised podocytes.
Animal models
Male Sprague–Dawley (SD) rats (Experimental Animal Centre, Southern Medical University) with an initial weight of 180–220 g were raised in a specific pathogen-free (SPF) laboratory animal room under standardised conditions with access to water and food ad libitum and injected intraperitoneally with streptozotocin (STZ) 60 mg/kg 16 h after fasting. Animals with serum glucose concentrations of >16.7 mmol/l measured with an electronic blood glucose metre (ACCU-CHEK Performa; Roche, Mannheim, Germany) 72 h after injection of STZ were included in the study as diabetic models.
Diabetic rats were randomly divided into four groups (n = 8 in each group) and received the following treatment for 16 weeks: group 3, iv injection of unmodified RSA at 40 mg/kg•d; group 4, iv injection of HOCl-RSA at 40 mg/kg•d; group 5, iv injection of unmodified RSA at 40 mg/kg•d and intraperitoneal injection of SS-31 at 3 mg/kg•d; group 6, iv injection of HOCl-RSA at 40 mg/kg•day and intraperitoneal injection of SS-31 at 3 mg/kg•d. Male SD rats, matched for body weight with the diabetic groups and intraperitoneally injected with equal volumes of sodium citrate buffer (pH 4.5), were used as controls: group 1 (n = 8, iv injection of PBS for 16 weeks) and group 2 (n = 8, iv injection of unmodified RSA at 40 mg/kg•d for 16 weeks).
At the end of treatments, the animals were anaesthetised and exsanguinated after collection of 24-h urine samples in metabolic cages. Blood samples were taken from the abdominal aorta. The kidneys were collected after perfusion through the abdominal aorta with ice-cold normal saline to remove circulating blood cells. All animal procedures were in accordance with guidelines set by the Ethics Committee of Laboratory Animals of Southern Medical University. All the animal experiments were approved by the Ethics Committee of Laboratory Animals of Southern Medical University.
Biochemical parameters
The serum and urine creatinine levels and 24-h urinary protein were measured by an automatic biochemical analyser (MINDRAY BS480; Mindray, Shenzhen, China). The creatinine clearance rate (Ccr) was calculated as described and factored for body weightCitation22.Ccr (ml/min•kg) = [urine creatinine (μmol/l) × 24-h urine output (ml)]/[serum creatinine (μmol/l) × 1440 (min)×rat body weight (kg)].
Renal morphologic observation
A transversal section at the level of the hilus was fixed in periodatelysine-paraformaldehyde solution and embedded in paraffin. Sections (4 µm thick) of the renal tissues were stained with Masson’s trichrome and Periodic Acid-Schiff (PAS) for morphologic observation.
Determination of podocyte number in the glomerulus with immunohistochemistry
Wilms’ tumour (WT)-1, a transcription factor located in podocyte nuclei, is regarded as a specific marker of podocytes. The podocyte number was determined by staining WT1 in renal tissues with immunohistochemistry as described previouslyCitation21. In brief, the renal cortex was fixed, dehydrated, paraffin embedded, and cut into 4 µm sections. WT1 expression was analysed with rabbit anti-rat WT1 (1:50; Santa Cruz Biotechnology, Santa Cruz, CA) as the primary antibody. Control experiments were conducted by substituting the primary antibodies with nonimmune rabbit IgG. The number of WT1 positive cells in 50 randomly selected glomeruli was counted, and the mean value per glomerulus was calculated. The results were expressed as cells/glomerulus.
Determination of podocyte apoptosis
The assessment of podocyte apoptosis in vivo was performed with double-immunofluorescence labelling, including WT1 and terminal deoxyuridine nick-end labelling (TUNEL) staining. In brief, frozen renal sections were incubated with a primary anti-WT1 polyclonal antibody (1:50; Santa Cruz Biotechnology) and then a fluorescent secondary antibody, Alexa Fluor 546 (1:1000; Invitrogen, Carlsbad, CA, USA). Afterwards, TUNEL staining was performed using the In Situ Cell Death Detection kit (Roche Molecular Biochemicals, Mannheim, Germany) according to the manufacturer’s protocol. The specimens were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; BestBio, Shanghai, China; blue) for 10 min, and the images were obtained with a confocal microscope (Ni-U; Nikon Corporation, Tokyo, Japan). The cells double labelled by WT1 (red) and TUNEL (green) were counted as apoptotic podocytes.
To identify apoptosis in cultured podocytes, podocytes were cultured in 6-well plates at 37 °C to differentiate for 12 days and incubated with 200 mg/l HOCl-MSA for 12, 24, and 48 h or 200 mg/l native MSA for 48 h. In other groups of cells, 1, 10, or 50 nM SS-31 was added 30 min before HOCl-MSA stimulation for 48 h. Cells were collected for apoptosis detection using an Annexin V-FITC Apoptosis Detection Kit (KeyGen, Nanking, China) according to the manufacturer’s instructions and analysed using flow cytometry (BD FACSCalibur System, Franklin Lakes, NJ) with excitation at 488 nm and emission at 525 nm.
Detection of oxidative stress parameters
The HOCl-alb concentrations in plasma and homogenates of renal tissue were quantified as described previouslyCitation20,Citation22.
Urinary 8-hydroxy deoxyguanosine (8-OHdG) was quantified with spectrophotometry method by using a commercial Rat 8-OHdG ELISA Kit (CusAb, Wuhan, China) according to the manufacturer’s instruction.
The levels of ROSin cultured podocytes was detected with a Reactive Oxygen Species Assay Kit (Beyotime). In brief, podocytes differentiating for 12 days at 37 °C in 6-well plates were pre-incubated for 30 min with 10 µmol/l 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA). The cells were then incubated with 200 mg/l HOCl-MSA for 15, 30, and 60 min or 200 mg/l native MSA for 60 min. In other groups of cells, 50 nM SS-31 was added 30 min before HOCl-MSA stimulation for 60 min. The cells were then washed and collected. One millilitre aliquots were removed for fluorescence intensity analysis on a flow cytometer (BD FACSCalibur System).
Immunoprecipitation and western blotting
To determine binding of Apaf-1 and caspase-9 in cultured podocytes, cells were stimulated with 200 mg/L HOCl-MSA or native MSA for 24 h. Another group of cells was pre-treated with 50 nM SS-31 for 30 min before HOCl-MSA stimulation. Cells were collected for protein extraction and detected by immunoprecipitation as described previouslyCitation23,Citation24. In brief, the cell lysates were preabsorbed with protein A/G agarose beads (Santa Cruz Biotechnology) and incubated with a mouse monoclonal antibody against caspase-9 (2 μg; Santa Cruz Biotechnology). The immunocomplexes were resolved with SDS-PAGE and transferred into nitrocellulose membranes. Immunoblotting was performed using a mouse anti-Apaf-1 (1:200; Santa Cruz Biotechnology) as the primary antibody and an HRP-conjugated secondary antibody. The membranes were detected by ECL. To determine the total caspase-9, the membranes were eluted and incubated with the anti-mouse caspase-9 (1:200) and then with the HRP-conjugated secondary antibody and detected by ECL.
For determination of the release of cytochrome c, mitochondrial and cytoplasmic fractions were isolated from either renal tissue homogenate or cultured podocytes as our previousCitation20 article. The concentration of cytochrome c in the mitochondrial and cytoplasmic fractions was analysed by western blot using a primary rabbit mAb reactive to cytochrome c (1:1000; Cell Signaling Technology, Inc., Danvers, MA).
The expression of caspase-3, caspase-7, PARP-1, and WT1 was measured by western blot using total proteins extracted from renal tissue homogenate or cultured podocytes. The primary antibodies were rabbit anti-caspase-3 (1:1000), rabbit anti-caspase-7 (1:1000), rabbit anti-PARP-1 (1:1000; all from Cell Signaling Technology, Inc.) and mouse anti-WT1 (1:200; Santa Cruz Biotechnology).
Bands were quantified by densitometry (Quantity One v4.52; BioRad, Milan, Italy).
Statistical analysis
Continuous variables, expressed as the means ± SD, were compared using one-way ANOVA. Pairwise comparisons were evaluated by the Least Significant Difference (LSD) or Dunnett’s T3 procedure when the assumption of equal variances did not hold. A two-tailed p value of 0.05 was considered statistically significant. Statistical analyses were conducted using SPSS 17.0 (SPSS Inc., Chicago, IL).
Results
Characteristics of the experimental rats
As shown in , diabetic rats had a significantly lower body weight and higher blood glucose concentrations (P < .001) compared with group 1 and group 2. However, HOCl-RSA and SS-31 administration had no effect on body weight and glycaemia (P > .05 vs Group 3).
Table 1. Biochemical parameters and urine examination.
Effects of HOCl-RSA and SS-31 administration on renal tissue damage, albuminuria and renal function
Renal pathological changes were observed using Masson’s trichrome and PAS staining. No glomerular and tubulointerstitial abnormalities were observed in control rats (Group 1 and Group 2). The glomerular volume, mesangial area, and basement membrane thickness were significantly increased, accompanied with detachment, necrosis and granular, and vacuolar degeneration in partial tubular epithelial cells in diabetes rats (Group 3). These parameters were significantly deteriorated and accompanied with segmental glomerular sclerosis by HOCl-RSA challenge in diabetic rats compared with those treated with native RSA (). Renal tissue damage was in parallel with the increase of urinary protein excretion and Ccr (P < .05, Group 3 vs Group 1 and Group 2, Group 4 vs Group 3; ).
Figure 1. Pathological features of rats in each group.
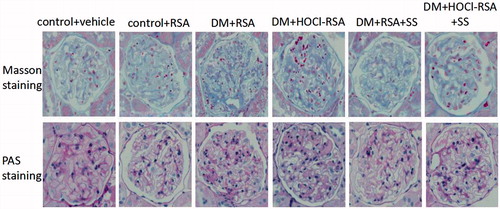
Administration of SS-31 prevented renal tissue damage and also the increase of proteinuria and Ccr in diabetic rats treated with either native RSA or HOCl-RSA (P < .01, Group 5 vs Group 3 and Group 4, Group 6 vs Group 3 and Group 4; ).
Effects of HOCl-RSA and SS-31 administration on podocyte loss and apoptosis
The glomerular podocyte number, as presented as WT1 positive cells, was significantly reduced in DM rats (p < .030, Group 3 vs Group 1 and Group 2). Repeated administration of HOCl-RSA to the DM rats further reduced the number of WT1 positive cells compared with those treated with native RSA (p < .011, Group 4 vs Group 3). SS-31 administration significantly ameliorated podocyte loss in the glomeruli of diabetic rats treated with either native RSA or HOCl-RSA (P < .05, Group 5 vs Group 3 and Group 4, Group 6 vs Group 3 and Group 4; ).
Figure 2. SS-31 attenuated podocyte loss in diabetic rats injected with native RSA or HOCl-RSA and prevented apoptosis in cultured podocytes. (A) Podocyte number in the glomerula. (B) Expression of WT1, the specific antigen of podocyte in renal tissues. Data are expressed as Mean ± SD. n = 8 in each group. ANOVA, p < .05. *p < .05 vs Group 1 and Group 2; #p < .05 vs Group 3; &p < .05 vs Group 4.
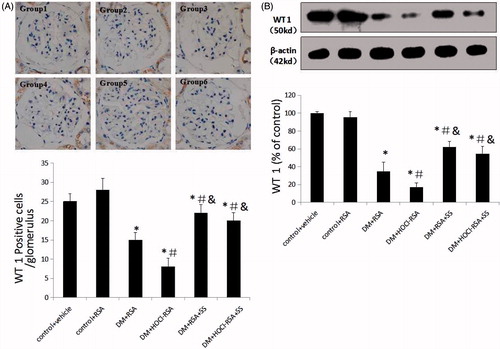
Similarly, expression of WT1, measured by western blotting, was significantly decreased in renal tissues in DM rats compared with that in the control group (p < .018 vs Group 1 and Group 2) and further decreased in the HOCl-RSA injected DM group. SS-31 treatment improved the WT1 expression in renal tissues in the DM group injected with HOCl-RSA or native RSA (p < .05, Group 5 vs Group 3 and Group 4, Group 6 vs Group 3 and Group 4; ).
To examine whether glomerular podocyte loss resulted from podocyte apoptosis and to delineate the relationship between apoptosis and changes in the podocyte number, we subsequently detected podocyte apoptosis in the glomeruli using double-immunofluorescence labelling, including WT1 and TUNEL, and found that the number of TUNEL-positive podocytes per glomerular cross-section significantly increased in DM rats treated with native or HOCl-modified RSA compared with control animals, which was ameliorated by SS-31 administration ().
Figure 3. (A) SS-31 attenuated podocyte apoptosis in diabetic rats injected with native RSA or HOCl-RSA. (B) HOCl-MSA induced apoptosis time-dependently prevented by SS-31 in cultured podocytes. Data are expressed as Mean ± SD. ANOVA, p < .05. *p < .05 vs Medium and MSA; #p < .05 vs Group HOCl-MSA 48h.
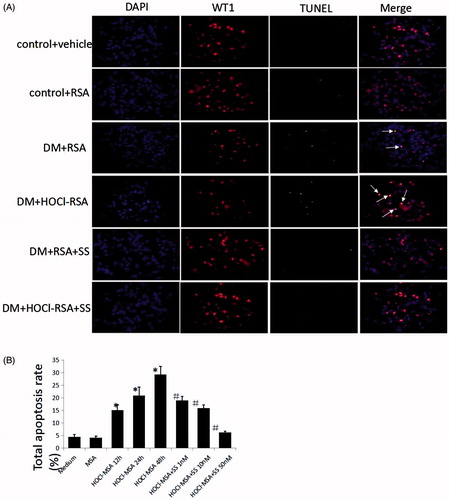
We detected podocyte apoptosis in vitro with Annexin V. HOCl-MSA-induced apoptosis of the cells in a time-dependent manner. After incubation of 200 mg/l HOCl-MSA with cells for 12, 24 or 48 h, the apoptosis rate of the cells was 15.07 ± 2.11%, 20.91 ± 3.31%, and 29.30 ± 3.24% (p < .001 compared with the medium group), respectively. The same doses of native MSA had no effect on the cells (p = .812 compared with the medium group). Doses of 1, 10, and 50 nM SS-31 dose-dependently inhibited HOCl-MSA-induced podocyte apoptosis. The rates of inhibition were 35.03%, 45.72%, and 78.70%, respectively (p < .001 compared with cells stimulated with HOCl-MSA for 48 h) ().
Effects of HOCl-RSA and SS-31 administration on oxidative parameters
The HOCl-alb concentrations reflect the redox state in diabetesCitation25,Citation26. We examined the HOCl-alb concentrations in plasma and renal tissue homogenate of animals. The plasma HOCl-alb concentrations were spontaneously elevated in diabetic rats compared with the controls (p < .05, Group 3 vs Group 1 and Group 2). The concentration of plasma HOCl-alb increased by more than 1-fold in HOCl-RSA-treated diabetic rats compared with RSA-treated diabetic animals (p = .007, Group 4 vs Group 3; ). Similarly, the HOCl-alb concentrations in the renal homogenates were elevated in diabetic rats (p < .05, Group 3 vs Group 1 and Group 2). Chronic administration of HOCl-RSA significantly enhanced the HOCl-alb concentrations in diabetic kidney (p = .004, Group 4 vs Group 3; ). The intervention of SS-31 significantly restored the increase of the HOCl-alb concentration in both plasma and renal tissues of diabetic rats (p < .05, Group 5 and Group 6 vs Group 3 and Group 4; .
Figure 4. Changes of the oxidative stress indexes. SS-31 decreased HOCl-alb concentration in plasma (A) and renal tissue (B) of diabetic rats injected with native RSA or HOCl-RSA. Data are expressed as Mean ± SD. n = 8 in each group. ANOVA, p < .05. n = 8 in each group. *p < .05 vs Group 1 and Group 2; #p < .05 vs Group 3; &p < .05 vs Group 4. (C) HOCl-MSA-induced ROS production (represented as fluorescent intensity) time-dependently prevented by SS-31 in cultured podocytes. Data are expressed as Mean ± SD. ANOVA, p < .05. *p < .05 vs Group MSA; #p < .05 vs Group HOCl-MSA 60 min.
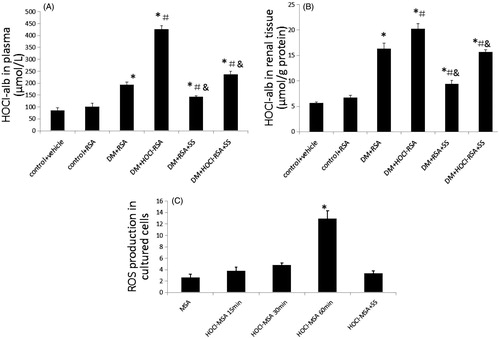
8-Hydroxy deoxyguanosine (8-OHdG) is an end product and marker of mitochondrial DNA oxidation and excreted in urine. The concentration of urinary 8-OHdG in diabetic rats increased compared with the control groups (p < .01, Group 3 vs Group 1 and Group 2). The concentration of urinary 8-OHdG in diabetic rats injected with HOCl-RSA further increased compared with rats injected with native RSA (p = .015, Group 4 vs Group 3). SS-31 administration significantly restored the increased concentration of 8-OHdG in urine in diabetic rats injected with HOCl-RSA or native RSA (p < .05, Group 5 and Group 6 vs Group 3 and Group 4; ).
In vitro, incubation of 200 mg/L HOCl-MSA with podocytes for 15, 30, and 60 min time-dependently induced an increase of intracellular ROS measured by the DCFH-DA fluorescent probe by 0.44 (p = .090), 0.82 (P = .006), and 3.92 times (p < .001) compared with native MSA-treated cells. Treatment with 50 nM SS-31 ameliorated the increase of intracellular ROS induced by HOCl-MSA stimulation for 60 min (p < .0001; ). DCFH-DA is prone to autoxidation, photo-oxidation, and autocatalysis thus leading to the bias of the experimental results. For example, auto-oxidation of DCFH-DA occurs when alkaline activation and dilution, in room air and under culture conditions. The rate of auto-oxidation increases in a linear fashion over time. About 20% of DCFH-DA undergo deacetylation in 1 hCitation27. That means about 80% DCFH-DA still hasn't stepped into the condition of auto-oxidation. We also took some methodologies into considerations. Such as stock and working solutions of DCFH-DA were made in a light-protected vessel and in ice. The working solution was prepared fresh and used only once. As incubation of cells with DCFH-DA for no shorter than the experimental groups in the control group (MSA group) showed fewer rate of DCFH-DA oxidation () in the present study, the influence of DCFH-DA auto-oxidation can be negligible.
Effects of HOCl-RSA and SS-31 administration on the release of cytochrome c from mitochondria to the cytoplasm
The release of cytochrome c (cyt c) from mitochondria to the cytoplasm reflected mitochondrial damage. In the present study, western blotting was used to detect the changes of cyt c protein concentration in mitochondria and cytoplasm either of renal tissues of rats or of cultured podocytes.
Compared with the control groups, the cyt c protein concentration significantly increased in the cytoplasm in renal tissues of diabetic rats, whereas the mitochondrial cyt c protein concentration was significantly reduced (p < .05 vs Group 1 and Group 2). Repeated injection of HOCl-RSA further increased the cyt c protein concentration in the cytoplasm and reduced that in the mitochondria in renal tissues of diabetic rats injected with native RSA (p = .008, Group 4 vs Group 3). By contrast, administration of SS-31 restored the increase of cyt c in cytoplasm and reduction of cyt c in mitochondria in renal tissues of DM rats injected with HOCl-RSA or native RSA (p < .05, Group 5 and Group 6 vs Group 3 and Group 4; ).
Figure 5. Content of cytochrome c in cytoplasm and mitochondria in vivo (A) and in vitro (B). Data are expressed as Mean ± SD. ANOVA, p < .05. (A) n = 8 in each group. *p < .05 vs Group 1 and Group 2; #p < .05 vs Group 3; &p < .05 vs Group 4. (B) *p < .05 vs Group MSA; #p < .05 vs Group HOCl-MSA 24 h.
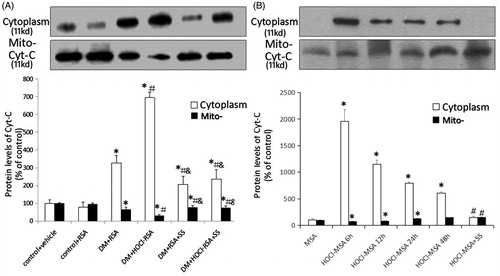
Cultured podocytes were stimulated with 200 mg/L HOCl-MSA for 6, 12, 24, and 48 h. Another group of cells was pre-treated with 50 nM SS-31 for 30 min before HOCl-MSA stimulation for 24 h. We found that HOCl-MSA promoted the increase of cyt c in cytoplasm and decrease in mitochondria of cells, which was the most obvious in the time point of 6 h (p < .05 vs native MSA-treated cells). The changes induced by HOCl-MSA were prevented by adding SS-31 to the cells before HOCl-MSA stimulation (p < .05 vs HOCl-MSA-treated cells for 24 h; ).
Effects of SS-31 on the binding of apaf-1 and caspase-9 induced by HOCl-RSA in cultured podocytes
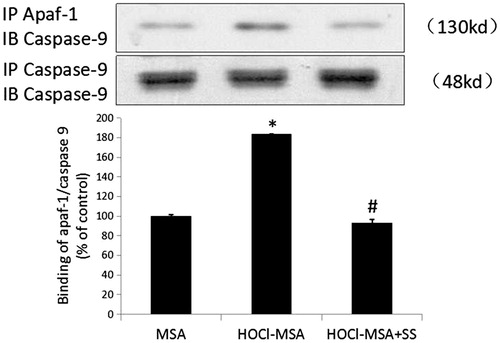
Following the release of cyt c into the cytoplasm, the caspase recruitment domain of apoptosis activated factor-1 (Apaf-1) was exposed and bound to caspase-9, which led to activation of caspase-3 and caspase-7, as well as cell apoptosis. We detected the combination of apaf-1 and caspase-9 with immunoprecipitation and western blotting and found that 200 mg/L HOCl-MSA stimulation for 24 h promoted the binding of Apaf-1 and caspase 9 in the cells (p < .0001 vs native MSA-treated cells), which was prevented by pre-addition of SS-31 to the cells (p = .001 vs HOCl-MSA-treated cells) ().
Figure 6. HOCl-MSA promoted binding of apaf-1 and caspase 9, which was prevented by SS-31 in cultured podocyte. Data are expressed as Mean ± SD. ANOVA, p < .05. *p < .05 vs Group MSA; #p < .05 vs Group HOCl-MSA.
Effects of HOCl-RSA and SS-31 administration on the expression of apoptosis-related proteins
Compared with control groups, protein expression of cleaved fragments of caspase-3, caspase-7, and PARP-1 (116 kD) in renal tissues in diabetic rats was significantly upregulated (p < .05 vs Group 1 and Group 2). Expression of cleaved fragments of caspase-3, caspase-7, and PARP-1 (116 kD) was further upregulated in diabetic rats injected with HOCl-RSA compared with those injected with native RSA (p < .01 vs Group3). SS-31 administration significantly restored the upregulation of cleaved fragments of caspase-3, caspase-7, and PARP-1 (116 kD) of diabetic rats injected with both HOCl-RSA and native RSA (p < .05 vs Group 3 and Group 4). The changes of pro-caspase-3, pro-caspase-7, and PARP-1 (89 kD) were opposite to those of cleaved-caspase-3, cleaved-caspase-7, and PARP-1 (116 kD) ().
Figure 7. Changes of caspase and PARP in vivo (A) and in vitro (B). Data are expressed as Mean ± SD. ANOVA, p < .05. (A) n = 8 in each group. *p < .05 vs Group 1 and Group 2; #p < .05 vs Group 3; &p < .05 vs Group 4. (B) *p < .05 vs Group MSA; #P < .05 vs Group HOCl-MSA 24 h.
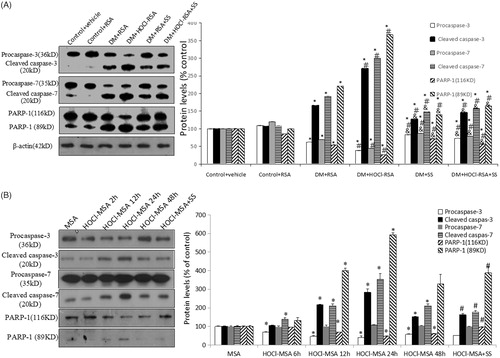
Stimulation of 200 mg/L HOCl-MSA for 6, 12, 24, and 48 h time-dependently upregulated protein expression of cleaved-caspase-3, cleaved-caspase-7, and PARP-1 (116 kD) in cultured podocytes (p < .05 vs native MSA-treated cells), which was prevented by pre-treatment of 50 nM SS-31 for 30 min before HOCl-MSA stimulation for 24 h (p = .018 vs HOCl-MSA-treated cells for 24 h). Changes of pro-caspase-3, pro-caspase-7, and PARP-1 (89 kD) were opposite to the cleaved-caspase-3, cleaved-caspase-7, and PARP-1 (116 kD) ().
Discussion
It is extremely important to study the mechanisms of the SS peptide for renal protection in DM, a specific mitochondrial targeted antioxidant, in view of the key roles of mitochondrial perturbation in the development of DKD. The present study, for the first time, provided evidence for SS peptide attenuation of podocyte apoptosis enhancement by oxidation-modified albumin in DM and in vitro through a mitochondria-mediated signalling pathway.
Our data indicated that SS-31 prevented podocyte loss and apoptosis along with an increase of urinary protein and aggravation of renal pathological injury (as evidenced by hypertrophy and basement membrane thickening or segmental sclerosis in glomeruli, as well as focal detachment, necrosis and granular and vacuolar degeneration in tubular epithelial cells), all of which were enhanced by HOCl-RSA administration in DM rats. SS-31 had no effect on blood glucose, indicating that the protection of SS-31 was independent of hypoglycaemic effects. The benefits of SS-31 were further confirmed by in vitro experimental data that showed that SS-31 dose-dependently prevented HOCl-MSA-induced podocyte apoptosis. Podocyte injury manifested as the fusion of the foot process, hypertrophy, detachment, loss or death, led to glomerular sclerosis and was an important cellular basis for the initiation and early development of DKD. The reduction of the podocyte number indicated the progression of glomerulopathyCitation28. Therefore, an effective therapy for podocyte injury might retard the progression of DKD at an early stage. Similar to our results, olmesartanCitation29, valsartan, and mycophenolate mofetilCitation30,Citation31 all prevented microalbuminuria and progression of DKD through inhibition of loss or apoptosis of podocytes in animals with DM. Astragaloside ameliorated glomerular sclerosis and renal inflammation by inhibiting podocyte apoptosis in a DM mouse modelCitation32, and metformin regulated human podocytes apoptosis under high glucose conditions in vitroCitation33.
The mechanisms involved in the protective effects of SS-31 on podocytes from reduction and apoptosis remained to be clarified. Simultaneous with deterioration and apoptosis in podocytes, proteinuria, and renal pathological injury, the content of HOCl-alb in plasma and renal tissues increased in rats after STZ injection and were enhanced by repeated intravenous administration of HOCl-RSA, which was consistent with our previous reportCitation22. Urinary 8-OHdG also increased in DM rats and was enhanced by HOCl-RSA administration. In vitro, HOCl-MSA induced an increase in ROS production in mouse podocytes in a time-dependent manner. These data indicated that HOCl-alb was spontaneously generated in DM, as reported previouslyCitation34,Citation35, and further enhanced oxidative stress and modification, thus forming a positive feedback loop and a vicious cycle of damage to podocytes and the glomerular filtration membrane. Although multiple pathways might result in ROS generation, recent studies indicate that mitochondria are a major source of ROS in many renal cells, including podocytesCitation36. ROS generated in mitochondria were 5–10 times higher than other cytoplasmic and nuclear componentsCitation9. Increased generation of mtROS had been observed in an experimental DM modelCitation37. mtROS emerged as a critical pathogenic factor that leads to DKDCitation38. In our results, SS-31, a tetrapeptide targeted in the inner membrane and clearing the local ROS of mitochondria, prevented HOCl-alb-enhanced oxidative stress in DM rats and in vitro, indicating that ROS were mainly derived from mitochondria and that SS-31 prevented injury to podocytes and renal tissues in DM by reducing mtROS and breaking the vicious cycle of oxidative stress.
Mitochondria are the main source and victims of ROS. cytochrome c is physically bound to the inner mitochondrial membrane by an association with cardiolipinCitation39. ROS result in cardiolipin oxidation, decreasing its binding affinity for cytochrome c and facilitating the detachment of cytochrome c from the inner mitochondrial membrane and release to the cytoplasmCitation40. The release of cyt c is a critical step in apoptosisCitation41,Citation42. cyt c combines with apoptosis activated factor-1 (Apaf-1) in the cytoplasm and facilitates the oligomerization of Apaf-1, which recruits caspase-9 and forms the cyt c-Apaf-1-caspase-9 complex, namely, the apoptosome. Caspase-9 is self-activated through alterations of its molecular conformation and is then cleaved, activating effector caspases, such as caspase-3 and -7, resulting in widespread proteolysis and commitment to cell deathCitation43. Thus, measures against cyt c release might be a key step in preventing apoptosis pathway activation. In our experiments, SS-31 alleviated the release of cyt c from mitochondria to the cytoplasm in DM rats injected with native RSA and HOCl-RSA. Accordingly, SS-31 alleviated the increase of protein expression of cleavage of caspase-3, caspase-7, and PARP-1, which are aggravated by HOCl-RSA in DM rats. In vitro, SS-31 successively inhibited HOCl-MSA-induced release of cyt c, the combination of Apaf-1 and caspase-9 and cleavage of caspase-3, caspase-7, and PARP-1 in cultured mouse podocytes.
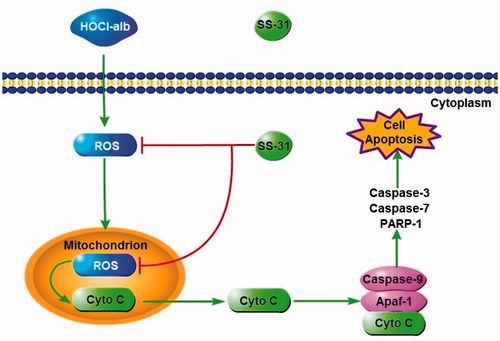
The present study established a cascade from HOCl-alb and oxidative stress, which culminated in mitochondria, to podocyte apoptosis in DM and provided new insight on the pathogenesis of DKD. The SS peptide inhibited mitochondrial oxidative damage and cyt c release through localisation in mitochondria and elimination of mtROS, prevented activation of apoptosis pathways (including binding of cyt c, Apaf-1, and caspase-9, the formation of apoptosome and activation of effector caspases) and podocyte injury and thus alleviated proteinuria and kidney disease in DM. Mitochondrial-targeted peptides might be a novel and effective therapy for prevention or retardation of the progression of renal complications in DM ().
Figure 8. A working model how SS-31 plays a protective role in podocyte by ameliorating podocyte apoptosis through a HOCl-alb-enhanced and mitochondria-dependent signalling pathway.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Conserva F, Gesualdo L, Papale M. A systems biology overview on human diabetic nephropathy: from genetic susceptibility to post-transcriptional and post-translational modifications. J Diabetes Res 2016;2016:7934504.
- Fox CS, Matsushita K, Woodward M, et al. Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without diabetes: a meta-analysis. Lancet 2012;380:1662–73.
- Pagtalunan ME, Miller PL, Jumping-Eagle S, et al. Podocyte loss and progressive glomerular injury in type ii diabetes. J Clin Invest 1997;99:342–8.
- Weil EJ, Lemley KV, Mason CC, et al. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int 2012;82:1010–7.
- Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006;55:225
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001;414:813–20.
- Witko-Sarsat V, Friedlander M, Capeillere-Blandin C, et al. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int 1996;49:1304–13.
- Tang DD, Niu HX, Peng FF, et al. Hypochlorite-modified albumin upregulates ICAM-1 expression via a MAPK-NF-kappab signaling cascade: protective effects of apocynin. Oxid Med Cell Longev 2016;2016:1852340.
- Small DM, Coombes JS, Bennett N, et al. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology (Carlton) 2012;17:311–21.
- Indo HP, Yen HC, Nakanishi I, et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J Clin Biochem Nutr 2015;56:1–7.
- Gnudi L, Coward RJ, Long DA. Diabetic nephropathy: Perspective on novel molecular mechanisms. Trends Endocrinol Metab 2016;27:820–30.
- Kumar A, Yerra VG, Malik RA. Comment on sharma. Mitochondrial hormesis and diabetic complications. Diabetes 2015;64:663–72. Diabetes 2015:64:e32–3; discussion e34.
- Myung SK, Ju W, Cho B, et al. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ 2013;346:f10.
- Walsh PC. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. J Urol 2005;174:1823–4.
- Zhao K, Zhao GM, Wu D, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 2004;279:34682–90.
- Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal 2008;10:601–19.
- Cai M, Li J, Lin S, et al. Mitochondria-targeted antioxidant peptide ss31 protects cultured human lens epithelial cells against oxidative stress. Curr Eye Res 2015;40:822–9.
- Zhao WY, Han S, Zhang L, et al. Mitochondria-targeted antioxidant peptide ss31 prevents hypoxia/reoxygenation-induced apoptosis by down-regulating p66shc in renal tubular epithelial cells. Cell Physiol Biochem 2013;32:591–600.
- Hou Y, Li S, Wu M, et al. Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am J Physiol Renal Physiol 2016;310:F547–59.
- Zhao H, Liu YJ, Liu ZR, et al. Role of mitochondrial dysfunction in renal fibrosis promoted by hypochlorite-modified albumin in a remnant kidney model and protective effects of antioxidant peptide SS-31. Eur J Pharmacol 2017;804:57–67.
- Zhou LL, Hou FF, Wang GB, et al. Accumulation of advanced oxidation protein products induces podocyte apoptosis and deletion through NADPH-dependent mechanisms. Kidney Int 2009;76:1148–60.
- Shi XY, Hou FF, Niu HX, et al. Advanced oxidation protein products promote inflammation in diabetic kidney through activation of renal nicotinamide adenine dinucleotide phosphate oxidase. Endocrinology 2008;149:1829–39.
- Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and DATP-dependent formation of apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997;91:479–89.
- Guo ZJ, Niu HX, Hou FF, et al. Advanced oxidation protein products activate vascular endothelial cells via a rage-mediated signaling pathway. Antioxid Redox Signal 2008;10:1699–712.
- Taylor EL, Armstrong KR, Perrett D, et al. Optimisation of an advanced oxidation protein products assay: its application to studies of oxidative stress in diabetes mellitus. Oxid Med Cell Longev 2015;2015:496271.
- Piwowar A, Knapik-Kordecka M, Warwas M. Markers of oxidative protein damage in plasma and urine of type 2 diabetic patients. Br J Biomed Sci 2009;66:194–9.
- Chen X, Zhong Z, Xu Z, et al. 2',7'-dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: forty years of application and controversy. Free Radic Res 2010;44:587–604.
- Brosius FC, Coward RJ. Podocytes, signaling pathways, and vascular factors in diabetic kidney disease. Adv Chronic Kidney Dis 2014;21:304–10.
- Gu J, Yang M, Qi N, et al. Olmesartan prevents microalbuminuria in db/db diabetic mice through inhibition of angiotensin ii/p38/sirt1-induced podocyte apoptosis. Kidney Blood Press Res 2016;41:848–64.
- Gao F, Yao M, Cao Y, et al. Valsartan ameliorates podocyte loss in diabetic mice through the notch pathway. Int J Mol Med 2016;37:1328–36.
- Lv W, Zhang Y, Guan G, et al. Mycophenolate mofetil and valsartan inhibit podocyte apoptosis in streptozotocin-induced diabetic rats. Pharmacology 2013;92:227–34.
- Guo H, Cao A, Chu S, et al. Astragaloside IV attenuates podocyte apoptosis mediated by endoplasmic reticulum stress through upregulating sarco/endoplasmic reticulum ca2+-ATPase 2 expression in diabetic nephropathy. Front Pharmacol 2016;7:500.
- Langer S, Kreutz R, Eisenreich A. Metformin modulates apoptosis and cell signaling of human podocytes under high glucose conditions. J Nephrol 2016;29:765–73.
- Korkmaz GG, Konukoglu D, Kurtulus EM, et al. Total antioxidant status and markers of oxidative stress in subjects with normal or impaired glucose regulation (IFG, IGT) in diabetic patients. Scand J Clin Lab Invest 2013;73:641–9.
- Gradinaru D, Borsa C, Ionescu C, Margina D. Advanced oxidative and glycoxidative protein damage markers in the elderly with type 2 diabetes. J Proteomics 2013;92:313–22.
- Casalena G, Krick S, Daehn I, et al. Mpv17 in mitochondria protects podocytes against mitochondrial dysfunction and apoptosis in vivo and in vitro. Am J Physiol Renal Physiol 2014;306:F1372–80.
- Chacko BK, Reily C, Srivastava A, et al. Prevention of diabetic nephropathy in Ins2(+/)−(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem J 2010;432:9–19.
- Jha JC, Banal C, Chow BS, et al. Diabetes and kidney disease: Role of oxidative stress. Antioxid Redox Signal 2016;25:657–84.
- Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis 2007;12:913–22.
- Yu L, Liu Y, Wu Y, et al. Smad3/nox4-mediated mitochondrial dysfunction plays a crucial role in puromycin aminonucleoside-induced podocyte damage. Cell Signal 2014;26:2979–91.
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for bcl-2 regulation of apoptosis. Science 1997;275:1132–6.
- Eisenreich A, Langer S, Herlan L, Kreutz R. Regulation of podoplanin expression by microrna-29b associates with its antiapoptotic effect in angiotensin ii-induced injury of human podocytes. J Hypertens 2016;34:323–31.
- Sanz AB, Santamaria B, Ruiz-Ortega M, et al. Mechanisms of renal apoptosis in health and disease. J Am Soc Nephrol 2008;19:1634–42.