
Abstract
The unregulated activation of STAT3 has been demonstrated to occur in many cancers and enhances tumour growth, migration, and invasion. Stimulation by cytokines, growth factors, and hormones triggers this activation by phosphorylating STAT3 at tyrosine 705. Novel imidazopyridine compounds were synthesized to evaluate the inhibition of STAT3 at Y705. Among the tested compounds, 16 reduced the level of phospho-STAT3, inhibited the downstream signalling cascade and subsequently attenuated the survival of hepatocellular carcinoma (HCC) cells. Further assays showed that the reduction effects of compound 16 on tyrosine 705 of STAT3 were attributed to up-regulation of protein tyrosine phosphatase SHP-1.
Keywords:
Introduction
Recent cancer drug discovery has focused on molecular target therapy to reduce nonspecific cytotoxicity in patients. Targeting oncogenes or activating tumour suppressor genes with small molecules is a new approach for cancer treatment. Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that plays a central role in tumour cell proliferation, survival, invasion, and immunosuppressionCitation1. The STAT3 pathway is activated by several receptors, including those for the interleukin-6 (IL-6) family cytokinesCitation2, G-protein-coupled receptors (GPCRs)Citation3,Citation4, and Toll-like receptors (TLRs)Citation5. Once the signal is activated, STAT3 is phosphorylated at the tyrosine 705 residue, resulting in automatic dimerization in the cytosol. The dimeric STAT3 translocates from the cytosol to the nucleus for the transcription of its target genes, such as Mcl-1, Bcl-xl, and VEGF-ACitation6–8 and further regulates cell proliferation, apoptosis, and migration. This phosphorylation activation of STAT3 is negatively regulated by protein tyrosine phosphatases, such as SHP-1, SHP-2, and PTP1B by targeting the phosphor group at tyrosine 705Citation9,Citation10. Therefore, the enhancement of protein tyrosine phosphatases is another approach for target therapy.
Potentiating the effect of traditional chemotherapy agents and targeting chemotherapy-induced signalling cascades is a practical approach for combating resistant cancer cells. In gastric cancer cells, inhibition of STAT3 phosphorylation re-sensitized the chemotherapeutic agent treatmentCitation11.
Recently, not only prodigiosin, a natural product extracted from Serratia marcescens, but also obatoclax, a synthetic oligopyrrole scaffold derived from prodigiosin, have been shown to regulate biological and pharmacological effects ()Citation12–14. The combination of DNA damaging doxorubicin and prodigiosin showed significant growth inhibition in human small-cell lung doxorubicin-resistant cancer cellsCitation15. Obatoclax, currently in the clinical trial stage, has also been applied to synergize with targeted agents such as bortezomibCitation16 and rituximabCitation17 in various cancers, and also combined with OlaparibCitation18 and LapatinibCitation19 in preclinical studies.
Figure 1. The structure of obatoclax, prodigiosin, SC-2001, and the core structure of pyrrole-benzimidazole.
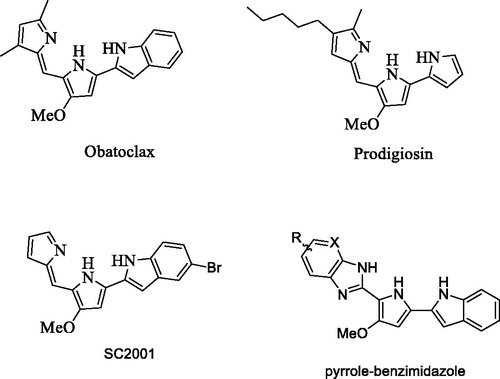
Previously, we designed and synthesized obatoclax derivatives by the replacement of the indole ring with thiophene and furan, resulting in a new chemical entity possibly suitable for the development of anticancer agentsCitation20. Moreover, these derivatives induced cell apoptosis through a novel mechanism by targeting the SHP-1/STAT3 pathway, a different mechanism from obatoclax to Mcl-1, Bcl-2, and Bcl-xL inhibitionCitation21. Therefore, our hypothesis is that pyrrole-benzimidazole could be a new pharmacophore for SHP-1 enhancement which represses STAT3 activation and induces cell apoptosis (). In this study, we designed a short synthetic scheme for pyrrole-benzimidazole and synthesized various pyrrole-benzimidazole-based compounds aiming to find compounds that enhance SHP-1 activity (Scheme 1). The structure–activity relationship of these compounds was investigated using hepatocellular carcinoma (HCC) cancer cells. We further studied the SHP-1 activity and p-STAT-3 inhibition by treating HCC cells with the effective compounds.
Materials and methods
Chemistry
Proton nuclear magnetic resonance (1H-NMR and 13C-NMR) spectra were recorded on Bruker Avance III (400 MHz, Bruker, Billerica, MA) instruments. Reaction progress was determined by thin layer chromatography (TLC) analysis on a silica gel 60 F254 plate (Merck, Darmstadt, Germany). Chromatographic purification was carried out on silica gel columns 60 (0.063–0.200 mm or 0.040–0.063 mm; Merck), basic silica gel. Commercial reagents and solvents were purchased from Aldrich (Darmstadt, Germany) and Acros Chemical Corporation (Bridgewater, NJ) without additional purification. High-resolution mass spectra were recorded on a FINNIGAN MAT 95S mass spectrometer.
General procedure for the synthesis of compound 5
Compound 3 (1.0 equiv.) and NaHSO3 (2.0 equiv.) were mixed in ethanol solution. After the mixtures were heated with a 50 Watt microwave at 90 °C for 10 min, the solution was evaporated with rotavapor. The crude mixtures were added with a solution of dimethylformamide and 1.2 equiv benzene-diamine. The reaction was stirred in 50 Watt microwave reactor at 125 °C for 30 min, poured into 20 ml water and extracted with ethyl acetate (30 ml) 3 times. The organic layer was collected, washed with brine, dried with MgSO4. The concentrated crude product was purified by silica gel with elute (ratio: ethyl acetate/hexane =1/6 to 1/2). The yield of coupling product in this procedure was 4–24%.
Tert-butyl-2-(5-(1H-benzo[d]imidazol-2-yl)-4-methoxy-1H-pyrrol-2-yl)-1H-indole-1-carboxylate (6)
Tert-butyl-2–(5-(1H-benzo[d]imidazol-2-yl)-4-methoxy-1H-pyrrol-2-yl)-1H-indol. 1H NMR (400 MHz, CDCl3): δ = 8.42 (s, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.52 (d, J = 7.2 Hz, 1H), 7.29 (t, J = 7.2 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 6.96 (t, J = 7.2 Hz, 1H), 6.83 (s, 1H), 6.73 (d, J = 7.2 Hz, 2H), 6.19 (s, 1H), 3.88 (s, 3H), 1.59 (s, 10H).
2–(5-(5-bromo-1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-1H-benzo[d] imidazole (7)
1H NMR (400 MHz, MeOH-d4): δ = 7.63 (d, J = 1.6 Hz, 1H), 7.52 (dd, J = 6.0, 3.2 Hz, 2H), 7.25 (d, J = 8.4 Hz, 1H), 7.1 5–7.18 (m, 3H), 6.67 (s, 1H), 6.49 (s, 1H), 4.02 (s, 3H) ppm. 13 C NMR (100 MHz, MeOH-d4): δ = 151.4, 146.5, 139.5, 136.8, 133.3, 131.9, 127.1, 125.1, 123.0, 122.8, 115.3, 115.1, 114.8, 114.1, 113.5, 113.1, 108.2, 97.6, 94.3, 58.5 ppm.
2–(5-(1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-1H-benzo[d]imidazole (8)
1H NMR (400 MHz, MeOH-d4): δ = 7.5 1–7.54 (m, 3H), 7.36 (d, J = 8.2 Hz, 1H), 7.1 7–7.19 (m, 2H), 7.10 (t, J = 8.2 Hz, 1H), 7.01 (t, J = 7.6 Hz, 1H), 6.73 (s, 1H), 6.51 (s, 1H), 4.05 (s, 3H) ppm. 13 C NMR (100 MHz, MeOH-d4): δ = 151.9, 146.8, 138.4, 132.0, 130.3, 128.2, 123.1, 122.8, 121.0, 120.7, 114.9, 111.8, 107.8, 98.4, 94.1, 58.7 ppm. HRMS calculated for C20H16N4O (M + H): 329.1402, Found: 329.1403.
2–(4-methoxy-1H, 1'H-[2, 2'-bipyrrol]-5-yl)-1H-benzo[d]imidazole (9)
1H NMR (400 MHz, MeOH-d4): δ = 7.4 7 ∼ 7.49 (m, 2H), 7.1 3 ∼ 7.15 (m, 2H), 6.78 (dd, J = 3.2, 1.6 Hz, 1H), 6.42 (dd, J = 3.2, 1.6 Hz, 1H), 6.23 (s, 1H), 6.16 (t, J = 3.2 Hz, 1H), 4.01 (s, 3H) ppm. HRMS calculated for C16H14N4O (M + H): 279.1246, Found: 279.1248.
6-bromo-2-(5-(5-bromo-1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-3H-imidazo[4,5-b]pyridine (10)
1H NMR (400 MHz, MeOH-d4): δ = 8.30 (s, 1H), 7.98 (s, 1H), 7.67 (d, J = 2.0 Hz, 1H), 7.30 (d, J = 8.8 Hz, 1H), 7.20 (dd, J = 8.8, 2.0 Hz, 1H), 6.77 (s, 1H), 6.54 (s, 1H), 4.07 (s, 3H) ppm. 13 C NMR (100 MHz, MeOH-d4): δ = 136.8, 132.8, 131.8, 129.0, 125.4, 123.1, 113.5, 113.2, 98.4, 96.7, 94.2, 58.5 ppm.
2–(5-(5-bromo-1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-3H-imidazo[4,5-b]pyridine (11)
1H NMR (400 MHz, MeOH-d4): δ = 8.24 (d, J = 3.6 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.65 (s, 1H), 7.26 (d, J = 8.8 Hz, 1H), 7.1 6–7.19 (m, 2H), 6.72 (s, 1H), 6.50 (s, 1H), 4.07 (s, 1H) ppm. 13 C NMR (100 MHz, MeOH-d4): δ = 153.1, 143.7, 137.1, 133.2, 132.1, 128.7, 125.6, 123.4, 120.1, 118.3, 113.8, 113.4, 107.9, 98.4, 94.4, 58.8 ppm. HRMS calculated for C19H14BrN5O (M + H): 408.0460, Found: 408.0459.
2–(5-(5-bromo-1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-5-fluoro-1H-benzo[d]imidazole (12)
1H NMR (400 MHz, MeOH-d4): δ = 7.60 (d, J = 1.6 Hz, 1H), 7.43 (dd, J = 8.8, 4.8 Hz, 1H), 7.22 (d, J = 8.8 Hz, 1H), 7.19 (dd, J = 9.2, 2.4 Hz, 1H), 7.14 (dd, J = 8.8, 1.6 Hz, 1H), 6.91 (td, J = 9.2, 2.4 Hz, 1H), 6.63 (s, 1H), 6.43 (s, 1H), 3.98 (s, 3H) ppm. 13 C NMR (100 MHz, MeOH-d4): δ = 161.9, 159.5, 151.8, 137.0, 133.5, 133.4, 132.1, 132.0, 127.5, 125.4, 123.2, 113.7, 113.3, 110.7, 110.5, 108.3, 97.9, 94.5, 58.7 ppm. HRMS calculated for C20H14BrFN4O (M–H): 423.0257, Found: 423.0243.
2–(5-(5-bromo-1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-1H-benzo[d] imidazole-5-carbonitrile (13)
1H NMR (400 MHz, MeOH-d4): δ = 7.86 (s, 1H), 7.66 (d, J = 2.0 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 7.19 (dd, J = 8.4, 2.0 Hz, 1H), 6.75 (s, 1H), 6.54 (s, 1H), 4.07 (s, 3H) ppm. HRMS calculated for C21H14BrN5O (M-H): 430.0303, Found: 430.0326
3–((2–(5-(5-bromo-1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-1H-benzo[d] imidazol-6-yl)oxy)aniline (14)
1H NMR (400 MHz, MeOH-d4): δ = 7.65 (d, J = 1.6 Hz, 1H), 7.48 (d, J = 8.8 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.18 (dd, J = 8.4, 1.6 Hz, 1H), 7.15 (d, J = 2.0 Hz, 1H), 7.03 (t, J = 8.0 Hz, 1H), 6.90 (dd, J = 8.8, 2.0 Hz, 1H), 6.69 (s, 1H), 6.52 (s, 1H), 6.43 (dd, J = 8.0, 2.0 Hz, 1H), 6.36 (t, J = 2.4 Hz, 1H), 6.30 (dd, J = 8.0, 2.4 Hz, 1H), 4.04 (s, 1H) ppm.
3–((2–(5-(5-bromo-1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-1H-benzo[d] imidazol-6-yl)oxy)benzonitrile (15)
1H NMR (400 MHz, MeOH-d4): δ = 7.65 (d, J = 2 Hz, 1H), 7.57 (d, J = 8.4 Hz, 1H), 7.49 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 7.6 Hz, 1H), 7.2 6 ∼ 7.28 (m, 3H), 7.24 (d, J = 2.4 Hz, 1H), 7.18 (dd, J = 8.8, 2 Hz, 1H), 6.94 (dd, J = 8.4, 2.4 Hz, 1H), 6.71 (s, 1H), 6.53 (s, 1H), 4.05 (s, 3H) ppm. 13 C NMR (100 MHz, MeOH-d4): δ = 152.2, 151.9, 137.0, 133.5, 132.1, 127.0, 125.4, 123.3, 123.1, 121.3, 120.5, 119.2, 116.0, 114.5, 113.7, 113.4, 98.0, 94.6, 58.8 ppm.
2–(5-(1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-6-bromo-3H-imidazo[4,5-b]pyridine (16)
1H NMR (400 MHz, MeOH-d4): δ = 8.25 (s, 1H), 7.91 (s, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.10 (t, J = 8.0 Hz, 1H), 7.02 (t, J = 8.0 Hz, 1H), 6.76 (s, 1H), 6.47 (s, 1H), 4.04 (s, 3H) ppm. 13 C NMR (100 MHz, MeOH-d4): δ = 153.7, 144.2, 138.5, 131.6, 130.3, 130.1, 123.1, 121.2, 120.8, 111.9, 107.2, 99.3, 94.0, 58.6 ppm. HRMS calculated for C19H14BrN5O (M-H): 406.0303, Found: 406.0302.
2–(5-(1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-3H-imidazo[4,5-b]pyridine (17)
1H NMR (400 MHz, MeOH-d4): δ = 8.23 (d, J = 4.4 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 7.6 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.17 (dd, J = 8.0, 4.8 Hz, 1H), 7.11 (t, J = 7.6 Hz, 1H), 7.01 (t, J = 7.6 Hz, 1H), 6.77 (s, 1H), 6.50 (s, 1H), 4.06 (s, 3H) ppm. 13 C NMR (100 MHz, MeOH-d4): δ = 153.2, 143.5, 138.5, 131.7, 130.3, 129.5, 122.9, 121.1, 120.7, 118.2, 111.8, 107.4, 98.9, 93.9, 58.7 ppm. HRMS calculated for C19H15N5O (M + H): 330.1355, Found: 330.1344.
2–(5-(1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-5-fluoro-1H-benzo[d] imidazole (18)
1H NMR (400 MHz, MeOH-d4): δ = 7.50 (d, J = 8.0 Hz, 1H), 7.43 (dd, J = 8.4, 4.8 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.20 (dd, J = 9.2, 2.4 Hz, 1H), 7.08 (td, J = 7.6, 1.2 Hz, 1H), 6.99 (td, J = 7.6, 1.2 Hz, 1H), 6.91 (td, J = 9.2, 2.4 Hz, 1H), 6.70 (s, 1H), 6.45 (s, 1H), 4.00 (s, 3H) ppm. 13 C NMR (100 MHz, MeOD-d4): δ = 161.9, 159.5, 152.1, 138.4, 131.9, 130.3, 128.4, 122.8, 121.0, 120.7, 111.8, 110.7, 110.5, 98.6, 94.0, 58.7 ppm. HRMS calculated for C20H15FN4O (M–H): 345.1152, Found: 345.1143.
3–((2-(5-(1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)-1H-benzo[d]imidazol-6-yl)oxy)aniline (19)
1H NMR (400 MHz, MeOH-d4): δ = 7.52 (d, J = 7.6 Hz, 1H), 7.41 (d, J = 8.4 Hz, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.15 (d, J = 2.4 Hz, 1H), 7.10 (t, J = 7.2 Hz, 1H), 6.9 9–7.07 (m, 2H), 6.90 (dd, J = 8.4, 2.4 Hz, 1H), 6.73 (s, 1H), 6.51 (s, 1H), 6.43 (dd, J = 8.0, 2.0 Hz, 1H), 6.36 (t, J = 2.4 Hz, 1H), 6.30 (dd, J = 8.0, 2.4 Hz, 1H), 4.05 (s, 3H) ppm.
6–bromo-2-(4-methoxy-1H, 1'H-[2, 2'-bipyrrol]-5-yl)-3H-imidazo[4,5-b] pyridine (20)
1H NMR (400 MHz, MeOH-d4): δ = 8.24 (s, 1H), 7.92 (d, J = 2.0 Hz, 1H), 6.82 (dd, J = 2.8, 1.2 Hz, 1H), 6.50 (dd, J = 3.6, 1.2 Hz, 1H), 6.26 (s, 1H), 6.18 (dd, J = 3.6, 2.8 Hz, 1H), 4.03 (s, 3H) ppm. 13 C NMR (100 MHz, MeOH-d4): δ = 154.2, 146.3, 131.6, 129.1, 125.7, 120.1, 112.9, 110.1, 106.8, 106.5, 105.4, 91.8, 89.8, 81.5, 58.7 ppm. HRMS calculated for C15H12BrN5O (M–H): 356.0147, Found: 356.0152.
2–(4-methoxy-1H, 1'H-[2, 2'-bipyrrol]-5-yl)-1H-benzo[d]imidazole-5-carbonitrile (21)
1H NMR (400 MHz, MeOH-d4): δ = 7.80 (s, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.45 (dd, J = 8.4, 1.6 Hz, 1H), 6.81 (dd, J = 3.2, 1.6 Hz, 1H), 6.47 (dd, J = 3.2, 1.6 Hz, 1H), 6.25 (s, 1H), 6.17 (t, J = 3.2 Hz, 1H), 4.03 (s, 3H) ppm.
(E)-N4-((5–(5-bromo-1H-indol-2-yl)-3-methoxy-1H-pyrrol-2-yl)methylene) pyrimidine-4,5-diamine (22)
Compound 3 (1.0 equiv.), pyrimidine-4,5-diamine (1.2 equiv) and NaHSO3 (2.0 equiv.) were mixed in the solution of ethanol. After the mixtures were heated with a 50 Watt microwave at 100 °C for 30 min, the solution was evaporated with rotavapor. The crude mixtures were poured into 20 ml water and extracted with ethyl acetate (30 ml) 3 times. The organic layer was collected, washed with brine, dried with MgSO4. The concentrated crude product was purified by silica gel with elute (ratio:ethyl acetate/hexane =1/1). Yield: 8.2%. 1H NMR (400 MHz, MeOH-d4): δ = 8.44 (d, J = 1.6 Hz, 1H), 8.17 (d, J = 1.6 Hz, 1H), 7.95 (d, J = 1.6 Hz, 1H), 7.67 (s, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.21 (d, J = 8.8 Hz, 1H), 6.86 (s, 1H), 6.45 (d, J = 1.6 Hz, 1H), 3.94 (s, 3H) ppm.
(Z)-3-(2-((1H-pyrrol-2-yl)methylene)-3-methoxy-2H-pyrrol-5-yl)pyridine (23)
Pd(PP3)4 (0.1 equiv) was added to a solution of 1.0 equiv (Z)-2-((1H-pyrrol-2-yl)methylene)-3-methoxy-2H-pyrrol-5-yl trifluoromethanesulfonate (0.086 g, 0.267 mmole), Na2CO3 (0.028 g, 0.264 mmole), and 1.2 equiv pyridin-3-ylboronic acid (0.039 g, 0.317 mmole) in 10% water/dioxane (5 ml) purged with nitrogen. After the mixtures were heated at 100 °C for 90 min, the reaction was quenched with water. The mixture was extracted with ethyl acetate (30 ml) 3 times. The organic layer was collected, washed with brine, and dried with MgSO4. The concentrated crude product was purified by silica gel with elute (ratio: ethyl acetate/hexane =1/1). Yield: 35.6%. 1H NMR (400 MHz, CDCl3): δ = 9.18 (d, J = 1.6 Hz, 1H), 8.61 (dd, J = 4.8, 1.6 Hz, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.36 (dd, J = 8.0, 4.8 Hz, 1H), 7.24 (s, 1H), 7.18 (s, 1H), 7.01 (s, 1H), 6.68 (d, J = 3.6 Hz, 1H), 6.29 (dd, J = 3.6, 2.8 Hz, 1H), 6.05 (s, 1H), 3.91 (s, 3H) ppm.
Biological assays
Cell culture
Hepatoma cells PLC/PRF/5 (PLC5) were purchased from ATCC (Manassas, VA) (CRL-8024) and maintained in DMEM supplemented with 10% FBS, 100 units/mL penicillin G, 100 mg/mL streptomycin sulphate and 25 mg/mL amphotericin B in a 37 °C humidified incubator in an atmosphere of 5% CO2 in air.
Cell viability assay
The effect of individual test agents on cell viability was assessed by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay in five replicates. Cells were seeded and incubated in 96-well, flat-bottomed plates for 24 h and were exposed to various concentrations (2.5, 5, 10, 20, 40 μM) of test agents dissolved in DMSO (final concentration, 0.1%) in media. Controls received DMSO vehicle at a concentration equal to that in drug-treated cells. For the MTT assay, 10 μL of MTT dye was directly added to the wells. After 2 h, media were removed and then the cells were lysed with 100 μL of DMSO. The absorbance at 570 nm and 630 nm (background) were read with a microplate reader.
Western blot
Lysates of PLC5 treated with compounds at the indicated concentrations for 24 h were analysed by western blot. p-STAT3 (#9145, 1:1000), STAT3 (#4904, 1:1000), Mcl-1 (#4572, 1:1000), survivin (#2803, 1:1000), JAK2 (#3230, 1:1000), GAPDH (#8884, 1:5000), and actin (#4967, 1:5000) antibodies were purchased from Cell Signaling (Danvers, MA). SHP-1 (ab32559, 1:1000) and cyclin D1 (ab24249, 1:1000) antibodies were purchased from Abcam (Cambridge, MA). PTP1B (2066–1, 1:2000) antibody was purchased from Epitomics (Cambridge, MA). p-JAK2(Tyr1007/1008) (44–42 G, 1:1000) antibody was purchased from ThermoFisher Scientific (Waltham, MA).
SHP-1 phosphatase activity
A RediPlate 96 EnzChek Tyrosine Phosphatase Assay Kit (R-22067) was used for SHP-1 activity assay (Molecular Probes, Carlsbad, CA). The method was as described previouslyCitation22.
STAT3 activity assay
PLC5 cells were treated with the indicated compounds in 10 μM for 24 h and then analysed by the PathScan Phospho-Stat3 (Tyr705) Sandwich ELISA Kit (Cell Signaling)
Gene knockdown using siRNA
Smart-pool small interfering RNAs (siRNAs), including the control (D-001810–10), SHP-1, were purchased from Dharmacon (Chicago, IL). The knockdown procedure was as described previouslyCitation23.
Statistical analysis
Data are expressed as mean ± SD. All statistical analyses were performed using SPSS for Windows version 12.0 software (SPSS Inc, Chicago, IL).
Results and discussion
Chemistry
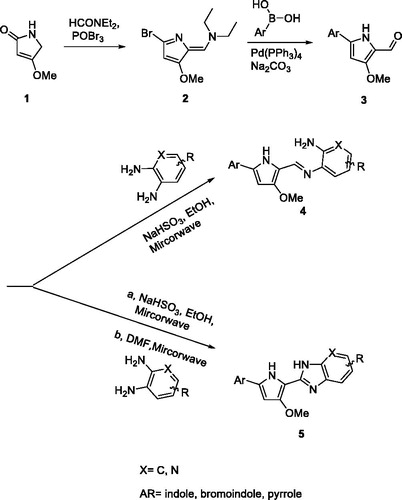
The synthesis of the pyrrole-benzimidazole-based compounds was initiated with 4-methoxy-1H-pyrrol-2-one and dimethyl amide in the presence of phosphoryl tribromide to generate two functional groups, enamine, and bromine in the pyrrole ring 2. The position of the brominated pyrrole ring was soon conjugated with aromatic boron acid in the presence of Pd(PPh3)4 by using the Suzuki coupling reaction and enamine was hydrolysed to the carbonyl group in acid conditions, resulting in the key intermediate 3. The condensation reaction of the carbonyl group of 3 and various aromatic diamines under the different conditions resulted in the final imine 4 or pyrrole-imidazopyridine 5 products.
Biological evaluation
All the new pyrrole-benzimidazole derivatives were analysed by MTT assay for growth inhibition against HCC cancer cells. The various substituents attached to the scaffold are summarized in . The growth inhibition of each of the compounds is shown with an IC50 value which was calculated by interpolation from the dose–response curve in MTT assay.
Table 1. Chemical structure of compounds 6–9 and IC50 of cell death in PLC5 cells.
Table 2. Chemical structure of compounds 7, and 10–15 and IC50 of cell death in PLC5 cells.
Table 3. Chemical structure of compounds 8, and 16–19 and IC50 of cell death in PLC5 cells.
Table 4. Chemical structure of compounds 9, 20, and 21 and IC50 of cell death in PLC5 cells.
Table 5. Chemical structure of compounds 9, 11, 16, 17, 22, and 23 and IC50 of cell death in PLC5 cells.
A set of pyrrole-benzimidazole derivatives was generated in which an indole, bromoindole, pyrrole, and Boc-indole connected to the methoxyl-pyrrole ring in the centre of the core moiety. In vitro test of cell death activity with these compounds against PLC5 cells showed differentiated activity with various substituents. Among these substituents, the indole ring 8 exhibited slightly better activity than bromo-indole 7 and pyrrole 9 (). In addition, the Boc-indole substituent 6 showed no activity against cell growth compared with the indole ring, suggesting the nitrogen atom plays an important role in the potency of cell growth. We next tested our hypothesis that the presence of various functional groups on benzimidazole might exhibit anti-cancer cell growth effects. A series of substituents, such as bromo, fluoro, cyano, and phenyl-cyano groups were thus introduced to the benzene and pyridine ring of benzimidazole to obtain a structure–activity relationship. These agents were tested in an MTT assay with PLC5 cells and the inhibition results are shown in . The pyridine ring of compound 11 demonstrated better activity than the benzene ring. In addition, Compound 13 with a cyano group connected to the benzene ring led to a reduction in inhibition of cell growth. Interestingly, the introduction of aniline 14 or phenyl-cyano 15 to benzimidazole resulted in no activity against PLC5 cells, implying that the linear electron-withdrawing group and phenyl substituent impede the anticancer activity. Analogues 17–19 that contained indole and imidazopyridine on both sides of pyrrole were also assayed for the inhibition of cell growth. As shown in , compounds 16 and 17 showed equal potency to compound 11. Compound 20, with imidazopyridine as a substituent to replace benzimidazole, led to a significant increase in anticancer activity. ( and ).
Mechanistic study of pyrrole-imidazopyridine in PLC 5 cells
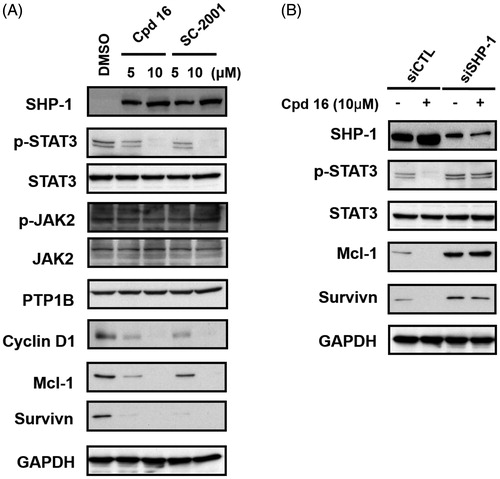
Previously, we have shown that indole-pyrrole reduces STAT3 phosphorylation in western blot assay and further represses cancer cell growthCitation20. To further study whether the newly synthesized compounds had STAT3 inhibition activity, we screened these compounds using a p-STAT3 ELISA kit. Compounds 11, 16, and 17 significantly repressed STAT3 activity on PLC 5 cells (). These results correlated with the potency of cell growth inhibition. We further analysed 6 compounds to PLC5 cells with western blot assay and studied the relationship of cell growth and the status of STAT3. As shown in , compounds 11, 16, and 17 resulted in a significant reduction of p-STAT3. In contrast, compounds 9, 22, and 23, which were showed no cell growth inhibition, showed no appreciable change in inhibition of STAT3 as compared with the control panel. We further examined SHP-1 activity, the negative regulator of STAT3, by analysing the dephosphorylating effect in PLC5 cells. Compounds 11, 16, and 17 increased SHP-1 activity two-fold compared with the control panel (). Meanwhile, we also explored the downstream signal cascade induced by compound 16 and SC-2001 in PLC5 cells. The downstream targets of STAT3, such as cyclin D1, survivin, and Mcl-1, were decreased in the treatment of compound 16 and SC-2001, indicating that SHP-1 phosphatase activation and further p-STAT3 reduction were the major effectors in regulating cell survival. In addition, both of compound 16 and SC-2001 had no effects on PTP-1B, p-JAK2, and JAK2 expressions (). Moreover, silencing SHP-1 with small-interference RNA (siRNA) abolished the effects of compound 16 on STAT3 de-phosphorylation and down-regulation of Mcl-1 and survivin ().
Figure 2. Effects of compounds on p-STAT3 inhibition. (A) PLC5 cells were exposed to the indicated compounds at a dose of 10 μM for 24 h and cell lysates were assayed by p-STAT3 ELISA kit. (B) PLC5 cells were exposed to the indicated compounds (cpd 16, 17, 22, 9, 23, 11) at 10 μM for 24 h and cell lysates were assayed by western blot.
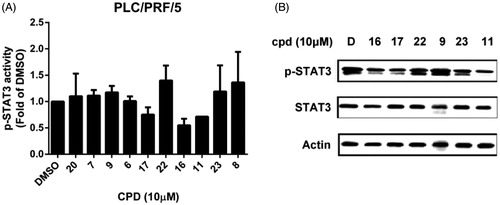
Figure 3. Effects of compounds on SHP-1 activity. PLC5 cells were exposed to cpd 11, 16, 17, and SC-2001 at 10 μM for 24 h and cell lysates were assayed by SHP-1 phosphatase activity kit.
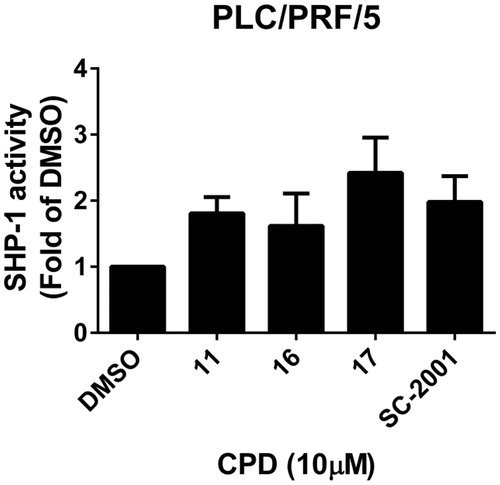
Figure 4. Effects of cpd 16 on protein levels of SHP-1, p-STAT3, and p-STAT3 relative downstream target in PLC5 cells. (A) Cells were exposed to cpd 16 and SC-2001 at the indicated doses for 24 h. Cell lysates were assayed by western blot. (B) PLC5 cells were transfected, respectively, with control siRNA or SHP-1 siRNA for 48 h. After transfection, the cells were treated w/wo cpd 16 (10 μM) for 24 h. The protein levels were analysed by western blot assay.
Scheme 1. General synthetic procedure for pyrrole-benzimidazole.
Discussion
In this study, we studied the pharmacological effect of imidazopyridine derivatives and explored compound-induced blockade of cell signals which regulated HCC cell growth. We first designed an efficient synthetic route for the imidazopyridine core structure and derived a series of analogues for the anticancer test in PLC5 cells. These synthetic routes simplify the complicated procedure of amplifying a large amount of imidazopyridine and imine-pyridine for future in vivo studies by using commercially available starting materials and reagents. Based on the structure–activity relationship for cell growth inhibition, imidazopyridine substituents connected to an indole–pyrrole core are crucial. Replacement of imidazopyridine with imine-pyridine led to the loss of activity.
SHP-1 is reported to be a phosphatase in hematopoietic cells and executed de-phosphorylation activity to its target proteins in some biological processes. STAT3 is a major target of SHP-1 and highly expressed in different cancer cell types, including HCCCitation24,Citation25. Some reports have indicated that small molecules can negatively regulate cell growth through activating SHP-1 and further eliminating the phosphor group from active p-STAT3Citation26–28. These phosphatase agonists provide a new direction for future clinical approaches instead of only kinase inhibition. Our discovery also provides evidence that imidazopyridine derivatives mediate both SHP-1 activation and also increase SHP-1 expression. Importantly, the reduction of cell survival genes, such as cyclin D1, survivin, and Mcl-1 regulated by SHP-1 were consistent with cell survival in the treatment of imidazopyridine derivatives.
In summary, this study showed that the structure of indole–pyrrole-imidazopyridine is a novel anticancer agent. We also demonstrated that these compounds can enhance SHP-1 expression and activation and consequently reduced the level of p-STAT3 in HCC cells. This indicates that the strategy of SHP-1 activation is a possible drug target in cancer patients who have no or low SHP-1 activity. In addition, these agents might be applied along with therapeutic cancer drugs to increase efficacy and reduce toxicity.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Yu H, Lee H, Herrmann A, et al. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 2014;14:736–46.
- Zhong Z, Wen Z, Darnell JE. Jr., Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994;264:95–8.
- Lee H, Deng J, Kujawski M, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med 2010;16:1421–8.
- Xin H, Lu R, Lee H, et al. G-protein-coupled receptor agonist BV8/prokineticin-2 and STAT3 protein form a feed-forward loop in both normal and malignant myeloid cells. J Biol Chem 2013;288:13842–9.
- Eyking A, Ey B, Runzi M, et al. Toll-like receptor 4 variant D299G induces features of neoplastic progression in Caco-2 intestinal cells and is associated with advanced human colon cancer. Gastroenterology 2011;141:2154–65.
- Nielsen M, Kaestel CG, Eriksen KW, et al. Inhibition of constitutively activated Stat3 correlates with altered Bcl-2/Bax expression and induction of apoptosis in mycosis fungoides tumor cells. Leukemia 1999;13:735–8.
- Bhattacharya S, Ray RM, Johnson LR. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem J 2005;392:335–44.
- Su JC, Mar AC, Wu SH, et al. Disrupting VEGF-A paracrine and autocrine loops by targeting SHP-1 suppresses triple negative breast cancer metastasis. Sci Rep 2016;6:28888.
- den Hertog J, Ostman A, Bohmer FD. Protein tyrosine phosphatases: regulatory mechanisms. FEBS J 2008;275:831–47.
- Yamada S, Shiono S, Joo A, Yoshimura A. Control mechanism of JAK/STAT signal transduction pathway. FEBS Lett 2003;534:190–6.
- Huang S, Chen M, Shen Y, et al. Inhibition of activated Stat3 reverses drug resistance to chemotherapeutic agents in gastric cancer cells. Cancer Lett 2012;315:198–205.
- Prabhu VV, Hong B, Allen JE, et al. Small-molecule prodigiosin restores p53 tumor suppressor activity in chemoresistant colorectal cancer stem cells via c-Jun-mediated ΔNp73 inhibition and p73 activation. Cancer Res 2016;76:1989–99.
- Mott JL, Bronk SF, Mesa RA, et al. BH3-only protein mimetic obatoclax sensitizes cholangiocarcinoma cells to Apo2L/TRAIL-induced apoptosis. Mol Cancer Ther 2008;7:2339–47.
- Nguyen M, Marcellus RC, Roulston A, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA 2007;104:19512–7.
- Llagostera E, Soto-Cerrato V, Joshi R, et al. High cytotoxic sensitivity of the human small cell lung doxorubicin-resistant carcinoma (GLC4/ADR) cell line to prodigiosin through apoptosis activation. Anticancer Drugs 2005;16:393–9.
- Goy A, Hernandez-Ilzaliturri FJ, Kahl B, et al. A phase I/II study of the pan Bcl-2 inhibitor obatoclax mesylate plus bortezomib for relapsed or refractory mantle cell lymphoma. Leuk Lymphoma 2014;55:2761–8.
- Goy A, Berger M, Ford P, et al. Sequential single-agent obatoclax mesylate (GX15-070MS) followed by combination with rituximab in patients with previously untreated follicular lymphoma. Leuk Lymphoma 2014;55:2932–4.
- Chen S, Wang G, Niu X, et al. Combination of AZD2281 (Olaparib) and GX15-070 (Obatoclax) results in synergistic antitumor activities in preclinical models of pancreatic cancer. Cancer Lett 2014;348:20–8.
- Tang Y, Hamed HA, Cruickshanks N, et al. Obatoclax and lapatinib interact to induce toxic autophagy through NOXA. Mol Pharmacol 2012;81:527–40.
- Su JC, Chen KF, Chen WL, et al. Synthesis and biological activity of obatoclax derivatives as novel and potent SHP-1 agonists. Eur J Med Chem 2012;56:127–33.
- Chen KF, Su JC, Liu CY, et al. A novel obatoclax derivative, SC-2001, induces apoptosis in hepatocellular carcinoma cells through SHP-1-dependent STAT3 inactivation. Cancer Lett 2012;321:27–35.
- Chen KF, Tai WT, Liu TH, et al. Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin Cancer Res 2010;16:5189–99.
- Tai WT, Cheng AL, Shiau CW, et al. Signal transducer and activator of transcription 3 is a major kinase-independent target of sorafenib in hepatocellular carcinoma. J Hepatol 2011;55:1041–8.
- Santoni M, Massari F, Del Re M, et al. Investigational therapies targeting signal transducer and activator of transcription 3 for the treatment of cancer. Exp Opin Investig Drugs 2015;24:809–24.
- Subramaniam A, Shanmugam MK, Perumal E, et al. Potential role of signal transducer and activator of transcription (STAT)3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochim Biophys Acta 2013;1835:46–60.
- Bi L, Yu Z, Wu J, et al. Honokiol inhibits constitutive and inducible STAT3 signaling via PU.1-induced SHP1 expression in acute myeloid leukemia cells. Tohoku J Exp Med 2015;237:163–72.
- Su JC, Chiang HC, Tseng PH, et al. RFX-1-dependent activation of SHP-1 inhibits STAT3 signaling in hepatocellular carcinoma cells. Carcinogenesis 2014;35:2807–14.
- Huang TT, Su JC, Liu CY, et al. Alteration of SHP-1/p-STAT3 signaling: a potential target for anticancer therapy. Int J Mol Sci 2017;18:1234.