
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A series of novel N9-heterobivalent β-carbolines has been synthesized. All the novel compounds were tested for their anticancer activity against six tumour cell lines in vitro. Among these molecules, compounds 5b, and 5w exhibited strong cytotoxic activities with IC50 value of lower than 20 μM. Acute toxicities and antitumor efficacies of the selected compounds in mice were also evaluated, compounds 5b and 5w exhibited that tumour inhibition rate of over 40% in the Sarcoma 180 and Lewis lung cancer animal models. Preliminary structure–activity relationships (SARs) analysis indicated that: (1) C1-methylation and C7-methoxylation were favorable for increased activities; (2) 3-Pyridyl or 2-thienyl group substituent into position-1 of the β-carboline core, and the aryl substituent into another β-carboline ring might be detrimental to cytotoxic effects of this class compounds. Investigation of the preliminary mechanism of action demonstrated that compound 5b had obvious angiogenesis inhibitory effects in the chicken chorioallantoic membrane (CAM) assay.
Introduction
The population growth and aging associated with some risk factors leveraged the incidence of new cases of cancer and related deaths in developed countries and in developing countriesCitation1. Cancer resistance to therapy is becoming a common phenomenon that threatens the current strategies adopted against this disease. For that reason we need to discover new anticancer agents. One of the successful and effective methods for the discovery of new anticancer drugs from natural products is to synthesize novel compounds through chemical structural modifications on the basis of leading compounds.
Peganum harmala L. have been traditionally used for hundreds of years to treat the alimentary tract cancers and malaria in Northwest China. Harmine, originally isolated from the seeds of Peganum harmala L. in 1847, is the most representative natural occurring β-carboline alkaloid, having a common tricyclic pyrido [3,4-b] indole ring structure. In the past several decades, it has confirmed that harmine was the important active ingredients to treat the alimentary tract cancersCitation2,Citation3. Recent reports demonstrated that harmine and its derivatives had remarkable antitumor activities, together with potential neurotoxicityCitation2–5. Moreover, it has been reported that harmine and its derivatives can exert antitumor activities through multiple mechanisms, such as DNA bindingCitation6–8, inhibition topoisomerases I and IICitation9,Citation10, CDK (cyclin-dependent kinase)Citation11,Citation12, PLK1 (polo-like kinase)Citation13, lipoxygenaseCitation14,Citation15 and IκB kinasesCitation16.
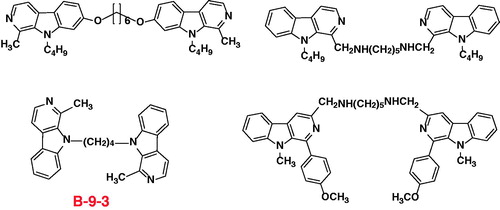
Previous investigations has shown that some dimer antitumor agents via an appropriate linker could lead to significantly improved antitumor activities (100- to 500-fold improvement over the corresponding monomers)Citation17–20. Therefore, bivalent β-carbolines were expected to exhibit more potent antitumor efficacies than monomers. Inspired by this information, our group reported the synthesis, in vitro evaluation, in vivo efficacies and structure-activity relationships for the novel homobivalent β-carbolines with an alkyl spacer or alkylamino spacer in positon-1, 3, 7, and 9 of the β-carboline nucleus, respectively ()Citation21–26. In these compounds, 1-Methyl-9-[4–(1-methyl-β-carboline-9-yl)butyl]-β-carboline (B-9–3)Citation21,Citation27 exhibited potent antitumor activity. It was a symmetric dimeric β-carboline compound that contains two molecules of harman bound to each other by a tetramethylene group. The pharmacological mechanisms showed that the angiogenesis inhibitor B-9–3 selectively induces apoptosis of endothelial cells, in part through disruption of VEGF-A/VEGFR2 signalingCitation28.
Figure 1. The chemical structure of the representative reported symmetric bivalent β-carbolines.
Our strategy was based on the modification of the prototype B-9–3, following this previous work, we have continued our search for novel antitumor agents endowed with better antitumor activities, and we provide detailed studies of structure–activity relationships (SARs) on the antitumor efficacies in vitro and in vivo of this class of compounds. Here, we designed and synthesized a series of novel N9-heterobivalent β-carboline derivatives as potent antitumor agents.
Materials and methods
Chemistry
All reagents were purchased from commercial suppliers and were dried and purified when necessary. The following intermediates 3a-3bCitation29, 3c-3eCitation21, 3fCitation30, 3gCitation31, 3hCitation32, 4g-4hCitation33 were prepared as previously described.
Melting points were determined in capillary tubes on an electrothermal X-5 apparatus and without correction. NMR spectra were recorded at room temperature on a Bruker Avance III HD 400 instrument at 400 MHz for 1H NMR and 100 MHz for 13C NMR. Chemical shifts (δ) are reported in ppm relatively to the residual solvent peak and the multiplicity of each signal is designated by the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Coupling constants (J) are quoted in Hz. High resolution mass spectra (HRMS) were recorded on Bruker ultrafleXtreme MALDI-TOF/TOF-MS and HCCA (alpha-cyano-4-hydroxycinnamic acid) is used as matrix. Elemental analyses (C, H, and N) were carried out on an Elementar Vario ELIII CHNS Elemental Analyzer. Column chromatography was performed with silica gel (200–300 mesh) and analytical TLC on silica gel 60-F254.
General procedure for the preparation of compounds 4a-h
A mixture of 3a (1.68 g, 10 mmol) and anhydrous DMF (50 ml) was stirred at room temperature for 0.5 h, then NaH (0.50 g, 20 mmol) and the 1,4-dibromobutane (20 mmol) were added. The mixture was stirred at room temperature. After that period, the mixture was monitored via TLC and at the end of the reaction the mixture was poured into H2O (150 ml) and extracted with ethyl acetate. The organic phase was collected and washed with water and brine, then dried under anhydrous sodium sulfate, filtered, and evaporated. The resulting oil was crystallized from ethyl ether or ethyl ether-petroleum ether to afford the compound 4a. Products 4b-h were prepared according to the same method of 4a.
9–(4-bromobutyl)-β-carboline (4a)
Colorless crystals, yield 87%, m.p. 285.3–286.8 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.10 (s, 1H), 8.40 (d, J = 5.2 Hz, 1H), 8.28 (d, J = 8.0 Hz, 1H), 8.14 (dd, J = 5.2, 0.8 Hz, 1H), 7.75 (d, J = 8.4 Hz, 1H), 7.60–7.64 (m, 1H), 7.27–7.31 (m, 1H), 4.56 (t, J = 6.8 Hz, 2H), 3.56 (t, J = 6.4 Hz, 2H), 1.90–1.97 (m, 2H), 1.81–1.88 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 141.13, 138.88, 136.47, 133.13, 128.77, 127.66, 122.44, 120.80, 119.92, 115.04, 110.62, 42.15, 35.09, 30.23, 27.87.
9–(4-bromobutyl)-1-methyl-β-carboline (4b)
Colorless crystals, yield 88%, m.p. 213.9–215.6 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.71 (dd, J = 6.4, 1.2 Hz, 1H), 8.54 (dd, J = 8.0, 1.2 Hz, 1H), 8.49 (dd, J = 6.4, 2.0 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.83–7.87 (m, 1H), 7.46–7.50 (m, 1H), 4.75 (t, J = 7.6 Hz, 2H), 3.70 (t, J = 6.4 Hz, 2H), 3.27 (s, 3H), 2.04–1.77 (m, 4H). 13C NMR (100 MHz, DMSO-d6): δ 144.33, 139.00, 133.74, 133.26, 132.07, 129.29, 123.86, 122.14, 119.67, 116.15, 111.78, 45.35, 44.31, 29.63, 28.29, 18.31.
9–(5-bromopentyl)-1-methyl-β-carboline (4c)
Colorless crystals, yield 90%, m.p. 187.7–189.5 °C. 1H NMR (400 MHz, CDCl3): δ 8.42–8.33 (m, 2H), 8.29 (dt, J = 8.0, 1.2 Hz, 1H), 7.82–7.86 (m, 1H), 7.66 (dt, J = 8.4, 0.8 Hz, 1H), 7.48–7.51 (m, 1H), 4.69 (t, J = 7.6 Hz, 2H), 3.52 (s, 3H), 1.82–1.98 (m, 4H), 1.60–1.68 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 144.30, 137.12, 133.87, 133.76, 132.22, 128.57, 123.11, 122.38, 119.70, 115.13, 110.68, 45.35, 44.43, 31.86, 30.29, 24.04, 18.07.
9–(6-bromohexyl)-1-methyl-β-carboline (4d)
Colorless crystals, yield 69%, m.p. 73.9–74.8 °C. 1H NMR (400 MHz, CDCl3): δ 8.33 (d, J = 5.2 Hz, 1H), 8.12 (d, J = 7.6 Hz, 1H), 7.84 (d, J = 5.2 Hz, 1H), 7.56–7.60 (m, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.25–7.29 (m, 1H), 4.54 (t, J = 7.6 Hz, 2H), 3.38 (t, J = 6.4 Hz, 2H), 3.05 (s, 3H), 1.81–1.89 (m, 4H), 1.40–1.55 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 141.51, 141.13, 137.94, 135.09, 129.14, 128.17, 121.55, 121.33, 119.66, 112.99, 109.67, 44.76, 33.56, 30.70, 27.90, 26.13, 23.55.
9–(4-bromobutyl)-1–(3-pyridyl)-β-carboline (4e)
Yellow crystals, yield 71%, m.p. 135.0–136.7 °C. 1H NMR (400 MHz, CDCl3): δ 8.67 (d, J = 6.0 Hz, 1H), 8.53 (d, J = 6.0 Hz, 1H), 8.37 (d, J = 8.0 Hz, 1H), 7.87–7.81 (m, 2H), 7.68–7.64 (m, 2H), 7.54–7.49 (m, 1H), 7.36–7.33 (m, 1H), 4.19 (t, J = 8.0 Hz, 2H), 3.32 (dt, J = 55.2, 6.4 Hz, 2H), 1.77–1.69 (m, 2H), 1.60–1.44 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 149.60, 149.24, 142.21, 139.90, 138.50, 137.27, 131.21, 129.04, 128.23, 125.31, 123.53, 121.85, 121.25, 120.45, 114.54, 110.17, 43.77, 32.59, 29.44, 27.35.
9–(4-bromobutyl)-1–(2-thienyl)-β-carboline (4f)
Light yellow crystals, yield 60%, m.p. 197.6–199.8 °C. 1H NMR (400 MHz, CDCl3): δ 8.55 (d, J = 5.2 Hz, 1H), 8.19 (d, J = 7.6 Hz, 1H), 8.03 (d, J = 5.2 Hz, 1H), 7.62–7.54 (m, 3H), 7.54–7.51 (m, 1H), 7.49–7.41 (m, 3H), 7.30 (t, J = 7.2 Hz, 1H), 4.03–3.78 (m, 2H), 3.20 (dt, J = 54.8, 6.4 Hz, 2H), 1.71–1.49 (m, 2H), 1.44–1.24 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 141.58, 140.89, 138.69, 138.31, 134.19, 131.47, 130.09, 129.47, 128.58, 126.95, 121.70, 121.26, 119.96, 114.31, 109.76, 43.23, 32.60, 29.77, 27.94.
General procedure for the preparation of compounds 5a-x
A solution of compound 3c (2 mmol) in anhydrous DMF (6 ml) was added slowly with stirring to a solution of 4a (3 mmol), NaH (0.25 g, 10 mmol), potassium iodide (1.68 g, 10 mmol) in anhydrous DMF (25 ml). The mixture was stirred at room temperature until the reaction is completed. Then the mixture was poured into ice-cold water. The reaction mixture was extracted with ethyl acetate (3 × 30 ml), washed with water and brine, dried over anhydrous Na2SO4, and filtered. Purification by column chromatography (DCM/MeOH 100:1 as the eluent) furnished the dimeric β-carboline 5a. Compounds 5b-x were synthesized using similar procedure as compound 5a.
1–(3-pyridyl)-9–(4-(β-carboline-9-yl)butyl)-β-carboline (5a)
This compound was obtained as colourless crystals in 65% yield, m.p. 173.3–175.1 °C. 1H NMR (400 MHz, CDCl3) δ: 8.77 (dd, J = 2.4, 0.8 Hz, 1H), 8.65 (d, J = 0.8 Hz, 1H), 8.61 (dd, J = 4.8, 1.6 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.47 (d, J = 5.2 Hz, 1H), 8.15–8.18 (m, 2H), 8.00 (dd, J = 5.2, 0.8 Hz, 1H), 7.98 (d, J = 5.2 Hz, 1H), 7.56–7.63 (m, 3H), 7.30–7.34 (m, 3H), 7.24 (d, J = 8.4 Hz, 1H), 7.11–7.15 (m, 1H), 4.14 (t, J = 7.2 Hz, 2H), 3.96 (t, J = 7.2 Hz, 2H), 1.38–1.41 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 150.00, 149.59, 142.09, 141.14, 140.25, 138.92, 138.05, 136.35, 136.06, 135.77, 134.09, 130.89, 128.85, 128.80, 123.04, 122.11, 121.81, 121.26, 120.90, 120.33, 120.02, 114.80, 114.22, 109.90, 109.37, 44.08, 42.65, 26.53, 26.01. HRMS calcd for C31H26N5 [M + H]+ 468.2183, found 468.2183. Anal. calcd for C31H25N5: C, 79.63; H, 5.39; N, 14.98; found C 79.16, H 5.49, N 14.38.
1–(3-pyridyl)-9–(4-(1-methyl-β-carboline-9-yl)butyl)-β-carboline (5b)
This compound was obtained as slight yellow crystals in 54% yield, m.p. 201.2–202.2 °C. 1H NMR (400 MHz, CDCl3) δ: 8.79 (dd, J = 2.4, 0.8 Hz, 1H), 8.65 (dd, J = 4.8, 1.6 Hz, 1H), 8.52 (d, J = 5.2 Hz, 1H), 8.33 (d, J = 5.2 Hz, 1H), 8.17–8.19 (m, 1H), 8.11 (d, J = 7.6 Hz, 1H), 7.98 (d, J = 5.2 Hz, 1H), 7.84 (d, J = 5.2 Hz, 1H), 7.69–7.71 (m, 1H), 7.52–7.61 (m, 2H), 7.27–7.35 (m, 3H), 7.18–7.24 (m, 2H), 4.23 (t, J = 7.2 Hz, 2H), 3.96 (t, J = 7.2 Hz, 2H), 2.81 (s, 3H), 1.37–1.45 (m, 2H), 1.28–1.35 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 150.06, 149.64, 142.18, 141.40, 140.66, 140.31, 139.05, 136.41, 135.83, 134.70, 134.12, 130.97, 129.35, 128.86, 128.39, 123.11, 121.85, 121.63, 121.34, 121.17, 120.38, 119.91, 114.23, 113.07, 109.95, 109.55, 44.13, 43.90, 29.70, 27.71, 26.07. HRMS calcd for C32H28N5 [M + H]+ 482.2339, found 482.2342. Anal. calcd for C32H27N5: C, 79.81; H, 5.65; N, 14.54; found C 79.39, H 5.37, N 14.75.
1–(3-pyridyl)-9–(5-(1-methyl-β-carboline-9-yl)pentyl)-β-carboline (5c)
This compound was obtained as yellow crystals in 62% yield, m.p. 189.4–190.5 °C. 1H NMR (400 MHz, CDCl3) δ: 8.82 (d, J = 2.0 Hz, 1H), 8.55 (d, J = 5.2 Hz, 1H), 8.51 (dd, J = 4.8, 1.6 Hz, 1H), 8.34 (d, J = 5.2 Hz, 1H), 8.19 (d, J = 7.6 Hz, 1H), 8.14 (d, J = 8.4 Hz, 1H), 8.02 (d, J = 5.2 Hz, 1H), 7.85–7.89 (m, 2H), 7.57–7.61 (m, 2H), 7.37 (d, J = 8.4 Hz, 1H), 7.29–7.33 (m, 3H), 7.23–7.25 (m, 1H), 4.35 (t, J = 7.6 Hz, 2H), 3.95 (t, J = 7.6 Hz, 2H), 2.96 (s, 3H), 1.47–1.55 (m, 2H), 1.35–1.42 (m, 2H), 0.86–0.94 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 150.12, 149.51, 142.03, 141.61, 140.69, 140.38, 138.84, 137.29, 136.56, 135.99, 134.87, 134.27, 130.85, 129.51, 128.80, 128.52, 123.01, 121.76, 121.67, 121.30, 121.16, 120.27, 119.98, 114.26, 113.15, 110.01, 109.69, 44.48, 44.24, 30.29, 28.56, 23.87, 23.07. HRMS calcd for C33H30N5 [M + H]+ 496.2496, found 496.2503. Anal. calcd for C33H29N5: C, 79.97; H, 5.90; N, 14.13; found C 79.11, H 5.88, N 13.90.
1–(3-pyridyl)-9–(6-(1-methyl-β-carboline-9-yl)hexyl)-β-carboline (5d)
This compound was obtained as yellow crystals in 66% yield, m.p. 141.3–142.9 °C. 1H NMR (400 MHz, CDCl3) δ: 8.89 (dd, J = 2.0, 0.8 Hz, 1H), 8.72 (dd, J = 4.8, 1.6 Hz, 1H), 8.56 (d, J = 5.2 Hz, 1H), 8.31 (d, J = 5.2 Hz, 1H), 8.17–8.19 (m, 1H), 8.08–8.10 (m, 1H), 8.02 (d, J = 5.2 Hz, 1H), 7.93–7.96 (m, 1H), 7.82 (d, J = 5.2 Hz, 1H), 7.53–7.60 (m, 2H), 7.30–7.41 (m, 4H), 7.24–7.28 (m, 1H), 4.39 (t, J = 7.2 Hz, 2H), 3.97 (t, J = 7.2 Hz, 2H), 2.97 (s, 3H), 1.58–1.66 (m, 2H), 1.29–1.37 (m, 2H), 1.07–1.15 (m, 2H), 0.81–0.90 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 150.16, 149.54, 142.11, 141.45, 141.02, 140.44, 138.79, 137.84, 136.65, 136.07, 134.98, 134.35, 130.86, 129.13, 128.73, 128.15, 123.07, 121.72, 121.53, 121.31, 121.26, 120.19, 119.66, 114.23, 112.98, 110.10, 109.60, 44.54, 44.39, 30.59, 28.66, 26.33, 23.44. HRMS calcd for C34H32N5 [M + H]+ 510.2652, found 510.2656. Anal. calcd for C34H31N5: C, 80.13; H, 6.13; N, 13.74; found C 79.72, H 6.14, N 13.32.
1–(2-thienyl)-9–(4-(β-carboline-9-yl)butyl)-β-carboline (5e)
This compound was obtained as yellow crystals in 56% yield, m.p. 141.6–143.1 °C. 1H NMR (400 MHz, CDCl3) δ: 8.72 (d, J = 0.8 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.46 (d, J = 5.2 Hz, 1H), 8.15–8.16 (m, 1H), 8.13–8.14 (m, 1H), 7.97 (dd, J = 5.2, 0.8 Hz, 1H), 7.95 (d, J = 5.2 Hz, 1H), 7.51–7.71 (m, 3H), 7.33–7.35 (m, 1H), 7.30–7.32 (m, 1H), 7.28–7.29 (m, 1H), 7.24–7.28 (m, 2H), 7.05 (dd, J = 4.0, 1.2 Hz, 1H), 6.94 (dd, J = 5.2, 3.6 Hz, 1H), 4.14 (t, J = 7.2 Hz, 2H), 4.13 (t, J = 7.2 Hz, 2H), 1.47–1.58 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 141.95, 141.05, 140.84, 138.62, 136.84, 136.22, 134.60, 132.44, 131.55, 130.86, 130.80, 128.78, 128.68, 128.54, 128.49, 128.21, 127.03, 126.58, 122.04, 121.73, 121.28, 121.02, 120.17, 119.79, 114.70, 114.23, 109.90, 109.34, 43.91, 42.68, 26.98, 26.28. HRMS calcd for C30H25N4S [M + H]+ 473.1794, found 473.1792. Anal. calcd for C30H24N4S: C, 76.24; H, 5.12; N, 11.86; S, 6.78; found C, 76.01; H, 5.61; N, 11.46; S, 6.34.
1–(2-thienyl)-9–(4-(1-methyl-β-carboline-9-yl)butyl)-β-carboline (5f)
This compound was obtained as yellow crystals in 47% yield, m.p. 214.8–216.0 °C. 1H NMR (400 MHz, CDCl3) δ: 8.50 (d, J = 5.2 Hz, 1H), 8.32 (d, J = 5.2 Hz, 1H), 8.14–8.17 (m, 1H), 8.12–8.06 (m, 1H), 7.96 (d, J = 5.2 Hz, 1H), 7.83 (d, J = 5.2 Hz, 1H), 7.56–7.60 (m, 1H), 7.51–7.55 (m, 1H), 7.35–7.37 (m, 1H), 7.33–7.34 (m, 1H), 7.28–7.31(m, 1H), 7.24–7.26 (m, 1H), 7.20–7.23 (m, 1H), 7.12 (dd, J = 4.0, 1.2 Hz, 1H), 6.99 (dd, J = 5.2, 3.6 Hz, 1H), 4.26 (t, J = 7.2 Hz, 2H), 4.13 (t, J = 7.2 Hz, 2H), 2.89 (s, 3H), 1.41–1.55 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 142.02, 141.36, 140.95, 140.86, 138.72, 137.86, 136.90, 134.85, 134.59, 130.89, 129.24, 128.70, 128.22, 127.07, 126.67, 121.76, 121.60, 121.36, 121.25, 120.22, 119.79, 114.23, 113.03, 109.94, 109.55, 44.04, 43.94, 27.94, 26.52, 23.35. HRMS calcd for C31H27N4S [M + H]+ 487.1951, found 487.1950. Anal. calcd for C31H26N4S: C, 76.51; H, 5.39; N, 11.51; S, 6.59; found C, 76.27; H, 5.59; N, 10.93; S, 6.38.
1–(2-thienyl)-9–(5-(1-methyl-β-carboline-9-yl)pentyl)-β-carboline (5g)
This compound was obtained as yellow crystals in 62% yield, m.p. 158.4–159.2 °C. 1H NMR (400 MHz, CDCl3) δ: 8.51 (d, J = 5.2 Hz, 1H), 8.34 (d, J = 5.2 Hz, 1H), 8.15–8.17 (m, 1H), 8.11–8.14 (m, 1H), 7.97 (d, J = 5.2 Hz, 1H), 7.86 (d, J = 5.2 Hz, 1H), 7.54–7.60 (m, 2H), 7.40 (d, J = 8.4 Hz, 1H), 7.27–7.32 (m, 3H), 7.21 (dd, J = 5.2, 1.2 Hz, 1H), 7.16 (dd, J = 3.2, 1.2 Hz, 1H), 6.91 (dd, J = 5.2, 3.6 Hz, 1H), 4.37 (t, J = 7.6 Hz, 2H), 4.12 (t, J = 7.6 Hz, 2H), 2.96 (s, 3H), 1.53–1.60 (m, 2H), 1.42–1.50 (m, 2H), 0.98–1.06 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 142.08, 141.45, 141.00, 140.94, 138.55, 137.76, 136.96, 134.97, 134.66, 130.82, 129.21, 128.62, 128.29, 128.23, 126.85, 126.51, 121.67, 121.55, 121.27, 121.25, 120.09, 119.75, 114.16, 113.02, 110.02, 109.66, 44.54, 44.06, 30.22, 28.89, 23.98, 23.40 . HRMS calcd for C32H29N4S [M + H]+ 501.2107, found 501.2113. Anal. calcd for C32H28N4S: C, 76.71; H, 6.24; N, 10.84; S, 6.20; found C, 76.24; H, 5.92; N, 10.45; S, 6.07.
1–(2-thienyl)-9–(6-(1-methyl-β-carboline-9-yl)hexyl)-β-carboline (5h)
This compound was obtained as yellow crystals in 57% yield, m.p. 238.5–239.6 °C. 1H NMR (400 MHz, CDCl3) δ: 8.52 (d, J = 5.2 Hz, 1H), 8.32 (d, J = 5.2 Hz, 1H), 8.15–8.17 (m, 1H), 8.09–8.11 (m, 1H), 7.97 (d, J = 5.2 Hz, 1H), 7.83 (d, J = 5.2 Hz, 1H), 7.53–7.59 (m, 2H), 7.45 (dd, J = 5.2, 1.2 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.24–7.30 (m, 3H), 7.13 (dd, J = 5.2, 3.6 Hz, 1H), 4.39 (t, J = 7.6 Hz, 2H), 4.14 (t, J = 7.6 Hz, 2H), 2.98 (s, 3H), 1.61–1.68 (m, 2H), 1.38–1.46 (m, 2H), 1.13–1.21 (m, 2H), 0.91–0.98 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 142.18, 141.47, 141.21, 141.01, 138.54, 137.75, 137.09, 134.98, 134.77, 130.86, 129.14, 128.57, 128.27, 128.16, 126.98, 126.65, 121.66, 121.53, 121.34, 121.23, 120.06, 119.66, 114.12, 112.99, 110.20, 109.62, 44.58, 44.17, 30.61, 28.96, 26.43, 26.34, 23.40 . HRMS calcd for C33H31N4S [M + H]+ 515.2264, found 515.2268. Anal. calcd for C33H30N4S: C, 77.01; H, 5.88; N, 10.89; S, 6.23; found C, 76.80; H, 5.76; N, 10.72; S, 5.97.
1–(2-chlorophenyl)-9–(4-(1-methyl-β-carboline-9-yl)butyl)-β-carboline (5i)
This compound was obtained as yellow crystals in 51% yield, m.p. 232.0–232.8 °C. 1H NMR (400 MHz, CDCl3) δ: 8.52 (d, J = 5.2 Hz, 1H), 8.34 (d, J = 5.2 Hz, 1H), 8.21–8.16 (m, 1H), 8.12–8.09 (m, 1H), 8.01 (d, J = 5.2 Hz, 1H), 7.85 (d, J = 5.2 Hz, 1H), 7.60–7.51 (m, 2H), 7.35–7.31 (m, 4H), 7.30–7.26 (m, 2H), 7.24–7.23 (m, 1H), 7.17–7.13 (m, 1H), 4.34–4.22 (m, 2H), 3.94–3.78 (m, 2H), 2.90 (s, 3H), 1.63–1.34 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 141.57, 141.41, 140.87, 140.83, 138.62, 138.44, 137.97, 134.89, 134.02, 133.93, 131.34, 130.13, 130.05, 129.45, 129.30, 128.63, 128.27, 126.72, 121.81, 121.64, 121.31, 120.05, 119.86, 114.34, 113.08, 109.62, 109.58, 44.06, 43.78, 28.23, 26.73, 23.45. HRMS calcd for C33H28ClN4 [M + H]+ 515.1997, found 515.2003. Anal. calcd for C33H27ClN4: C, 76.96; H, 5.28; N, 10.88; found C 76.39, H 5.37, N 10.57.
1–(2-chlorophenyl)-9–(5-(1-methyl-β-carboline-9-yl)pentyl)-β-carboline (5j)
This compound was obtained as yellow crystals in 70% yield, m.p. 169.1–171.2 °C. 1H NMR (400 MHz, CDCl3) δ: 8.52 (d, J = 5.2 Hz, 1H), 8.36 (d, J = 5.2 Hz, 1H), 8.17–8.19 (m, 1H), 8.13–8.15 (m, 1H), 8.01 (d, J = 5.2 Hz, 1H), 7.87 (d, J = 5.2 Hz, 1H), 7.55–7.59 (m, 2H), 7.41 (dd, J = 7.6, 1.6 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.30–7.33 (m, 3H), 7.24–7.28 (m, 1H), 7.09–7.13 (m, 1H), 6.98–7.02 (m, 1H), 4.37 (t, J = 7.6 Hz, 2H), 3.76–3.93 (m, 2H), 2.97 (s, 3H), 1.35–1.56 (m, 4H), 0.88–1.02 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 141.58, 141.41, 140.97, 140.86, 138.72, 138.27, 137.92, 134.99, 134.09, 133.94, 131.43, 130.06, 129.85, 129.30, 129.17, 128.54, 128.23, 126.56, 121.71, 121.59, 121.29, 121.22, 119.91, 119.82, 114.30, 113.04, 109.68, 109.65, 44.52, 43.80, 30.20, 28.98, 24.03, 23.50. HRMS calcd for C34H30ClN4 [M + H]+ 529.2154, found 529.2161. Anal. calcd for C34H29ClN4: C, 77.19; H, 5.53; N, 10.59; found C 76.74, H 5.32, N 10.82.
1–(4-methoxyphenyl)-9–(4-(1–(3-pyridyl)-β-carboline-9-yl)butyl)-β-carboline (5k)
This compound was obtained as yellow solid in 57% yield, m.p. 241.2–242.4 °C. 1H NMR (400 MHz, CDCl3) δ: 8.67 (d, J = 2.0 Hz, 1H), 8.62 (dd, J = 4.8, 1.6 Hz, 1H), 8.51 (d, J = 5.2 Hz, 1H), 8.47 (d, J = 5.2 Hz, 1H), 8.17 (m, 2H), 7.97 (d, J = 5.2 Hz, 1H), 7.91 (d, J = 5.2 Hz, 1H), 7.54–7.59 (m, 3H), 7.29–7.35 (m, 2H), 7.13–7.23 (m, 5H), 6.87 (s, 1H), 6.84 (s, 1H), 3.78 (s, 3H), 3.70–3.75 (m, 2H), 3.61–3.67 (m, 2H), 0.78–0.85 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 159.68, 149.96, 149.45, 143.69, 142.07, 141.98, 140.22, 138.83, 138.42, 136.39, 135.75, 133.95, 133.70, 132.04, 130.74, 130.44, 130.40, 128.73, 128.40, 122.96, 121.75, 121.65, 121.34, 121.19, 120.20, 119.95, 114.16, 113.43, 113.32, 110.01, 109.95, 55.34, 43.73, 43.41, 25.96, 25.80. HRMS calcd for C38H32N5O [M + H]+ 574.2601, found 574.2606. Anal. calcd for C38H31N5O: C, 79.56; H, 5.45; N, 12.21; found C 79.51, H 5.66, N 11.72.
1–(3,4-dimethoxyphenyl)-9–(4-(1–(3-pyridyl)-β-carboline-9-yl)butyl)-β-carboline (5l)
This compound was obtained as yellow crystals in 63% yield, m.p. 239.8–240.5 °C. 1H NMR (400 MHz, CDCl3) δ: 8.68 (d, J = 2.0 Hz, 1H), 8.61 (dd, J = 4.8, 1.6 Hz, 1H), 8.51 (d, J = 5.2 Hz, 1H),8.48 (d, J = 5.2 Hz, 1H), 8.18 (d, J = 7.6 Hz, 1H), 8.16 (d, J = 7.6 Hz, 1H),7.97 (d, J = 5.2 Hz, 1H),7.95 (d, J = 5.2 Hz, 1H), 7.55–7.60 (m, 3H), 7.31–7.35 (m, 2H), 7.13–7.20 (m, 3H), 6.96 (d, J = 2.0 Hz, 1H), 6.85 (d, J = 8.0 Hz, 1H), 6.79 (d, J = 8.0 Hz, 1H), 3.84 (s, 3H), 3.80 (s, 3H), 3.76 (t, J = 6.4 Hz, 2H), 3.66 (t, J = 6.4 Hz, 2H), 0.82–0.92 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 149.95, 149.44, 149.20, 148.54, 143.62, 142.00, 141.87, 140.20, 138.83, 138.28, 136.36, 135.75, 133.98, 133.77, 132.17, 130.74, 130.46, 128.79, 128.49, 122.97, 121.73, 121.68, 121.34, 121.19, 120.23, 120.04, 114.19, 113.52, 112.45, 110.61, 110.09, 109.89, 55.99, 55.96, 43.82, 43.46, 26.05, 25.89. HRMS calcd for C39H34N5O2 [M + H]+ 604.2707, found 604.2705. Anal. calcd for C39H33N5O2: C, 77.59; H, 5.51; N, 11.60; found C 77.36, H 5.57, N 11.51.
1–(3,4,5-trimethoxyphenyl)-9–(4-(1–(3-pyridyl)-β-carboline-9-yl)butyl)-β-carboline (5m)
This compound was obtained as yellow crystals in 49% yield, m.p. 231.0–232.3 °C. 1H NMR (400 MHz, CDCl3) δ: 8.71 (dd, J = 2.0, 0.8 Hz, 1H), 8.60 (dd, J = 4.8, 1.6 Hz, 1H), 8.51 (d, J = 0.8 Hz, 1H), 8.49 (d, J = 0.8 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 8.14 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 5.2 Hz, 1H), 7.96 (d, J = 5.2 Hz, 1H), 7.57–7.62 (m, 3H), 7.30–7.36 (m, 2H), 7.21 (d, J = 8.4 Hz, 1H), 7.17 (d, J = 8.4 Hz, 1H), 7.11–7.15 (m, 1H), 6.67 (s, 2H), 3.80 (s, 3H), 3.78 (s, 6H), 3.76 (t, J = 6.4 Hz, 2H), 3.68 (t, J = 7.2 Hz, 2H), 0.95–0.98 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 153.03, 149.94, 149.43, 143.58, 141.94, 141.68, 140.15, 138.78, 138.28, 138.24, 136.34, 135.79, 135.24, 133.98, 133.74, 130.72, 130.39, 128.97, 128.57, 122.98, 121.69, 121.67, 121.31, 121.11, 120.28, 120.11, 114.20, 113.79, 110.13, 109.86, 106.54, 60.93, 56.24, 43.91, 43.54, 26.09, 25.94. HRMS calcd for C40H36N5O3 [M + H]+ 634.2813, found 634.2817. Anal. calcd for C40H35N5O3: C, 75.81; H, 5.57; N, 11.05; found C 75.79, H 5.56, N 11.32.
1–(2-chlorophenyl)-9–(4-(1–(3-pyridyl)-β-carboline-9-yl)butyl)-β-carboline (5n)
This compound was obtained as yellow crystals in 58% yield, m.p. 177.6–178.9 °C. 1H NMR (400 MHz, CDCl3) δ: 8.73 (dd, J = 2.4, 0.8 Hz, 1H), 8.66 (dd, J = 4.8, 1.6 Hz, 1H), 8.53 (d, J = 5.2 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.17–8.21 (m, 2H), 8.00 (d, J = 2.4 Hz, 1H), 7.99 (d, J = 2.4 Hz, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.60–7.63 (m, 1H), 7.57–7.59 (m, 1H), 7.26–7.37 (m, 4H), 7.22–7.25 (m, 2H), 7.13–7.20 (m, 3H), 3.72 (t, J = 7.2 Hz, 2H), 3.48–3.64 (m, 2H), 0.84–0.93 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 149.99, 149.55, 142.03, 141.51, 140.52, 140.19, 138.80, 138.34, 138.14, 136.48, 135.71, 134.02, 134.00, 133.71, 131.32, 130.86, 130.04, 129.92, 129.23, 128.76, 128.62, 126.60, 123.02, 121.81, 121.72, 121.24, 121.09, 120.31, 120.02, 114.31, 114.24, 109.97, 109.71, 43.72, 43.19, 26.30, 26.08. HRMS calcd for C37H29ClN5 [M + H]+ 578.2106, found 578.2111. Anal. calcd for C37H28ClN5: C, 76.87; H, 4.88; N, 12.11; found C 76.38, H 4.85, N 11.75.
1–(4-methoxyphenyl)-9–(4-(1–(2-thienyl)-β-carboline-9-yl)butyl)-β-carboline (5o)
This compound was obtained as yellow crystals in 64% yield, m.p. 232.5–233.3 °C. 1H NMR (400 MHz, CDCl3) δ: 8.50 (d, J = 5.2 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.16 (t, J = 7.6 Hz, 2H), 7.96 (d, J = 5.2 Hz, 1H), 7.94 (d, J = 5.2 Hz, 1H), 7.55–7.59 (m, 2H), 7.29–7.38 (m, 5H), 7.23 (d, J = 8.4 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.00–7.02 (m, 1H), 6.96–6.98 (m, 1H), 6.90–6.94 (m, 2H), 3.74–3.84 (m, 7H), 0.95–1.01 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 159.87, 142.11, 141.94, 140.82, 138.52, 136.77, 134.54, 133.87, 130.72, 130.55, 128.60, 128.52, 128.10, 126.92, 126.55, 121.77, 121.69, 121.38, 121.25, 120.10, 120.03, 114.18, 113.58, 113.48, 110.04, 109.93, 55.37, 43.60, 43.59, 26.18, 26.10. HRMS calcd for C37H31N4OS [M + H]+ 579.2213, found 579.2209. Anal. calcd for C37H30N4OS: C, 76.79; H, 5.23; N, 9.68; S, 5.54; found C, 76.10; H, 5.50; N, 9.29; S, 5.16.
1–(3,4-dimethoxyphenyl)-9–(4-(1–(2-thienyl)-β-carboline-9-yl)butyl)-β-carboline (5p)
This compound was obtained as yellow crystals in 49% yield, m.p. 217.2–218.4 °C. 1H NMR (400 MHz, CDCl3) δ: 8.51 (d, J = 5.2 Hz, 1H), 8.47 (d, J = 5.2 Hz, 1H), 8.18 (d, J = 7.6 Hz, 1H), 8.14 (d, J = 7.6 Hz, 1H), 7.96 (d, J = 5.2 Hz, 1H), 7.94 (d, J = 5.2 Hz, 1H), 7.55–7.59 (m, 2H), 7.22 (d, J = 8.4 Hz, 1H), 7.19 (d, J = 8.4 Hz, 1H), 7.03 (d, J = 2.0 Hz, 1H), 6.98 (dd, J = 3.2, 1.2 Hz, 1H), 6.96 (dd, J = 8.0, 2.0 Hz, 1H), 6.92 (dd, J = 5.2, 3.2 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 3.86 (s, 3H), 3.82(s, 3H),3.78–3.85 (m, 4H), 0.98–1.08 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 149.24, 148.63, 143.72, 141.92, 141.90, 140.81, 138.53, 138.26, 136.75, 134.54, 133.98, 132.30, 130.69, 130.51, 128.65, 128.41, 128.04, 126.86, 126.50, 121.77, 121.73, 121.68, 121.46, 121.23, 120.10, 119.98, 114.20, 113.57, 112.47, 110.63, 110.11, 109.88, 55.99, 55.97, 43.66, 43.59, 26.29, 26.22. HRMS calcd for C38H33N4O2S [M + H]+ 609.2319, found 609.2314. Anal. calcd for C38H32N4O2S: C, 74.98; H, 5.30; N, 9.20; S, 5.27; found C, 74.47; H, 5.24; N, 9.36; S, 5.02.
1–(3,4,5-trimethoxyphenyl)-9–(4-(1–(2-thienyl)-β-carboline-9-yl)butyl)-β-carboline (5q)
This compound was obtained as yellow crystals in 65% yield, m.p. 226.5–227.8 °C. 1H NMR (400 MHz, CDCl3) δ: 8.50 (d, J = 5.2 Hz, 1H), 8.46 (d, J = 5.2 Hz, 1H), 8.19 (d, J = 7.6 Hz, 1H), 8.12 (d, J = 7.6 Hz, 1H), 7.99 (d, J = 5.2 Hz, 1H), 7.92 (d, J = 5.2 Hz, 1H), 7.56–7.60 (m, 2H), 7.28–7.36 (m, 2H), 7.17–7.24 (m, 3H), 6.96 (dd, J = 7.6, 1.2 Hz, 1H), 6.88 (dd, J = 5.2, 3.6 Hz, 1H), 6.71 (s, 2H), 3.80–3.87 (m, 4H), 3.82 (s, 3H), 3.79 (s, 6H), 1.03–1.12 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 153.07, 143.44, 141.91, 141.87, 140.71, 138.45, 138.40, 137.94, 136.61, 134.50, 133.86, 130.70, 130.66, 128.83, 128.62, 128.05, 126.82, 126.48, 121.80, 121.64, 121.39, 121.15, 120.17, 120.15, 114.22, 113.88, 110.19, 109.84, 106.65, 60.95, 56.24, 43.68, 26.36, 26.27. HRMS calcd for C39H35N4O3S [M + H]+ 639.2424, found 639.2422. Anal. calcd for C39H34N4O3S: C, 73.33; H, 5.37; N, 8.77; S, 5.02; found C, 73.37; H, 5.59; N, 8.65; S, 4.81.
1–(2-chlorophenyl)-9–(4-(1–(2-thienyl)-β-carboline-9-yl)butyl)-β-carboline (5r)
This compound was obtained as yellow crystals in 52% yield, m.p. 225.4–227.2 °C. 1H NMR (400 MHz, CDCl3) δ: 8.52 (s, 1H), 8.50 (s, 1H), 8.16–8.20 (m, 2H), 8.01 (d, J = 5.2 Hz, 1H), 7.97 (d, J = 5.2 Hz, 1H), 7.56–7.62 (m, 2H), 7.27–7.38 (m, 7H), 7.19–7.25 (m, 2H), 7.06 (dd, J = 3.6, 1.2 Hz, 1H), 7.03 (dd, J = 5.2, 3.2 Hz, 1H), 3.88 (t, J = 7.2 Hz, 2H), 3.53–3.71 (m, 2H), 1.01–1.13 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 141.97, 141.52, 140.78, 140.60, 138.51, 138.41, 138.11, 136.77, 134.53, 134.06, 133.87, 131.34, 130.86, 130.07, 130.05, 129.33, 128.62, 128.54, 128.25, 127.03, 126.66, 126.63, 121.75, 121.29, 121.18, 120.19, 119.96, 114.33, 114.22, 109.97, 109.68, 43.59, 43.29, 26.42, 26.40. HRMS calcd for C36H28ClN4S [M + H]+ 583.1718, found 583.1720. Anal. calcd for C36H27ClN4S: C, 74.15; H, 4.67; N, 9.61; S, 5.50; found C, 73.73; H, 4.53; N, 9.63; S, 5.19.
7-methoxy-1-methyl-9–(4-(1-methyl-β-carboline-9-yl)butyl)-β-carboline (5s)
This compound was obtained as colorless crystals in 74% yield, m.p. 188.7–189.4 °C. 1H NMR (400 MHz, DMSO-d6) δ: 8.23 (s, 1H), 8.21 (s, 1H),8.16 (d, J = 5.2 Hz, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.99 (d, J = 5.2 Hz, 1H), 7.87 (d, J = 5.2 Hz, 1H), 7.70 (d, J = 8.8 Hz, 1H), 7.54–7.58 (m, 1H), 7.22–7.26 (m, 1H), 7.20 (d, J = 2.0 Hz, 1H), 6.86 (dd, J = 8.8, 2.0 Hz, 1H), 4.58–4.62 (m, 4H), 3.87 (s, 3H), 2.94 (s, 3H), 2.89 (s, 3H), 1.82–1.86 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ: 160.38, 142.59, 141.14, 140.96, 140.39, 137.69, 137.49, 134.37, 134.23, 128.33, 127.97,127.94, 122.27, 121.36, 120.36, 119.36, 114.09, 112.83, 112.12, 110.27, 108.91, 93.74, 55.48, 43.67, 43.57, 27.55, 27.37, 22.98, 22.86. HRMS calcd for C29H29N4O [M + H]+ 449.2336, found 449.2340. Anal. calcd for C29H28N4O: C, 77.65; H, 6.29; N, 12.49; found C 77.39, H 5.97, N 12.38.
7-methoxy-1-methyl-9–(5-(1-methyl-β-carboline-9-yl)pentyl)-β-carboline (5t)
This compound was obtained as colorless crystals in 67% yield, m.p. 176.8–177.6 °C. 1H NMR (400 MHz, CDCl3) δ: 8.33 (d, J = 5.2 Hz, 1H), 8.29 (d, J = 5.2 Hz, 1H), 8.11 (d, J = 7.6 Hz, 1H), 7.97 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 5.2 Hz, 1H), 7.74 (d, J = 5.2 Hz, 1H), 7.52–7.56 (m, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.25–7.29 (m, 1H), 6.89 (dd, J = 8.4, 2.0 Hz, 1H), 6.77 (d, J = 2.0 Hz, 1H), 4.49 (t, J = 7.2 Hz, 2H), 4.42 (t, J = 7.2 Hz, 2H), 3.89 (s, 3H), 2.99 (s, 3H), 2.97 (s, 3H), 1.81–1.91 (m, 4H), 1.41–1.50 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 160.92, 143.09, 141.45, 141.04, 140.26, 138.15, 138.09, 135.17, 135.01, 129.58, 129.14, 128.21, 122.48, 121.53, 121.29, 119.73, 115.21, 112.97, 112.31, 109.56, 108.60, 93.51, 55.72, 44.55, 30.83, 30.56, 24.42, 23.58, 23.33. HRMS calcd for C30H31N4O [M + H]+ 463.2492, found 463.2497. Anal. calcd for C30H30N4O: C, 77.89; H, 6.54; N, 12.11; found C, 77.42; H, 6.33; N, 12.07.
7-methoxy-1-methyl-9–(4-(1–(3-pyridyl)-β-carboline-9-yl)butyl)-β-carboline (5u)
This compound was obtained as colorless crystals in 70% yield, m.p. 163.8–164.5 °C. 1H NMR (400 MHz, CDCl3) δ: 8.79 (d, J = 2.0 Hz, 1H), 8.65 (dd, J = 4.8, 1.6 Hz, 1H), 8.51 (d, J = 5.2 Hz, 1H), 8.29 (d, J = 5.2 Hz, 1H), 8.15–8.17 (m, 1H), 7.94–7.96 (m, 2H), 7.71 (d, J = 5.2 Hz, 1H), 7.65–7.68 (m, 1H), 7.56–7.60 (m, 1H), 7.30–7.34 (m, 2H), 7.23–7.25 (m, 1H), 6.89 (dd, J = 8.4, 2.0 Hz, 1H), 6.60 (d, J = 2.0 Hz, 1H), 4.16 (t, J = 7.2 Hz, 2H), 3.95 (t, J = 7.2 Hz, 2H), 3.90 (s, 3H), 2.77 (s, 3H), 1.48–1.35 (m, 2H), 1.35–1.27 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 160.85, 150.02, 149.66, 142.77, 142.20, 140.30, 140.13, 139.02, 138.36, 136.42, 135.80, 135.01, 134.09, 130.92, 129.43, 128.81, 123.15, 122.45, 121.79, 121.30, 120.33, 115.16, 114.17, 112.29, 109.94, 108.54, 93.55, 55.73, 44.12, 43.84, 27.37, 26.02, 23.30. HRMS calcd for C33H30N5O [M + H]+ 512.2445, found 512.2453. Anal. calcd for C33H29N5O: C, 77.47; H, 5.71; N, 13.69; found C, 76.67; H, 5.75; N, 13.05.
7-methoxy-1-methyl-9–(5-(1–(3-pyridyl)-β-carboline-9-yl)pentyl)-β-carboline (5v)
This compound was obtained as colorless crystals in 62% yield, m.p. 183.6–184.9 °C. 1H NMR (400 MHz, CDCl3) δ: 8.82 (dd, J = 2.0, 0.8 Hz, 1H), 8.54 (d, J = 5.2 Hz, 1H), 8.53 (dd, J = 5.2, 1.6 Hz, 1H) 8.30 (d, J = 5.2 Hz, 1H), 8.18 (d, J = 7.6 Hz, 1H), 8.02 (d, J = 5.2 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.85–7.88 (m, 1H), 7.75 (d, J = 5.2 Hz, 1H), 7.56–7.60 (m, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.30–7.34 (m, 1H), 7.24–7.28 (m, 1H), 6.90 (dd, J = 8.4, 2.0 Hz, 1H), 6.72 (d, J = 2.0 Hz, 1H), 4.27 (t, J = 7.6 Hz, 2H), 3.94 (t, J = 7.6 Hz, 2H), 3.91 (s, 3H), 2.90 (s, 3H), 1.45–1.53 (m, 2H), 1.33–1.42 (m, 2H), 0.87–0.90 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 160.95, 150.08, 149.51, 143.06, 142.01, 140.36, 140.19, 138.81, 138.05, 136.60, 135.99, 135.11, 134.25, 130.83, 129.58, 128.81, 123.05, 122.48, 121.74, 121.26, 120.26, 115.15, 114.25, 112.34, 110.00, 108.66, 93.56, 55.76, 44.43, 44.25, 30.09, 28.61, 23.82, 23.22 . HRMS calcd for C34H32N5O [M + H]+ 526.2601, found 526.2609. Anal. calcd for C34H31N5O: C, 77.69; H, 5.94; N, 13.32; found C, 77.76; H, 5.95; N, 13.74.
7-methoxy-1-methyl-9–(4-(1–(2-thienyl)-β-carboline-9-yl)butyl)-β-carboline (5w)
This compound was obtained as colorless crystals in 68% yield, m.p. 189.3–190.1 °C. 1H NMR (400 MHz, CDCl3) δ: 8.50 (d, J = 5.2 Hz, 1H), 8.28 (d, J = 5.2 Hz, 1H), 8.14–8.16 (m, 1H), 7.96 (d, J = 1.6 Hz, 1H), 7.95 (d, J = 2.0 Hz, 1H), 7.72 (d, J = 5.2 Hz, 1H), 7.55–7.60 (m, 1H), 7.34–7.38 (m, 2H), 7.29–7.33 (m, 1H), 7.13 (dd, J = 3.6, 1.2 Hz, 1H), 7.02 (dd, J = 5.2, 3.6 Hz, 1H), 6.88 (dd, J = 8.4, 2.0 Hz, 1H), 6.66 (d, J = 2.0 Hz, 1H), 4.21 (t, J = 7.2 Hz, 2H), 4.17–4.08 (t, J = 7.2 Hz, 2H), 3.90 (s, 3H), 2.86 (s, 3H), 1.42–1.54 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 160.86, 142.92, 142.05, 140.94, 140.18, 138.72, 138.18, 136.91, 135.10, 134.60, 130.91, 129.54, 128.69, 128.26, 127.12, 126.70, 122.49, 121.75, 121.35, 120.22, 115.24, 114.21, 112.33, 109.94, 108.45, 93.68, 55.75, 43.94, 29.69, 27.70, 26.51, 23.28. HRMS calcd for C32H29N4OS [M + H]+ 517.2057, found 517.2063. Anal. calcd for C32H28N4OS: C, 74.39; H, 5.46; N, 10.84; S, 6.21; found C, 74.21; H, 5.42; N, 10.65; S, 6.17.
7-methoxy-1-methyl-9–(5-(1–(2-thienyl)-β-carboline-9-yl)pentyl)-β-carboline (5x)
This compound was obtained as yellow crystals in 71% yield, m.p. 196.1–197.4 °C. 1H NMR (400 MHz, CDCl3) δ: 8.50 (d, J = 5.2 Hz, 1H), 8.30 (d, J = 5.2 Hz, 1H), 8.13–8.16 (m, 1H), 7.94–7.98 (m, 2H), 7.74 (d, J = 5.2 Hz, 1H), 7.55–7.59 (m, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.30 (m, 1H), 7.23 (dd, J = 5.2, 1.2 Hz, 1H), 7.16 (dd, J = 3.6, 1.2 Hz, 1H), 6.92 (dd, J = 5.2, 3.6 Hz, 1H), 6.89 (dd, J = 8.4, 2.0 Hz, 1H), 6.72 (d, J = 2.0 Hz, 1H), 4.28 (t, J = 7.6 Hz, 2H), 4.10 (t, J = 7.6 Hz, 2H), 3.90 (s, 3H), 2.92 (s, 3H), 1.50–1.58 (m, 2H), 1.40–1.48 (m, 2H), 0.96–1.04 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 160.86, 143.01, 142.10, 141.02, 140.33, 138.56, 138.18, 136.98, 135.20, 134.67, 130.84, 129.48, 128.66, 128.33, 126.89, 126.56, 122.44, 121.67, 121.28, 120.12, 115.23, 114.18, 112.32, 110.04, 108.52, 93.62, 55.73, 44.54, 44.09, 29.72, 28.94, 23.97, 23.38. HRMS calcd for C33H31N4OS [M + H]+ 531.2213, found 531.2213. Anal. calcd for C33H30N4OS: C, 74.69; H, 5.70; N, 10.56; S, 6.04; found C, 74.06; H, 6.08; N, 10.49; S, 6.44.
Biological evaluation
In vitro cell growth inhibition assay
Target compounds were assayed by the MTT method for cytotoxic activity, as described previouslyCitation5. The panel of cell lines included gastric carcinoma (BGC-823), liver carcinoma (HepG2), breast carcinoma (MCF-7), colon carcinoma (HT-29), esophageal carcinoma (Eca-109), and Lewis lung carcinoma (LLC). Growth inhibition rates were calculated with the following equitation: Inhibition ratio (%)= . Half maximal inhibitory concentration (IC50) of each compound was calculated using software Graph-Pad Prism (version 6.0).
Assay of acute toxicities
Specific pathogen-free KM mice (6–8 weeks old) weighing 19–22 g were housed in a mouse room at 24 ± 2 °C and 60–70% humidity with 12 h light/dark cycles. The mice were provided rodent laboratory chow pellets and tap water for a week to adapt to the environment of the mouse room. The experimental protocol was approved by the Institutional Animal Ethical Committee, and all of the animals were provided by Laboratory Animal Center of Xinjiang Uygur Autonomous Region. Prior to each experiment, mice were fastened overnight and allowed free access to water. Various doses of the asymmetric dimeric β-carboline derivatives, ranging from 5.0 to 500 mg kg−1 dissolved in 0.5% carboxymethyl cellulose sodium (CMC-Na) salt solution, were given intraperitoneally to different groups of healthy KM mice, and each group contained 10 mice (5 males and 5 females). After the administration of the compounds, the mice were observed continuously for the first 2 h for any gross behavioral changes and deaths, then intermittently for the next 24 h, and occasionally thereafter for 14 days, and for the onset of any delayed effects. All of the animals were killed on the 14th day after drug administration, and they were checked macroscopically for possible damage to the heart, liver, and kidneys. Mice that experienced immediate death following drug administration were also examined for any possible organ damage. LD50 values were calculated graphically as describedCitation34.
In vivo antitumor activity
Sarcoma180 and Lewis lung cancer cell lines were provided by Shanghai Institute of Pharmaceutical Industry. Mice were inoculated with Sarcoma180 and Lewis lung cancer tumour cells. After 7 days, the tumours were removed and the cells were harvested. Mice received subcutaneous injections of viable tumour cells (2 × 106 cells/mouse) in the armpit. Each compound was administered via i.p. injection to different groups of mice (each group contained 10 female mice) 24 h after the inoculation at a dosage about one-fifth of the LD50 value once a day for seven consecutive days. This dose was the maximum tolerated dose for most of the compounds based on our preliminary studies. CTX at 30 mg kg−1 was used as the positive control and the vehicle as the negative control. The weight of the animals was recorded every three days. All of the animals were killed on the 21st day after tumour inoculation, and the tumours were excised and weighed. The inhibition rate was calculated as follows:
where T is the average tumor weight of the treated group and C is the average tumor weight of the negative control group.
CAM assay in vivo
Anti-angiogenic activity of the selected compounds 5b and 5w were investigated in vivo using a CAM assay. Five-day-old fertilized eggs were obtained from a local hatchery. We injected 5 ml of albumin, and the eggs were incubated horizontally to allow the CAM to detach from the shell to produce a sham chamber. Compounds 5b and 5w were prepared in gelatin sponge discs (5 × 5 × 5 mm3) at the concentration of 0.5, 5.0, and 50 µM/disc, respectively. CA4P was used as the positive control drug. Discs containing the vehicle only (DMSO) were used as the negative control. A small window opening was made in the shell, and the discs were directly applied onto the CAM. The square opening was covered with sterilized surgical tape and the embryos were incubated for 48 h at 38.5 °C. The CAMs were photographed under a dissecting microscope and blood vessels in each CAM were counted. The results are presented as a mean percentage of inhibition to the control ± SD, (n = 3).
Results and discussion
Chemistry
The synthesis of the desired N9-heterobivalent β-carbolines (5a-r) were performed in four steps starting from L-tryptophan as outlined in Scheme 1–2. The tetrahydro-β-carbolines 2a-h was prepared by condensation of L-tryptophan with appropriate aldehyde via the Pictet-Spengler condensation, and followed by oxidation and decarboxylation to afford the intermediate 1-substituted-β-carbolines 3a-hCitation21,Citation29. Then the N9 of 3a-d was alkylated by the action of sodium hydride (NaH) in N,N-dimethylformamide (DMF) followed by the addition of the appropriate dibromo alkane to obtain intermediates 4a-f. Finally, 4a-f with appropriate 1-substituted-β-carbolines 3c-h in the presence of sodium hydride in DMF at room temperature to afford the target compounds 5a-r in 47–74% yield.
Scheme 1. Synthetic route to compounds 3a-h.
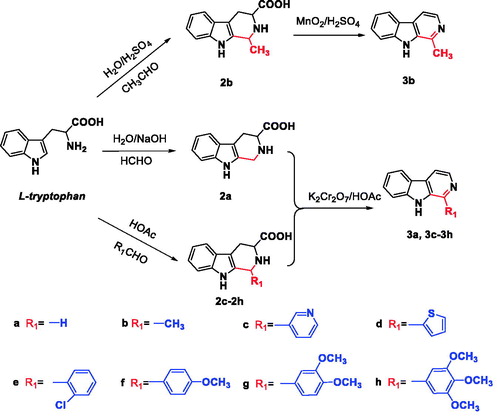
Scheme 2. Synthesis of the N9-heterobivalent β-carbolines 5a-r.
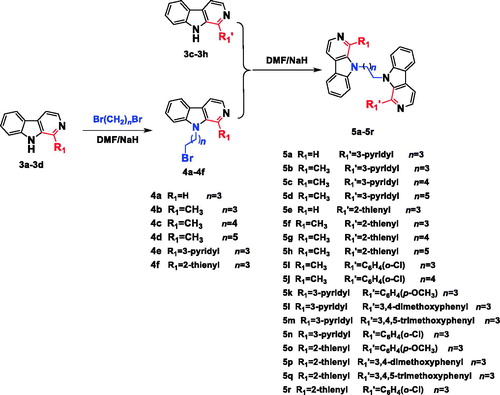
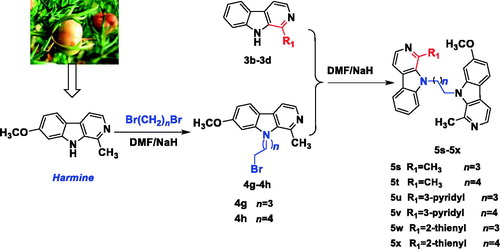
In Scheme 3, the intermediates 4g-f were synthesized from harmine and 1,4-dibromobutane or 1,5-dibromopentane using the same method of compound 4a, and finally compounds 5s-x were obtained from the similar method of 5a.
Scheme 3. Synthesis of the N9-heterobivalent β-carbolines 5s-x.
The chemical structures of all the novel N9-heterobivalent β-carbolines were characterized by 1H NMR, 13C NMR, HRMS and the elemental analysis.
In vitro cytotoxicity and structure–activity relationships
All the 24 novel synthesized N9-heterobivalent β-carbolines (5a-x) were screened for their in vitro cytotoxic activities against six different cancer cell lines, namely BGC-823 (gastric carcinoma), HepG2 (liver carcinoma), MCF-7 (breast carcinoma), HT-29 (colon carcinoma), Eca-109 (esophageal carcinoma) and LLC (Lewis lung carcinoma). Cisplatin and B-9–3 were used as the reference control and the results were expressed as IC50 values and summarized in . The IC50 values were the average of at least three independent experiments.
Table 1. Cytotoxic activity of N9-heterobivalent β-carbolines in vitro
As illustrated in , compounds 5b and 5w displayed a broad spectrum of cytotoxic activities with IC50 value of lower than 20 μM against the tested six tumor cell lines, while compounds 5c, 5s, 5t and 5v only exhibited strong cytotoxic effects with IC50 value of lower than 20 μM against three or four tumour cell lines. Interestingly, compounds 5d and 5j were selectively active against Eca-109 and LLC cell lines with IC50 value of lower than 20 μM but fail to show cytotoxic effects in other cell lines. Similarly, compound 5u displayed selective activities against HT-29 and LLC cell lines. Moreover, compound 5f only exhibited strong cytotoxic effects with IC50 value of lower than 20 μM against LLC cell lines. compounds 5g and 5i showed weak cytotoxic activities with IC50 values in the range of 18.8–97.6 μM. Unfortunately, compounds 5k–5r were weak or inactive against all tumour cell lines tested.
When N9-heterobivalent β-carbolines had the same linker, we examined the influence of the substituents in position-1 of the β-carboline core on the cytotoxic activities. In order to enhance the range of substituents, we designed compounds have methyl and different pattern of substitution with an aryl ring substituted by electron withdrawing (Cl) and donating (OCH3) groups in position-1 of β-carboline. For example, compound 5a, 5b, and 5k–5n, all have a 3-pyridyl group in R1 of one β-carboline ring, while in another β-carboline core, unlike compound 5a, the substituted group of the position-1’ were methyl (5b), 4-methoxyphenyl (5k), 3,4-dimethoxyphenyl (5l), 3,4,5-trimethoxyphenyl (5m), 2-chlorophenyl (5n), respectively. Of these six dimeric β-carbolines, the compound 5b displayed higher cytotoxic activities against BGC-823, HepG2, MCF-7, HT-29, Eca-109 and LLC with IC50 values of 12.1, 15.9, 8.4, 12.6, 10.5, and 12.4 μM, respectively. Additionally, among these six N9-heterobivalent β-carbolines 5e, 5f, and 5o–5r bearing a 2-thienyl group in R1 of one β-carboline core, compound 5f (R1’ = methyl) showed the highest cytotoxic activities against the test cell lines except HepG2. These results suggested that 3-pyridyl or 2-thienyl group substituent into position-1 of the β-carboline core, and in another β-carboline ring, the methyl substituent into R1’ facilitated cytotoxic potency, and the aryl substituent into R1’ might be detrimental to cytotoxic effects.
Next, we examined the influence of the spacer length of N9-heterobivalent β-carbolines on cytotoxic activities. According the previous investigation, the synthesized N9-heterobivalent β-carbolines were connected at the indolo-N by an alkyl chain, which ranged from 4 to 6 carbon atoms. Comparing the structure of 5b–5d, which bearing different carbon atoms linker and 3-pyridyl group in position-1 of the β-carboline core, compound 5b bearing 4-carbon atoms showed a broad spectrum of cytotoxic activities with IC50 value of lower than 20 μM against six tumor cell lines, and all the three compounds show selective cytotoxicities with IC50 value of lower than 20 μM against LLC. Similarly, further introduction of 2-thienyl group in position-1 of the β-carboline ring resulted in dimeric derivatives 5f–5h. Among these derivatives, the IC50 values of compound 5f against BGC, MCF7, and LLC cells is 38.8, 46.6, and 6.1 μM, respectively, compound 5 g displayed IC50 values for HepG2 and HT-29 of 18.8 and 32.3 μM, respectively, and compound 5h only exhibited higher cytotoxic effects with IC50 value of 30.2 μM against Eca-109 than other two compounds. Comparing the structure of 5i with 5j, compound 5i showed weak or moderate cytotoxic activities against all tumor cell lines tested, and compound 5j was selectively active against Eca-109 and LLC cell lines with IC50 value of lower than 20 μM but fail to show cytotoxic effects in BGC and HepG2 at the concentration of 100 μM. Comparing the structure of 5u with 5v, there is an extra methoxyl group attached to position-7 of the β-carboline, compound 5v demonstrated the higher cytotoxic activities against the tested tumor cell lines than 5u (except for HepG-2 cell line). Similarly, comparing the structure 5w with 5x, compound 5w displayed the higher cytotoxic activities against the tested tumor cell lines than 5x (except for LLC cell line). These results suggested that the length of the spacer had no obvious relationship with the cytotoxic activities against the tumor cell lines.
An overview of the cytotoxic activities data of all new synthesized N9-heterobivalent β-carbolines. 11 compounds were found to exhibit selective activity against LLC cell lines with IC50 value of lower than 20 μM. Moreover, Compound 5b, 5s displayed higher cytotoxic activities against at least four tumour cell lines than the prototype B-9–3, and compound 5c, 5t, 5v, 5w exhibited higher cytotoxic effects against three tumour cell lines than B-9–3.
In summary, a total analysis of the cytotoxic activities of N9-heterobivalent β-carbolines in vitro clearly suggest that: (1) C1-methylation and C7-methoxylation were favorable for increased activities; (2) 3-Pyridyl or 2-thienyl group substituent into position-1 of the β-carboline core, and the aryl (electron withdrawing and donating groups) substituent into position-1’ of another β-carboline ring might be detrimental to cytotoxic effects of this class of compounds.
Assessment of acute toxicity
The LD50 values of the selected N9-heterobivalent β-carbolines in mice after intraperitoneal (i.p.) administration are shown in . All of the tested dimeric β-carbolines resulted in acute toxic manifestation but they did not cause any obvious neurotoxic effects, including tremors, twitch, jumping, and supination. The animals showed a decrease in locomotive activity after the administration of various bivalent β-carbolines. Death occurred mostly in the high dosage group within 4–8 h after injection. All of the surviving animals returned to normal within the next day. Autopsies of the animals that died during the course of experiment and the necropsy findings in the surviving animals at the end of the experimental period (14 days) revealed no obvious changes in any of the organs.
Table 2. Acute toxic effects of N9-heterobivalent β-carbolines in mice and antitumor activities of these compounds against mice bearing Sarcoma 180 and Lewis lung cancer
Of all of the investigated asymmetric dimeric β-carbolines, compounds 5b and 5c, which had no substituent in position-7, and bearing different carbon atoms linker and 3-pyridyl group in position-1 of the β-carboline core, demonstrated weaker acute toxicities with the LD50 values of 150 mg kg−1 and 175 mg kg−1, respectively. Compounds 5t and 5w, which have a harmine molecular linked with another 1-substituted β-carboline ring, displayed remarkable acute toxicity with the LD50 values of 35 mg kg−1 and 50 mg kg−1, respectively. These results suggested that the methoxy substituent in position-7 of β-carboline nucleus played a vital role in determining the remarkable neurotoxic effects.
Evaluation of antitumor activity of N9-heterobivalent β-carbolines in vivo
Based on the in vitro assay results, we further tested the antitumor activity of four N9-heterobivalent β-carbolines in vivo against mice bearing Sarcoma 180 and Lewis lung cancer, respectively, and the reference drugs Cyclophosphamide (CTX). Our previous investigation demonstrated that mice bearing Lewis lung cancer were more susceptible to β-carbolines than other animal models; therefore, these animal models were selected and evaluated in the present investigation.Citation5 The tumor inhibition rates of all of the investigated asymmetric dimeric β-carbolines were summarized in .
As shown in , all the tested N9-heterobivalent β-carbolines displayed moderate to strong antitumor activities in animal model. Interestingly, compounds 5b and 5c, which having the same substituent and different carbon atoms linker. Compound 5b, which bearing 4-carbon atoms, exhibited the potent antitumor agent with the tumor inhibition rate of 43.6 and 41.9% against Sarcoma 180-bearing mice and Lewis lung cancer-bearing mice, respectively, and compound 5c displayed moderate antitumor activity with the tumor inhibition rate of 33.6% against Sarcoma 180-bearing mice at doses of 35 mg kg−1. Compound 5t, the harmine and harman linked with five methylene units, showed moderate antitumor activity against mice with Sarcoma 180 with the tumour inhibition rate of 37.2% at doses of 7 mg kg−1. Particularly, compound 5w was found to be the remarkable antitumor agent with the tumor inhibition rate of 47.1 and 42.3% against mice bearing Sarcoma 180 and Lewis lung cancer, respectively. These results implied that the harmine or harman linked with another of β-carboline nucleus (3-pyridyl or 2-thienyl substituent into position-1) enhanced their antitumor activities.
In vivo anti-angiogenic effect of compounds 5b and 5w
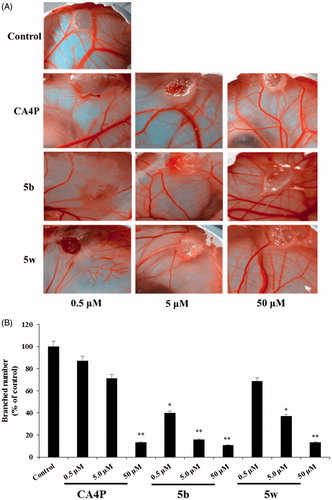
The most potent compounds, 5b and 5w, were selected to evaluate anti-angiogenic activity by chicken chorioallantoic membrane (CAM) assay. The inhibitory effects of compounds 5b and 5w on the angiogenesis of CAM are shown in . The anti-angiogenetic activity of compounds 5b and 5w were semi-quantitatively analyzed using Graph Pad Prism 5.0 (shown in ). The results showed that compound 5b (p < 0.05) could inhibit the angiogenesis of CAM. The anti-angiogenetic activity of compound 5b was comparable to the CA4P in vivo CAM assay at the same dose (50 μM).
Figure 2. Compounds 5b and 5w inhibited angiogenesis. (A) In vivo anti-angiogenic effect of compounds 5b and 5w in chick chorioallantoic membrane (CAM) assay. positive control (CA4P, 0.5–50 mg/mL) and vehicle control (0.1% DMSO). (B) Quantification graphs of the inhibitory effects of compounds 5b and 5w on angiogenesis and migration. *p < 0.05, **p < 0.01.
Conclusions
In this study, we synthesized 24 new, N9-heterodimeric β-carboline derivatives and focused on compounds with 4 − 6 carbon linkers between the indole nitrogen. All of the compounds were screened for their in vitro cytotoxic activity against BGC-823, HepG2, MCF-7, HT-29, Eca-109 and LLC cancer cell lines. The results showed that compounds 5b, and 5w exhibited strong cytotoxic activities with IC50 value of lower than 20 μM against the six tested tumor cell lines. In addition, four asymmetric dimeric β-carbolines were selected for evaluation in vivo against mice bearing Sarcoma 180 and Lewis lung cancer, compounds 5b and 5w exhibited potent antitumor efficacies with tumour inhibition rate of over 40% in the tested animal models. Moreover, the pharmacological mechanisms showed that compound 5b could retard in the CAM assay, and anti-angiogenetic potency was more potent than the reference drug CA4P. Preliminary structure-activity analysis indicated that: (1) C1-methylation and C7-methoxylation were favorable for increased activities; (2) 3-Pyridyl or 2-thienyl group substituent into position-1 of the β-carboline core, and the aryl (electron withdrawing and donating groups) substituent into position-1’ of another β-carboline ring might be detrimental to cytotoxic effects of this class of compounds. Although most N9-heterodimeric β-carbolines presented here showed modest cytotoxic activities, the investigations of these structural modifications and preliminary SARs would be helpful to further design and develop more potent compounds.
9_9_-asymmetric_bivalent_carbolines_supporting_information.pdf
Download PDF (6.9 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Smith BD, Smith GL, Hurria A, et al. Future of cancer incidence in the United States: burdens upon an aging, changing nation. J Clin Oncol 2009;27:2758–65.
- Ishida J, Wang HK, Bastow KF, et al. Antitumor agents 201. Cytotoxicity of harmine and beta-carboline analogs. Bioorg Med Chem Lett 1999; 9:3319–24.
- Cao RH, Chen Q, Hou XR, et al. Synthesis, acute toxicities and antitumor effects of novel 9-substituted β-carboline derivatives. Bioorg Med Chem 2004; 12:4613–23.
- Cao RH, Guan XD, Shi BX, et al. Design, synthesis and 3D-QSAR of beta-carboline derivatives as potent antitumor agents . Eur J Med Chem 2010; 45:2503–15.
- Cao RH, Fan WX, Guo L, et al. Synthesis and structure-activity relationships of harmine derivatives as potential antitumor agents. Eur J Med Chem 2013; 60:135–43.
- Shankaraiah N, Siraj KP, Nekkanti S, et al. DNA-binding affinity and anticancer activity of β-carboline–chalcone conjugates as potential DNA intercalators: molecular modelling and synthesis. Bioorg Chem 2015; 59:130–9.
- TAIRA Z, KANZAWA S, DOHARA C, et al. Intercalation of six β-carboline derivatives into DNA. J Toxicol Environ Health 1997; 43:83–91.
- Cao RH, Peng WL, Chen HS, et al. DNA binding properties of 9-substituted harmine derivatives. Biochem Biophys Res Commun 2005; 338:1557–63.
- Kamal A, Sathish M, Nayak VL, et al. Design and synthesis of dithiocarbamate linked β-carboline derivatives: DNA topoisomerase II inhibition with DNA binding and apoptosis inducing ability. Bioorg Med Chem 2015; 23:5511–26.
- Figueiredo PO, Perdomo RT, Garcez FR, et al. Further constituents of Galianthe thalictroides (Rubiaceae) and inhibition of DNA topoisomerases I and IIa by its cytotoxic β-carboline alkaloids. Bioorg Med Chem Lett 2014; 24:1358–61.
- Song YC, Kesuma D, Wang J, et al. Specific inhibition of cyclin-dependent kinases and cell proliferation by harmine. Biochem Biophys Res Commun 2004; 317:128–32.
- Li Y, Liang FS, Jiang W, et al. DH334, a beta-carboline anti-cancer drug, inhibits the CDK activity of budding yeast . Cancer Biol Ther 2007; 6:1193–9.
- Zhang J, Li Y, Guo L, et al. DH166, a beta-carboline derivative, inhibits the kinase activity of PLK1. Cancer Biol Ther 2009; 8:2374–83.
- Insaf F, Mohamed AB, Aymen BN, et al. Synthesis, cytotoxic, anti-lipoxygenase and anti-acetylcholinesterase capacities of novel derivatives from harmine. J Enzyme Inhib Med Chem 2016;31:23–33.
- Insaf F, Jalloul B, Mansour Z, et al. Synthesis of new isoxazoline derivatives from harmine and evaluation of their anti-Alzheimer, anti-cancer and anti-inflammatory activities. J Enzyme Inhib Med Chem 2015; 30:371–6.
- Castro AC, Dang LC, Soucy F, et al. Novel IKK inhibitors: β-carbolines. Bioorg Med Chem Lett 2003; 13:2419–22.
- Posner GH, D'Angelo J, O'Neill PM, et al. Anticancer activity of artemisinin-derived trioxanes. Expert Opin Ther Pat 2006; 16:1665–72.
- Alagbala AA, McRiner AJ, Borstnik K, et al. Biological mechanisms of action of novel C-10 non-acetal trioxane dimers in prostate cancer cell lines. J Med Chem 2006; 49:7836–42.
- Posner GH, McRiner AJ, Paik IH, et al. Anticancer and antimalarial efficacy and safety of artemisinin-derived trioxane dimers in rodents. J Med Chem 2004; 47:1299–301.
- Jung M, Lee S, Ham J, et al. Antitumor activity of novel deoxoartemisinin monomers, dimers, and trimer. J Med Chem 2003; 46:987–94.
- Shi BX, Cao RH, Fan WX, et al. Design, synthesis and in vitro and in vivo antitumor activities of novel bivalent β-carbolines. Eur J Med Chem 2013;60:10–22.
- Wu QF, Bai ZS, Ma Q, et al. Synthesis and biological evaluation of novel bivalent β-carbolines as potential antitumor agents. Med Chem Commun 2014; 5:953–8.
- Guo L, Cao RH, Fan WX, et al. Design, synthesis and in vitro antitumor activities of novel bivalent β-carbolines. Chem J Chin Univer 2013;60:10–99.
- Guo L, Chen W, Fan WX, et al. Synthesis and preliminary evaluation of novel alkyl diamine linked bivalent β-carbolines as angiogenesis inhibitors. Med Chem Commun 2016; 7:2177–83.
- Chen W, Zhang GX, Guo L, et al. Synthesis and biological evaluation of novel alkyl diamine linked bivalent β-carbolines as angiogenesis inhibitors. Eur J Med Chem 2016; 124:249–61.
- Sun RQ, Liu R, Zhou C, et al. Synthesis and biological evaluation of piperazine group-linked bivalent β-carbolines as potential antitumor agents. Med Chem Commun 2015; 6:2170–4.
- Daoud A, Song J, Xiao FY, et al. B-9-3, a novel β-carboline derivative exhibits anti-cancer activity via induction of apoptosis and inhibition of cell migration in vitro. Eur J Pharmacol 2014;724:219–30.
- Ma Q, Chen W, Chen W. Anti-tumor angiogenesis effect of a new compound: B-9-3 through interference with VEGFR2 signaling. Tumour Biol 2016; 37:6107–16.
- Xinjiang Huashidan Pharmaceutical Research Co., Ltd. EP 1634881 A1, 2006.
- Reddy POV, Mishra S, Tantak MP, et al. Design, synthesis and in vitro cytotoxicity studies of novel β-carbolinium bromides. Bioorg Med Chem Lett 2017; 27:1379–84.
- Hadjaz F, Yous S, Lebegue N, et al. A mild and efficient route to 2-benzyl tryptamine derivatives via ring-opening of β-carbolines. Tetrahedron 2008; 64:10004–8.
- Wu QF, Cao RH, Feng MX, et al. Synthesis and in vitro cytotoxic evaluation of novel 3,4,5-trimethoxyphenyl substituted β-carboline derivatives. Eur J Med Chem 2009;44:533–40.
- Du HT, Tian S, Chen JC, et al. Synthesis and biological evaluation of N9-substituted harmine derivatives as potential anticancer agents. Bioorg Med Chem Lett 2016;26:4015–9.
- Kassa J, Vachek J. A comparison of the efficacy of pyridostigmine alone and the combination of pyridostigmine with anticholinergic drugs as pharmacological pretreatment of tabun-poisoned rats and mice. Toxicology 2002; 177:179–85.