
Abstract
In this study, we examined the anti-Helicobactor pylori effects of the main protoberberine-type alkaloids in Rhizoma Coptidis. Coptisine exerted varying antibacterial and bactericidal effects against three standard H. pylori strains and eleven clinical isolates, including four drug-resistant strains, with minimum inhibitory concentrations ranging from 25 to 50 μg/mL and minimal bactericidal concentrations ranging from 37.5 to 125 μg/mL. Coptisine’s anti-H. pylori effects derived from specific inhibition of urease in vivo. In vitro, coptisine inactivated urease in a concentration-dependent manner through slow-binding inhibition and involved binding to the urease active site sulfhydryl group. Coptisine inhibition of H. pylori urease (HPU) was mixed type, while inhibition of jack bean urease was non-competitive. Importantly, coptisine also inhibited HPU by binding to its nickel metallocentre. Besides, coptisine interfered with urease maturation by inhibiting activity of prototypical urease accessory protein UreG and formation of UreG dimers and by promoting dissociation of nickel from UreG dimers. These findings demonstrate that coptisine inhibits urease activity by targeting its active site and inhibiting its maturation, thereby effectively inhibiting H. pylori. Coptisine may thus be an effective anti-H. pylori agent.
Introduction
Helicobacter pylori, a gram-negative, spiral-shaped, microaerophilic bacillus, is the major pathogen underlying multiple digestive system diseases, including gastritis, ulcers, gastric carcinoma, and related gastroduodenal disordersCitation1–3. It affects approximately 50% of the population worldwideCitation4 and is classified as a class I carcinogen by the World Health OrganizationCitation5. The first-line therapy for H. pylori eradication usually involves combination of a proton-pump inhibitor (e.g. lansoprazole) and two antibiotics (e.g. clarithromycin and metronidazole or amoxicillin). Although it is somewhat efficaciousCitation6,Citation7, there are substantial challenges associated with this regime, including poor patient compliance, antibiotic resistance, possible recurrence of infection, and the development of other gastrointestinal diseases due to disturbances of the gut microbiomeCitation8,Citation9. Identification of novel drug candidates, especially those with natural origins, is therefore a topic of ongoing researchCitation10–12.
Urease (EC 3.5.1.5; urea amidohydrolase), which is critical to the colonisation and virulence of H. pylori, is one of the most important targets for the treatment of H. pylori infectionCitation13. Urease is a nickel-dependent metalloenzyme that catalyses the hydrolysis of urea to ammonia, thus neutralising gastric acid and creating an alkaline local environment required for the survival of H. pyloriCitation14. Urease therefore plays a crucial role in the pathogenesis of gastric and peptic ulcers, gastric cancer, and gastric adenocarcinomaCitation15, and strategies that inhibit urease are essential for the treatment of H. pylori infection.
The binuclear nickel ions (Ni2+) in the active site and sulfhydryl groups situated in the mobile flap are essential for urease’s catalytic activityCitation16,Citation17. Indeed, hydroxamic acids, phosphoramidates, and urea derivatives interacting with the metallocentreCitation18–Citation20, and quinones and polyphenols binding to the sulfhydryl groups of the urease active siteCitation21,Citation22, constitute the current major structural classes of urease inhibitors. Moreover, it has been reported that GTP hydrolysis and the formation of a pre-activation complex consisting of apourease and four urease accessory proteins, UreE, UreF, UreG, and UreH (an ortholog of UreD found in other species), are required for carbamylation of Lys219 and insertion of two nickel ions into its active site, which contribute to the maturation of ureaseCitation23–25. UreG, a SIMIBI (signal recognition, MinD, and BioD) class GTPase, is recruited by the UreF–UreH complex and is responsible for coupling GTP hydrolysis and regulating nickel delivery during the process of urease activationCitation26. The biological functions of UreG are frequently regulated by dimerisationCitation27. Agents targeting the essential catalytic mechanism of urease could result in its inactivation and therefore provide an invaluable contribution to the treatment of H. pylori infectionCitation13.
Rhizoma Coptidis (the rhizoma of Coptis chinensis Franch., called Huanglian in Chinese), which is officially listed in the Chinese Pharmacopoeia, is one of the most commonly used traditional Chinese medicines for the treatment of H. pylori-related gastrointestinal diseases. Protoberberine alkaloids, such as berberine, coptisine, palmatine, epiberberine, and jatrorrhizine, which share a common core structure, are considered the major active components of Rhizoma Coptidis. Recent research on Coptidis alkaloids has focussed primarily on berberine, the most abundant alkaloid in Rhizoma Coptidis. Reports indicate that Rhizoma Coptidis extract usually has stronger anti-H. pyloriCitation28, anti-enzymeCitation29, gastroprotectiveCitation30, and anti-cancer effectsCitation31 than pure berberine, indicating that components other than berberine also play an important role in the extract’s biological activities.
Our previous studies have indeed indicated that Rhizoma Coptidis and its major active alkaloids, palmatine and epiberberine, exhibit excellent anti-H. pylori activity and/or inhibit urease, possibly by binding to the urease active site and decreasing the reactivity of active-site thiols in the vicinity of nickel ionsCitation29,Citation32,Citation33. However, berberine fails to show any inhibitory activity against ureaseCitation29. Previous reports indicate that these alkaloids might possess significantly different efficacy in their biological properties, despite their structural similarities. And the characteristic structure of alkaloids seems to be critical to their respective activitiesCitation34–37. Coptisine (structure shown in ), an active protoberberine alkaloid present in Rhizoma Coptidis, has the same isoquinoline parent structure as alkaloids (berberine, palmatine, epiberberine, and jatrorrhizine) but differs in the appended groups (namely with two methylenedioxy groups). Coptisine exhibits anti-enzymeCitation38, antimicrobialCitation35, anti-tumourCitation39, and gastroprotective effectsCitation40. However, comparisons of coptisine with other four alkaloids in their biological activities are rarely explored. In this study, we also found that coptisine had the strongest anti-H. pylori effects among the five major Rhizoma Coptidis alkaloids. As a follow-up study, we characterised the mechanisms underlying the anti-H. pylori activity of coptisine to better understand the anti-H. pylori effect of Rhizoma Coptidis alkaloids.
Figure 1. Structures of the five main active alkaloids in Rhizoma Coptidis.
Materials and methods
Chemicals and reagents
Berberine, palmatine, coptisine, jatrorrhizine, and epiberberine were obtained from Chengdu Purechem-Standard Co., LTD. (Sichuan, China); all purities were above 98%. Acetohydroxamic acid (AHA; C2H5NO2, purity: 98%), jack bean urease (JBU; type III with activity of 31.66 U/mg solid), urea, dithiothreitol (DTT), L-cysteine (L-cys), glutathione (GSH), sodium fluoride (NaF), and HEPES (Amresco >99%) were purchased from Sigma Aldrich (Steineheim, Germany). Boric acid (BA) was purchased from Aladdin Chemistry Company. Clarithromycin (CLR) and metronidazole (MET) were obtained from Toku-E, Japan. Brain-heart infusion (BHI), Mueller-Hinton agar, and Columbia agar base were obtained from OXOID, USA. Foetal bovine serum (FBS) was purchased from Gibco Rockville, MD, USA. Sheep blood was purchased from Pingrui Biotechnology, China. Other chemicals and analytical reagents were obtained from Guangzhou Chemical Reagent Factory (Guangdong, China). Coptisine stock solution was diluted in 20 mM HEPES (pH 7.5) buffer containing 0.3% dimethyl formamide, which does not affect urease activity at concentrations less than 1%Citation41.
H. pylori strains and preparation of crude H. pylori urease (HPU)
Fourteen H. pylori strains were used in this study. NCTC 11637 and NCTC 26695 strains were purchased from American Type Culture Collection. Sydney Strain-1 (SS1) was kindly provided by Prof. Richard Ferrero of Monash University, Australia. Clinical isolate ICDC 111001 was provided by Prof. Zhang Jian-zhong of the Chinese Center for Disease Control and Prevention, China. Ten clinical isolates (Hp1868, Hp1869, Hp1870, Hp1871, Hp1872, Hp1873, Hp1874, Hp1875, Hp1876, and Hp1877) were obtained from Renji Hospital, Shanghai Jiaotong University.
All strains were cultured in Columbia agar base containing 7% sheep blood at 37 °C in a micro-aerophilic environment (5% O2, 10% CO2, and 85% N2). After 3 days, H. pylori was scraped and suspended in phosphate buffered saline (PBS). Turbidimetry was employed to adjust the concentrations of different H. pylori strains (turbidity equivalent to 1.0 McFarland standard). Crude HPU was obtained according to the method described by Matsubara et al.Citation42
Minimal inhibitory concentration assay
In this study, different concentrations (12.5, 25, 50, and 100 μg/mL) of berberine, palmatine, epiberberine, jatrorrhizine, and coptisine were used to evaluate antimicrobial activity against the standard H. pylori strain NCTC 11637. The agar dilution method was then used to analyse the antibacterial effects of coptisine against two H. pylori standard strains (SS1 and NCTC 26695) and eleven clinical isolates (ICDC 111001, HP 1868, HP 1869, HP 1870, HP 1871, HP 1872, HP 1873, HP 1874, HP 1875, HP 1876, and HP 1877). In this assay, 100 μL of H. pylori suspension (McFarland = 1.0) was added to the test compound-containing or DMSO-containing (control) Columbia blood agar plate. CLR and MET were used as positive controls in this experiment. Minimum inhibitory concentration (MIC) was defined as the lowest concentration at which no strain growth was observed by visual examination after 3 days. This experiment was repeated twice.
Minimal bactericidal concentration assay
The broth dilution method was used to determine the minimal bactericidal concentrations (MBCs) of coptisine (25, 37.5, 50, 62.5, 75, 100, and 125 μg/mL) against three H. pylori standard strains (SS1, NCTC 11637, and NCTC 26695) and eleven clinical isolates (ICDC 111001, HP 1868, HP 1869, HP 1870, HP 1871 and HP 1872, HP 1873, HP 1874, HP 1875, HP 1876, and HP 1877). In brief, H. pylori suspension (McFarland = 1.0) was exposed to DMSO (control) and coptisine at 2–4 times MIC in BHI with 10% FBS. After shaking at 120 rpm in the tri-gas incubator for 6 h, 100 μL samples were removed and cultured in Columbia blood agar baseCitation43. MBC was defined as a 99.9% decrease in viability compared with the control. The experiment was repeated twice.
pH shock experiment in vivo
H. pylori cells (SS1) were harvested, and the concentration was adjusted to Mc = 4 with saline. Then, bacterial samples were mixed with AHA (positive control, 5 mM) or one of the five alkaloids from Rhizoma Coptidis (coptisine, berberine, jatrorrhizine, epiberberine, and palmatine) in 100 mM HCl (pH = 1) or PBS (pH = 7.2) at 37 °C with or without 5 mM urea. After 30 min of incubation, bacteria were collected by centrifugation at 2000g for 10 min and resuspended in the same volume of PBS. A dilution series (1:1–1:10000) was made from the samples, and the dilutions were plated on solid agar for 5 days to form single colony. Samples from urea-free wells were used as controlsCitation44. Results are expressed as CFU/mL; the experiment was repeated in triplicate. Inhibition rate was calculated using the following equation: Inhibition rate = (Urea group bacterial mean − X group bacterial mean)/Urea group bacterial mean ×100%, where X represents each experimental group.
Measurement of standard urease activity
This assay was performed using the modified Berthelot method (phenol-hypochlorite) in 96 well platesCitation45. At the beginning of each reaction, different concentrations of urease solution and 150 mM urea as a substrate were mixed in 20 mM HEPES buffer (pH 7.5) at 37 °C. After a 20-min reaction, urease activity was tested by measuring the concentration of ammonia produced by the reaction at 595 nm using the microplate reader (Thermo Fisher Multiskan FC, Instruments, Inc., USA) at ambient temperature. The activity of HPU was determined to be 17.0 U/mg by comparing it to JBU, which had a known activity of 31.66 U/mg solid. One unit (U) of urease activity was defined as the amount of urease required to catalyse 1 μmol urea per min. These experiments were repeated in triplicate.
Urease inhibition assay in vitro
Coptisine’s ability to inhibit urease activity was determined by measuring amounts of ammonia produced using the modified Berthelot method described above. The assay mixture, which contained 200 μL of enzyme and 200 μL of coptisine solution (0.16–2.51 mM) in 200 μL of 20 mM HEPES buffer (pH 7.5), was incubated for 20 min at 37 °C in 96-well plates. The reaction was then initiated by adding urea solution (150 mM) to determine residual urease activity. AHA was used as a standard urease inhibitor. Urease activity in the absence of inhibitors was used as a positive control to define 100% activity. Residual activity (RA %) for each assay was calculated using the following equation: RA % = (activity with inhibitors/activity without inhibitors) × 100%. The assay was repeated in triplicate.
Determination of urease inhibition type
Using the methods described above, the type of enzyme inhibition induced by coptisine was determined by examining changes in the Michaelis constant (KM) and maximum velocity (vmax) values from Lineweaver–Burk plots of 1/v versus 1/urea. The inhibition constant was determined using secondary plots of the apparent KM/vmax or 1/vmax versus the concentration of coptisine. The assays were performed in triplicate.
Progress curve analysis
Reaction progress curves for urea hydrolysis in the non-pre-incubated and pre-incubated systems were generated by plotting the concentration of ammonia generated as a function of the incubation time P(t) in the absence or presence of coptisine. Coptisine and urease were mixed immediately before the reaction in the non-pre-incubated system and were mixed and incubated for 20 min before urea was added to initiate the reaction in the pre-incubated system. Urease activity relative to the standard assay was determined at different time intervals.
Effects of thiol compounds on coptisine-modified urease
Different incubation times and orders of component addition were used to examine the effects of thiol-containing compounds (1.25 mM DTT, GSH, or L-cys, freshly prepared) on urease inhibited by coptisine. Briefly, mixtures of urease, thiol compounds, and coptisine (2.5 mM) were incubated at 37 °C for different amounts of time (5–30 min). Additionally, two of the reactants (urease, thiol compounds, and coptisine) were pre-incubated at 37 °C for 20 min followed by addition of the third compound and 20 additional min of incubation in separate assays. Urease activity was measured relative to the standard assay.
Effects of inorganic reagents on coptisine-modified urease
BA and NaF were used to analyse the influence of inorganic reagents on urease in the presence and absence of coptisine. Briefly, coptisine was added to a mixture containing urease and the inorganic reagent (5 mM BA or NaF) after a 20-min co-incubation at 37 °C. After another 20-min co-incubation, the mixture was added to the standard assay system and absorbance was measured at 595 nm. The assay was repeated three times.
Remobilisation of coptisine-inhibited urease
The ability of DTT (freshly prepared) to reactivate coptisine-inhibited urease was investigated in this assay. Urease was pre-incubated with coptisine until enzyme activity decreased to approximately 20% of baseline. DTT (1.25 mM) was then added and incubated with the coptisine-deactivated urease for different amounts of time. Urease activity before and after the addition of DTT was measured using the standard assay.
UV-visible spectroscopy
Binding of Ni2+ and thiols to coptisine was examined using UV-visible spectroscopy on a TU-1810PC UV-visible spectrophotometer (Purkinje, Beijing, China) at ambient temperature (∼25 °C). UV-visible spectra were scanned from 200 to 600 nm. For Ni2+ and L-cys titration experiments, coptisine solutions were prepared in a titration buffer (20 mM HEPES, pH 7.5) with or without different concentrations of nickel chloride and L-cys. The spectrum obtained from buffer without nickel chloride or L-cys was used for baseline correction.
Molecular docking study
The molecular docking study was performed as previously describedCitation46. AutoDock version 4.2 and the AutoDock Tools (ADT 1.5.6) graphic workstation were used to analyse the mode and free energy values of binding between urease and coptisine. The three-dimensional structures of JBU (PDB ID: 3LA4, resolution: 2.05 Å) and HPU (PDB ID: 1E9Y, resolution: 3.00 Å) were downloaded from the RCSB Protein Data Bank, and the energy-efficient three-dimensional shape of coptisine was examined using chem3D Ultra 8.0 software. A grid map was generated to evaluate the possible binding sites of coptisine on urease. The size of the cubic grid box was set to 60 Å (x, y, and z) with a spacing of 0.375 Å, and the midpoint of the two nickel ions was defined as the centre of the grid box. Before the initiation of the experiment, water molecules were removed from and polar hydrogen atoms were added to urease using AutoDock tools. Binding free energy was computed using the Lamarckian genetic algorithm (LGA)Citation47; coptisine–urease complexes with the lowest free energy of combination were considered the most favourable structures. Distances (Å) between hydrogen bond-forming residues were also measured.
UreG activity inhibition assay
UreG was expressed and purified as described previouslyCitation26. Briefly, the UreG expression vector was constructed by sub-cloning the ureG gene into an in-house pHisSUMO vector, which contains the coding sequence of a HisSUMO tag on a pRSETA vector (Invitrogen). UreG was expressed as a HisSUMO-tagged fusion protein using transformed Escherichia coli. HisSUMO-UreG-expressing bacterial cells were lysed via sonication in buffer B (20 mM Tris/HCl, pH 7.5, 200 mM NaCl, 1 mM TCEP, and 40 mM imidazole) and loaded onto a 5 mL HisTrap column (GE Healthcare) equilibrated with buffer B. After washing with 10 column volumes of buffer B, HisSUMO-UreG was eluted with 300 mM imidazole in buffer B. The HisSUMO tag was cleaved using small ubiquitin-like modifier protease SENP1C and separated from UreG by a second pass through the HisTrap column. UreG was further purified by size exclusion chromatography using HiLoad 26/60 Superdex 75 columns (GE Healthcare) in buffer B without imidazole. Purified UreG was dialysed again with 5 mM EDTA to remove any bound metal before further experimentation.
The assay was performed in 96-well plate. Briefly, 100 μM GTPγS (positive control) or coptisine was added to the reaction buffer containing 5 μM UreG monomer, 2.5 μM NiSO4, and 5 mM NaHCO3. 300 μM GTP was then added after 60, 48, 36, 24, or 12 min to start the reaction at 37 °C. Buffer A (150 mM KCl, 2 mM MgSO4, 100 mM Tris, pH adjusted to 7.5 with HCl) was added to bring the final reaction volume to 200 µL. At the end of the reaction, 50 μL malachite green solution was added to measure the amount of phosphate produced in the reactionCitation48, and the plate was read at 630 nm after 5 min of incubation at room temperature. Relative activity values were calculated compared to UreG group (drug-free) reactions using 100 µM coptisine or GTPγS. The IC50 value was then calculated based on GTP hydrolysis rates in the presence of coptisine at concentrations of 1.25–500 μM using KaleidaGraph 4.0.
Fluorescence emission spectra of coptisine titration with GTP
To determine whether coptisine affected GTP during coptisine-induced UreG inhibition, the interaction between GTP and coptisine was monitored using fluorescence emission spectra. Fluorescence data were collected on a SpectraMax i3x multimode plate reader (Molecular Devices, Sunnyvale, CA, USA). Coptisine (500 μM) was dissolved in buffer A (150 mM KCl, 2 mM MgSO4, and 100 mM Tris, pH 7.5) in the absence or presence of GTP at the desired concentrations (100, 200, 300, 400, or 500 μM). The solutions were incubated at 37 °C in the dark for 60 min before fluorescence measurement. The excitation wavelength was 380 nm.
UreG dimer formation inhibition assay
Size exclusion chromatography technology was used to analyse the effects of coptisine on UreG dimer formation. Experiment was carried out in the buffer containing 5 μM UreG monomer and 2.5 μM NiSO4, and divided into the following three groups: 300 μM GTP (positive control), buffer A (negative control), and 300 μM GTP +500 μM coptisine. After reaction for 60 min, samples were then injected into a Superdex 75 analytical gel filtration column pre-equilibrated with buffer A.
Nickel uptake assay
UreG dimers were obtained using GTPγS as described previouslyCitation26. Briefly, the UreG dimer was purified by incubating apo-UreG at 3 mg/mL on ice with 1 mM GTP, 2 mM MgSO4, and 0.5 mM NiSO4 for 15 min. Nickel-charged UreG dimers were then separated from excess GTP and nickel using a Sephadex G25 desalting column. UreG dimers (2.5 μM) were injected into dialysis bag (6–8 kDa) and mixed with different concentrations of coptisine (25, 50, or 100 μM) or DMSO (control). The dialysis bag was sealed and placed in beakers containing UreG dimer buffer at 37 °C in a water bath. After 30 min, dialysis bags were placed in fresh buffer and the sample was incubated for 2 h at 4 °C with continuous shaking to initiate dialysis. The dialysis process was repeated three times. Samples were then removed and nickel concentrations were measured using atomic absorption spectroscopy (AA-7000, SHIMADZU). Nickel concentration was determined by comparing measurements against a standard curve of known nickel concentrations.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 5 (GraphPad Software, Inc.) and SPSS 13.0 (SPSS, Inc.) softwares. Data were shown as means ± standard deviation (SD). Statistical differences between groups were calculated by one-way analysis of variance (ANOVA) followed by Dunnett's test. Statistical probability of 0.05 or p < 0.01 was considered significant.
Results
MIC of Rhizoma Coptidis alkaloids against H. pylori
An antibacterial study revealed that the main alkaloids in Rhizoma Coptidis inhibited growth of the standard H. pylori strain NCTC 11637 at pH 7.2 to varying degrees (). Coptisine, with an MIC of 25 μg/mL, had a two to fourfold more potent anti-H. pylori effect than the other four alkaloids tested (berberine, palmatine, epiberberine, and jatrorrhizine). In comparison, the standard antibacterial agents, CLR and MET exhibited appreciable anti-H. pylori activity, with MICs of 2 × 1.0−2 and 50 × 1.0−2 μg/mL, respectively.
Table 1. MICs of the main alkaloids in Rhizoma Coptidis and the reference drugs against H. pylori standard strain (NCTC 11637).
MICs and MBCs of coptisine against H. pylori
As shown in , MICs of coptisine against the three standard H. pylori strains SS1, NCTC 26695, and NCTC 11637 were all 25 μg/mL, while the MICs of coptisine against eleven clinical isolates obtained from three sites in China were either 25 or 50 μg/mL. Moreover, the MBCs of coptisine against the three H. pylori standard strains and eleven clinical isolates ranged from 37.5 to 125 μg/mL. An agent is considered bactericidal if the MBC is not more than four times the MICCitation49. The MBC/MIC values for coptisine ranged from 1.25 to 2.5, indicating that coptisine exhibited bactericidal effects against H. pylori. Importantly, coptisine showed potent antibacterial and bactericidal effects against isolates with CLR resistance (HP 1870 and HP 1876) and MET resistance (HP 1870, HP 1876, HP 1869, and HP 1871)Citation43, with MICs of 25, 50, 50, 25 μg/mL, and MBCs of 37.5, 75, 62.5, 37.5 μg/mL, respectively.
Table 2. MICs and MBCs of coptisine against different H. pylori strains under pH = 7.2.
Coptisine inhibits urea-dependent survival of H. pylori under acidic conditions
After the pH shock (incubation at pH 1 for 30 min), 472 ± 57 CFU/mL of H. pylori survived in the medium with 5 mM urea (), while the bacteria did not survive without the addition of urea (control). On the other hand, coptisine (50, 100, and 200 μM) potently inhibited the urea-dependent survival of H. pylori under acidic (pH = 1), but not neutral (pH = 7.2), conditions in a dose-dependent manner, with inhibition rates of 72.7%, 94.7%, and 98.5%, respectively. In comparison, AHA, a well-known urease inhibitorCitation50, resulted in 100% inhibition under acidic, but not neutral, conditions. However, the other Rhizoma Coptidis alkaloids (berberine, palmatine, epiberberine, and jatrorrhizine) failed to inhibit H. pylori after 30 min under either acidic or neutral conditions, even at concentrations of 100 μM. These results suggest that the anti-H. pylori effect of coptisine in vivo is a result of urease inhibition.
Table 3. Effect of the main alkaloids from Rhizoma Coptidis on urease activity in vivo (n = 3).
Coptisine inhibits urease activity in vitro
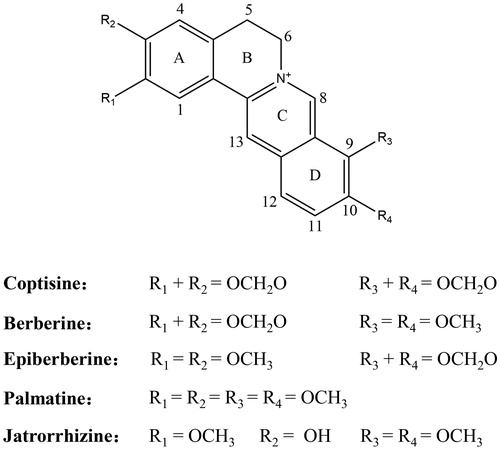
As shown in , coptisine-induced inhibition of urease was dose-dependent, with IC50 values of 1.78 ± 0.06 and 0.26 ± 0.02 mM for HPU () and JBU (), respectively. The IC50 values for AHA, the positive control, were 0.09 ± 0.01 and 0.02 ± 0.01 mM for HPU () and JBU (), respectively.
Figure 2. Coptisine (Cop)-induced inhibition of HPU (A) and JBU (B). Effects of AHA on HPU (C) and JBU (D). Results are expressed as means ± SD of three independent experiments.
Inhibition type analysis
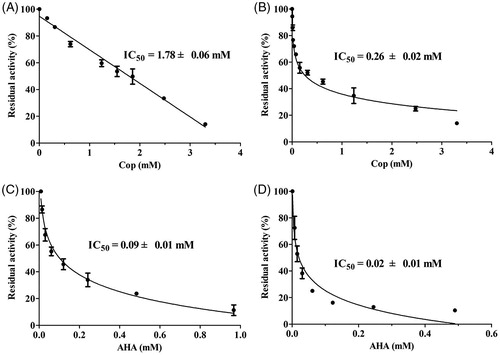
The mechanism by which coptisine inhibits urease was investigated by measuring variations in the Michaelis constant (KM) and maximum velocity (vmax) values using Lineweaver–Burk plots. Several straight lines intersected at the same point in the second quadrant, while KM increased and vmax decreased gradually after the addition of various concentrations of coptisine, suggesting that coptisine was a mixed-type inhibitor for HPU (). Furthermore, the equilibrium constants for binding of coptisine to the free enzyme (Ki) and the enzyme substrate complex (Kis) were 0.05 and 1.48 mM, respectively, and were calculated based on the slope or the vertical intercept versus the inhibitor concentration, respectively ().
Figure 3. Determination of inhibition type and inhibition constant of coptisine (Cop) on urease. Lineweaver–Burk plots of 1/v versus 1/urea in HPU (A) and JBU (B) in the absence or presence of 1.00, 2.00, or 4.00 mM Cop. (a, c) The inhibition constant Ki was determined from the plot of slope versus Cop concentration. (b, d) The inhibition constant Kis was determined from the plot of intercept versus Cop concentration.
The plot of 1/v versus 1/urea consisted of several straight lines that intersected each other at the same point on the X-axis, suggesting that the value of KM did not change much, while the vmax value gradually decreased, as the coptisine concentration increased for JBU (). This might indicate that coptisine non-competitively inhibits JBU. The inhibition constant Ki and Kis values were 0.17 and 0.25 mM, respectively ().
Reaction progress curves
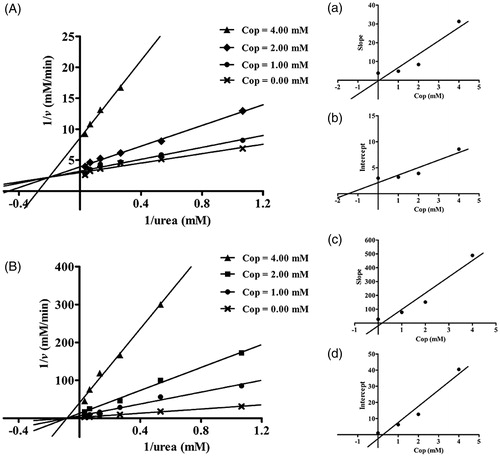
As shown in , inhibitor concentration and incubation time had obvious effects on binding rate between coptisine and HPU and JBU. Furthermore, there were no significant differences between the non-pre-incubated system and the pre-incubated system. The reaction progress curves for HPU and JBU were consistent with the slow-binding inhibition characterised in a previous reportCitation51. A curve-fitting programme was used for these data points to measure progress of the slow-binding reaction according to the equation: P(t) = vs × t + (v0 − vs) (l − e−kapp × t) kapp−1, where P(t) is the amount of accumulated product at time t, v0 and vs are the initial and steady-state reaction velocities, respectively, and kapp denotes the apparent velocity constant.
Figure 4. Reaction progress curves of urease-catalysed hydrolysis of urea in the presence or absence of coptisine (Cop). Reaction progress curves in the non-pre-incubated (A: HPU; B: JBU) and pre-incubated systems (C: HPU; D: JBU). Curves were obtained by measuring concentration of ammonia versus incubation time. The concentrations of Cop were 0, 0.63, 1.25, and 2.50 mM.
As shown in , reaction progress for coptisine-HPU binding exhibited a characteristic concave downward shape, indicating that binding involved the rapid formation of a collision complex (EI) then which underwent a slow conversion into a more stable final complex EI*. The initial velocity (v0) of urea hydrolysis decreased to a much slower steady-state velocity (vs) according to the apparent first-order velocity constant kapp. However, in the reaction system with coptisine and JBU, the linear curves demonstrated that the equilibrium among the enzyme, inhibitor and EI* complex was characterised by the steady-state velocity (vs) ().
Thiol-containing compounds protect urease against coptisine-induced inhibition
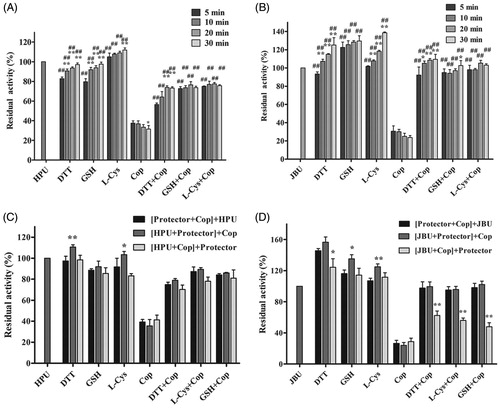
Thiol-containing compounds exert protective effects by interacting with the sulfhydryl group (–SH) of urease to restrict the ability of inhibitors to access the active siteCitation52,Citation53. We therefore performed an assay to investigate the potential effects of thiol-containing compounds (DTT, L-cys, and GSH) on coptisine-treated urease. As shown in , residual enzyme activity of coptisine-treated urease was higher in the presence of thiol-containing compounds than in the thiol-free system, suggesting that the thiol compounds had protected urease against coptisine-induced inhibition. Furthermore, this protective effect was stronger for JBU than for HPU. However, the incubation time had limited effects in this urease activity assay ( (HPU) and 5(B) (JBU)). Furthermore, while the order in which urease, inhibitor, and thiols were added did not significantly affect HPU (), residual enzyme activity was the lowest for JBU when thiols were added after coptisine (). Thus, inhibition of urease by coptisine might involve binding with the active site sulfhydryl group.
Figure 5. Effects of incubation time and order of component addition on coptisine (Cop)-modified HPU (A and C) and JBU (B and D). Urease activity was measured after incubation for 5, 10, 20, or 30 min. The compounds in brackets were pre-incubated for 20 min, and an additional 20-min incubation was performed after the last compound (outside of the brackets) was added. 1.25 mM thiol-containing compound and 2.5 mM Cop were used. 1.25 mM thiol compound without coptisine served as controls. Results are shown as means ± SD of three independent tests. One-way ANOVA revealed significant differences compared to the first column for each group and the Cop alone treatment group; *p < 0.05, **p < 0.01, and ##p < 0.01.
Effects of inorganic reagents on coptisine-modified urease
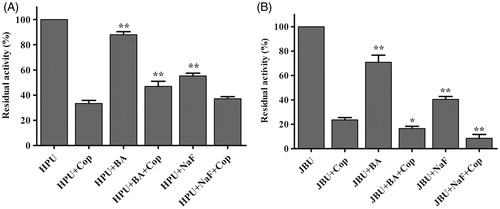
Studies have shown that the inorganic compounds BA and NaF, two well-known competitive urease inhibitors, inhibit urease activity by binding to the nickel ions (Ni2+) in ureaseCitation19, Citation54. Here, coptisine, NaF, and BA exhibited varying suppressive effects on HPU and JBU, and coptisine was the strongest urease inhibitor. Moreover, residual HPU activity was higher, whereas residual JBU activity was lower, in the mixed system containing urease, inorganic reagents, and coptisine compared to coptisine without inorganic reagents (). This result suggested that the inorganic compounds (BA and NaF) had a protective effect on HPU against coptisine inhibition, which indicated that the binding of coptisine to the active site might involve interactions with the Ni2+ ion. In contrast, a synergic effect was observed for JBU, suggesting that the inhibitory mechanism might involve blockage of its sulfhydryl group.
Figure 6. Effects of inorganic compounds on coptisine (Cop)-treated urease. 2.5 mM Cop and 5 mM inorganic compound (BA or NaF) were used. Results are expressed as means ± SD of three replicates. One-way ANOVA revealed significant differences compared with the Cop alone treatment group; *p < 0.05, **p < 0.01.
UV-visible spectroscopy
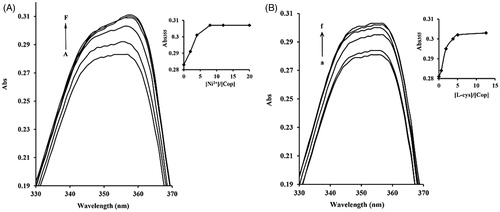
The most pivotal catalytic components of ureases are the cysteinyl residues and Ni2+ ions located in the active site. We therefore examined whether coptisine affected Ni2+ and L-cys using UV-visible spectroscopy. The maximum optical properties of bands at ∼355 nm for coptisine increased with the further addition of Ni2+ and L-cys, and levelled off at a Ni2+: coptisine molar ratio of 8.0 and a L-cys: coptisine ratio of almost 5.0, indicating that Ni2+ and L-cys differentially bind to coptisine ().
Figure 7. Ni2+ and L-cys binding to coptisine (Cop) measured using UV-visible spectroscopy. 16 μM Cop in the absence or presence of different molar equivalents of Ni2+ (1 mM NiCl2) (A) or L-cys (1 mM) (B) in 20 mM HEPES buffer, pH 7.5. A-F, 0.0 to 320 μM of Ni2+; A-F, 0.0 to 200 μM of L-cys. The inset shows the titration curves for binding of Ni2+ or L-cys to Cop at 355 nm. Wavelength (nm) is represented on the x-axis and absorption (Abs) is represented on the y-axis.
Reactivation of coptisine-inhibited urease
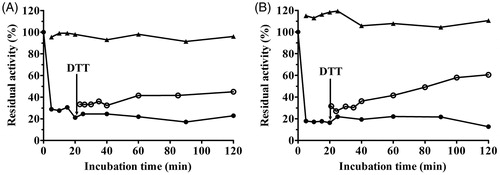
A DTT assay was performed to further explore the effect of sulfhydryl compounds on urease and the stability of the urease–coptisine complex. Administration of 1.25 mM DTT without coptisine served as a control. As shown in , activity of coptisine-inhibited urease was at least partially restored by 1.25 mM DTT, increasing from 20% of its initial activity after a 20-min incubation to nearly 45% and 60% after 120 min for HPU and JBU, respectively. This result revealed that coptisine-induced inhibition of urease was reversible. This restoration of activity confirmed the results of the urease protection experiment with thiol-containing reagents above, further indicating that the sulfhydryl groups in the urease active site are crucial to the inactivation of urease by coptisine.
Figure 8. Remobilisation of coptisine (Cop)-deactivated HPU (A) and JBU (B) by 1.25 mM DTT. Urease activity was inhibited by 3.0 mM Cop (•) and was restored by the addition of DTT (^) after 20 min. DTT (▲) without Cop served as the control.
Molecular docking study
We used an automated molecular docking simulation to investigate potential binding modes between coptisine and HPU and JBU. Mean free energy of binding values, which were calculated using the LGA, were −6.60 kcal/mol for HPU and −6.86 kcal/mol for JBU. Optimal binding modes for coptisine are shown in both cartoon form () and in enzyme surface depictions (). For HPU (), the coptisine 3-O-CH2- group formed hydrogen bond interactions with the NH group of His-221 at a distance of 2.2 Å and metal-ligand with the two nickel ions at distances of 3.2 and 3.4 Å, respectively. The benzene rings of coptisine might form hydrophobic interactions with Ala-169, Ala-365, Met-366, His-322, Cys-321, and His-323.
Figure 9. Interactions between coptisine (Cop) and HPU (A) and JBU (B) in molecular docking simulations. Ni2+ ions are represented by small yellow spheres. Surface representation of the active site flap of HPU (C) and JBU (D) in the presence of coptisine (colours represent atoms; carbon is yellow, nitrogen is blue, oxygen is red, hydrogen is grey, sulphur is orange. Yellow dashes represent hydrogen bonds between the receptor and ligand).
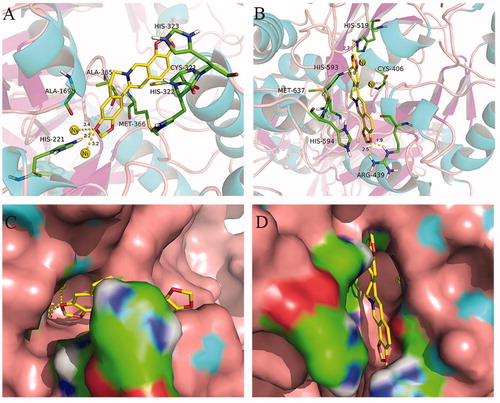
For JBU, autodocking results suggested that coptisine tightly anchored the helix-turn-helix motif over the active site cavity through N-H⋯O hydrogen bonding interactions, which prevented the mobile flap from returning to the closed position. Briefly, the coptisine 10-O-CH2- group interacted with both NH groups of Arg-439 at distances of 1.9 and 2.5 Å, respectively. The coptisine 3-O-CH2- and 2-O-CH2- groups also formed hydrogen bonding interactions with the NH groups of His-519 and His-593 at distances of 2.7 and 1.9 Å, respectively. Additionally, the isoquinoline ring of coptisine might form hydrophobic interactions with Met-637, His-594, and Cys-406 ().
Coptisine inhibits UreG activity
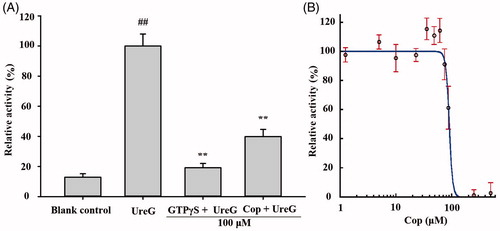
Urease maturation in H. pylori involves nickel delivery to the metallo-active centre and is highly dependent on the cooperation of at least four urease accessory proteins (UreE, UreG, UreF, and UreH). Among these, UreG plays a vital role as a nickel chaperone and GTPase enzyme during urease maturation. A previous experiment has demonstrated that nickel induces GTP-dependent dimerisation of UreG, and the UreG dimer can be hydrolysed in the presence of bicarbonate, resulting in phosphate releaseCitation26. As shown in , the UreG dimerisation reaction was inhibited by 100 μM coptisine, with an IC50 value of 89.86 ± 3.26 μM (), indicating that coptisine could inhibit the function of prototypical accessory protein UreG during urease maturation.
Figure 10. Coptisine (Cop) inhibits UreG activity. (A) 100 μM GTPγS (positive control) or Cop (100 μM) was added to reaction buffer containing 5 μM UreG monomer, 2.5 μM NiSO4, and 5 mM NaHCO3. 300 μM GTP was then added after 60, 48, 36, 24, or 12 min to start the reaction at 37 °C. The group without drug (GTPγS or Cop) was labelled UreG, while the group without UreG monomer and drug was designated the blank control. UreG activity in the absence of inhibitors was defined as 100% activity for comparison. (B) The IC50 value of coptisine was calculated using the initial GTP hydrolysis rate of Cop at concentrations ranging from 1.25 to 500 μM. Results are shown as means ± SD of triplicate tests. UreG activity in the absence of inhibitors was used to define 100% activity for comparison. ##p < 0.01 and **p < 0.01 suggested significant differences compared with blank control and UreG, respectively.
Fluorescence spectroscopy
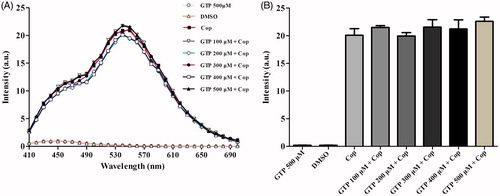
Fluorescence spectroscopy was used to investigate potential interactions between coptisine and GTP. Briefly, coptisine was incubated with different concentrations of GTP, and resulting fluorescence was measured using a fluorescence microplate. As shown in , the intensities of the coptisine fluorescence emission spectra were independent of GTP concentration, and the maximum emission peak was observed at ∼540 nm. The results suggest that coptisine has no influence on GTP.
Figure 11. Fluorescence emission spectra of coptisine (Cop) in different concentrations of GTP solution. (A) Cop (500 μM) was dissolved in buffer A (150 mM KCl, 2 mM MgSO4, and 100 mM Tris, pH 7.5) in the absence or presence of GTP at the desired concentration (100, 200, 300, 400, or 500 μM). The solutions were incubated at 37 °C for 60 min before fluorescence measurement. The excitation wavelength was 380 nm. (B) Maximum fluorescence intensity in the absence or presence of various concentrations of GTP was measured at 540 nm.
Coptisine inhibits UreG dimer formation
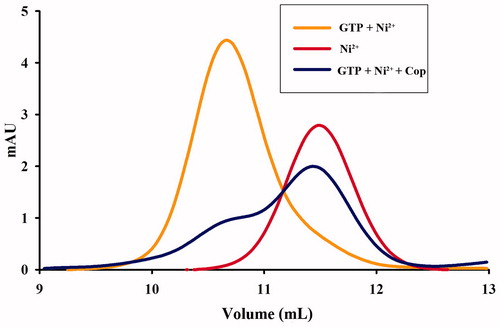
Nickel and GTP induce the formation of nickel-charged UreG dimers, which are composed of two UreG monomers and one nickel ion. We therefore examined the effect of coptisine on UreG dimer formation. As shown in , UreG dimers (GTP + Ni2+) and UreG monomers (Ni2+) were observed in two distinct peaks that differed approximately twofold in molecular weight. UreG dimerisation was obviously inhibited by treatment with 500 μM coptisine, suggesting that coptisine interferes with urease maturation by blocking UreG dimer formation.
Figure 12. Coptisine (Cop) inhibits formation of UreG dimers. UreG monomers were treated with GTP + Ni2+ (orange), Ni2+ (GTP-free, red), or GTP + Ni2+ + Cop (blue).
Coptisine promotes dissociation of nickel from UreG dimers
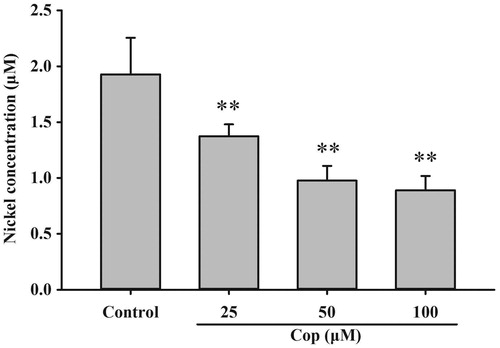
The urease active site contains two nickel ions that are necessary for catalysis. To become enzymatically active, apo-urease undergoes post-translational carboxylation of an active site lysine residue followed by the insertion of nickel ions. The UreG dimer plays a key role in the delivery of nickel ions during urease maturation. An assay was performed to examine the effect of coptisine on nickel delivery and UreG dimer function. Coptisine (25, 50, and 100 μM) markedly decreased bound nickel concentrations of UreG dimers in a dose-dependent manner during the dialysis process compared to the control (all p < 0.01). This observation indicates that coptisine promotes the dissociation of nickel from UreG dimers ().
Figure 13. Coptisine (Cop) inhibits nickel ion transfer from UreG dimers. UreG dimer (2.5 μM) was mixed with different concentrations of coptisine (25, 50, and 100 μM) and DMSO (control). Results are shown as means ± SD of triplicate tests. **p < 0.01 compared to the control group.
Discussion
Coptisine, an alkaloid found in Rhizoma Coptidis, possesses the same tetracyclic structure as alkaloids (berberine, epiberberine, palmatine, and jatrorrhizine) but contains different substituents on ring A or ring D, namely two methylenedioxy groups. However, comparisons of the biological activities of coptisine and other four alkaloids are lacking. In the present study, the inhibitory effects of coptisine on H. pylori and the potential underlying mechanisms were investigated.
MIC and MBC assays showed that coptisine exhibited varying antibacterial and bactericidal effects towards all tested H. pylori strains, including three reference strains (NCTC 26695, NCTC 11637, and SS1) and eleven clinical isolates (including four antibiotic-resistant strains). Among the five alkaloids tested, MIC values indicated that coptisine inhibited H. pylori to the greatest degree, followed by berberine, epiberberine, palmatine, and jatrorrhizine. However, another study found that berberine was the most effective at inhibiting Bacillus shigae and E. coli, followed by coptisine and palmatineCitation55,Citation56.
All five of the tested alkaloids share the same isoquinoline parent structure characterised by quaternary nitrogen and aromaticity, which might contribute to their anti-H. pylori activities. However, the specific groups located in the A (C2 and C3) and D (C9 and C10) rings, which differ among the alkaloids, seemed to play a more crucial role in their anti-H. pylori activities. Indeed, each alkaloid was crucial to their respective activities based on its characteristic structureCitation36. The biological activities of structurally related isoquinoline alkaloids of protoberberine class exhibited significant differences, such as vascular smooth muscle cell inhibitionCitation36, hydroxyl radical scavenging activitiesCitation34, antimicrobial activityCitation35, anti-tumour activityCitation37 and so on. Our finding that coptisine has the strongest anti-H. pylori effect indicates that its methylenedioxy functional groups in the A and D rings, which are more lipophilic than methoxy groups, may be crucial for enhanced H. pylori inhibition. However, further investigation of larger numbers of samples should be performed to confirm this structure-activity relationship.
Urease, which neutralises the acidic environment of the human stomach, is essential for the survival and pathogenesis of H. pylori. Previous studies have shown that coptisine inhibits the activity of various enzymes, such as elastaseCitation57, acetylcholinesteraseCitation38, CYP2D6Citation58, indoleamine 2,3-dioxygenaseCitation59, aldose reductaseCitation60, monoamine oxidaseCitation61, and bacterial collagenaseCitation62. Here, the pH shock experiment revealed that coptisine potently inhibited H. pylori under acidic, but not neutral, conditions within 30 min, suggesting that coptisine likely inhibited urease activity. In contrast, berberine, palmatine, epiberberine, and jatrorrhizine did not effectively inhibit H. pylori under either acidic or neutral conditions ().
The in vitro assay indicated that coptisine inhibited both HPU and JBU in a dose-dependent manner. We previously found that epiberberine and palmatine inhibited urease activity in vitro more effectively than coptisineCitation32,Citation33. However, H. pylori both contains internal (intracellular) and secretes external (extracellular) ureases, and internal urease in particular is vital for acid neutralisation and H. pylori growth in acidic environmentsCitation63. Epiberberine and palmatine might therefore inhibit only extracellular, and not intracellular, urease within 30 min, while coptisine might exert its antibacterial and bactericidal effects against H. pylori by inhibiting both intracellular and extracellular urease in vivo and in vitro. It is possible that the more hydrophobic groups present in the isoquinoline structure of coptisine facilitated its diffusion across H. pylori cell membranes, allowing it to inhibit intracellular urease. Additional studies are needed to investigate the mechanisms that might underlie this process.
Urease was first crystalised from jack beans (Canavalia ensiformis, JBU); because JBU is the best-characterised urease, it frequently serves as a model urease in studies of inhibitory mechanisms. We therefore compared the inhibitory effects of coptisine on HBU and JBU. Our kinetic study using the Lineweaver–Burk plot revealed that coptisine administration resulted in mixed-type inhibition of HPU, which agreed with previous studies of coptisine-induced inhibition patterns in bacterial collagenase from Clostridium histolyticum and in porcine pancreatic elastaseCitation57,Citation62. In contrast, coptisine non-competitively inhibited JBU, which was also the case in our earlier reports on Rhizoma Coptidis extract and the alkaloids epiberberine and palmatineCitation29,Citation32,Citation33. These kinetic mechanism differences might arise from structural differences between HPU and JBU. However, further in-depth investigation of these differences is needed.
Urease is a thiol-rich, nickel-dependent metalloenzyme. Ni2+ ions and –SH groups, especially the multiple cysteinyl residues located on the mobile flap that closes over the active site, are essential for the catalytic activity of all ureasesCitation64. Our results demonstrated that the sulfhydryl reagents DTT, GSH, and L-cys protected against coptisine-induced inhibition of HPU and JBU, and DTT restored urease activity, indicating that coptisine binds to the active site sulfhydryl group to inhibit urease activity. These observations agreed with our previous findings that non-competitive binding and blockage of JBU sulfhydryl groups by Rhizoma Coptidis extract, epiberberine, and palmatine contributed to urease inhibitionCitation29,Citation32,Citation33. Interestingly, the typical competitive urease inhibitors BA and NaF protected against coptisine-induced inhibition of HPU, but had a synergistic effect on coptisine-induced inhibition of JBU. This indicates that the active site Ni2+ may be essential for coptisine-induced inhibition of HPU, which was supported by the kinetic analysis, molecular docking simulation, and spectrophotometric studies.
Molecular docking analysis revealed that coptisine formed a hydrogen bond interaction with the NH group of His-221 and a metal-ligand with the active site Ni2+ in HPU. In addition, the benzene ring of coptisine might form hydrophobic interactions with Ala-169, Ala-365, Met-366, His-322, Cys-321, and His-323 of HPU (). However, coptisine only formed hydrogen bond or hydrophobic interactions with amino acid residues Arg-439, His-519, His-593 Met-637, His-594, and Cys-406 of JBU (). Previous studies have shown that these amino acid residues, which are located on the mobile flap that opens and closes during the urease catalytic cycle, are responsible for hydrogen bondingCitation65–67. Coptisine tightly anchored the helix-turn-helix motif over the active site cavity through N-H⋯O hydrogen bonding interactions, which prevented the mobile flap from returning to the closed position, thereby inhibiting urease activity. These observations together with the protective effects of active site binding inhibitors in the reactivation assay strongly support the involvement of the active site Ni2+ and sulfhydryl groups in coptisine-induced inhibition of HPU while sulfhydryl groups of JBU. The ultraviolet absorption assay revealed that coptisine chelated Ni2+ and bound to sulfhydryl groups, which was congruent with a previous study indicating that coptisine alters enzyme activity at least in part by chelating ferrous metal ionsCitation34. Taken together, our results suggest that coptisine inhibits urease not only by binding to active site sulfhydryl groups, but also via a specific interaction with the Ni2+ ion.
Because the catalytic activity of urease requires the presence of Ni2+ ions in the active site, H. pylori requires efficient acquisition of Ni2+ ions to surviveCitation68,Citation69. In vivo, urease is synthesised as an inactive apoenzyme, and maturation of urease involves delivery of nickel to the metallo-active centre, which is highly dependent on the cooperation of at least four accessory proteins (UreE, UreG, UreF, and UreH)Citation23. UreF and UreH form a complex that recruits UreG to the pre-activation complex. Formation of the UreG/UreF/UreH complex is a critical step in urease maturation and formation of the nickel-charged UreG dimer, which reverts to its monomeric form and releases nickel into the urease active site upon GTP hydrolysisCitation26. The UreG protein contains a highly conserved nickel-binding motif that is essential for nickel delivery and is required to complete biosynthesis of nickel enzymesCitation25,Citation70. UreG couples GTP hydrolysis to urease activation, and it is thought to catalyse the formation of carboxyphosphate, which results in carbamylation of the metal-binding lysine in the urease active siteCitation15,Citation24. In this study, coptisine inhibited activity of the prototypical urease accessory protein UreG () and suppressed nickel-charged UreG dimer formation (), without influence on GTP (). Furthermore, coptisine dissociated nickel from the UreG dimer (), which might be associated with its ability to chelate nickel as observed in the spectrophotometric study. Together, these results suggest that coptisine interferes with the assembly of the pre-activation complex and delivery of Ni2+ ions during HPU maturation.
Previous reports have suggested that coptisine promotes gastric mucosal protection more effectively than cimetidine or sucralfateCitation40,Citation71. Acute and sub-chronic toxicity assays indicate that coptisine has a more favourable safety profile than its congener berberine, an effective agent commonly used for the treatment of gastrointestinal disorders in ChinaCitation72,Citation73. At present, the first-line monotherapies for H. pylori infection typically have either direct anti-H. pylori effects (e.g. antibiotics) or gastroprotective properties (e.g. proton-pump inhibitor). However, none of the standard therapies demonstrate anti-H. pylori effects together with urease inhibition and anti-ulcer activity. Because of its anti-H. pylori, anti-HPU, and gastroprotective properties, coptisine may be a safe and effective therapeutic agent for the treatment of H. pylori infection.
Conclusion
In this study, we found that coptisine effectively inhibited H. pylori growth and urease activity at least in part by interacting with the nickel metallocentre and sulfhydryl groups of the urease active site. Coptisine also inhibited assembly of the pre-activation complex during HPU maturation by interacting with both the prototypical urease accessory protein UreG and nickel ions, potentially contributing to its anti-H. pylori effect. These findings suggest that coptisine might be a promising novel candidate H. pylori and urease inhibitor, and that the traditional Chinese medicine Rhizoma Coptidis might be effective for treating H. pylori-related gastrointestinal diseases.
Acknowledgements
We appreciate the vital help of Prof. Zhang Jian-zhong of the Chinese Center for Disease Control and Prevention (China) and Prof. Richard Ferrero of Monash University (Australia).
Disclosure statement
The authors declare that there are no conflicts of interest.
Additional information
Funding
Related Research Data
References
- Atherton JC. The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu Rev Pathol 2006;1:63–96.
- Franco AT, Johnston E, Krishna U, et al. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res 2008;68:379–87.
- Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984;1:1311–15.
- Cid TP, Fernández MC, Martinez SB, et al. Pathogenesis of Helicobacter pylori Infection. Helicobacter 2013;18:12–7.
- Listed N. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monogr Eval Carcinog Risks Hum 1994;61:1–241.
- Glupczynski Y, Burette A. Drug therapy for Helicobacter pylori infection: problems and pitfalls. Am J Gastroenterol 1990;85:1545–51.
- Marshall BJ. Treatment strategies for Helicobacter pylori infection. Gastroenterol Clin North Am 1993;22:183–98.
- Adamek RJ, Suerbaum S, Pfaffenbach B, et al. Primary and acquired Helicobacter pylori resistance to clarithromycin, metronidazole, and amoxicillin-influence on treatment outcome. Am J Gastroenterol 1998;93:386–9.
- Ng KM, Ferreyra JA, Higginbottom SK, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 2013;502:96–9.
- Babincova M, Schronerova K, Sourivong P. Antiulcer activity of water extract of Scoparia dulcis. Fitoterapia 2008;79:587–8.
- Falcao HS, Mariath IR, Diniz MF, et al. Plants of the American continent with antiulcer activity. Phytomedicine 2008;15:132–46.
- Shaikh RU, Dawane AA, Pawar RP, et al. Inhibition of Helicobacter pylori and its associate urease by labdane diterpenoids isolated from Andrographis paniculata. Phytother Res 2016;30:412–17.
- Follmer C. Ureases as a target for the treatment of gastric and urinary infections. J Clin Pathol 2010;63:424–30.
- Zeer-Wanklyn CJ, Zamble DB. Microbial nickel: cellular uptake and delivery to enzyme centers. Curr Opin Chem Biol 2017;37:80–8.
- Zambelli B, Musiani F, Benini S, et al. Chemistry of Ni2+ in urease: sensing, trafficking, and catalysis. Acc Chem Res 2011;44:520–30.
- Krajewska B, Zaborska W. Jack bean urease: the effect of active-site binding inhibitors on the reactivity of enzyme thiol groups. Bioorg Chem 2007;35:355–65.
- Kumar S, Kayastha AM. Soybean (Glycine max) urease: significance of sulfhydryl groups in urea catalysis. Plant Physiol Bioch 2010;48:746–50.
- Benini S, Rypniewski WR, Wilson KS, et al. The complex of Bacillus pasteurii urease with acetohydroxamate anion from X-ray data at 1.55 A resolution. J Biol Inorg Chem 2000;5:110–18.
- Dixon NE, Blakeley RL, Zerner B. Jack bean urease (EC 3.5.1.5). III. The involvement of active-site nickel ion in inhibition by beta-mercaptoethanol, phosphoramidate, and fluoride. Can J Biochem 1980;58:481–8.
- Bano B, Kanwal, Khan KM, et al. Synthesis, in vitro urease inhibitory activity, and molecular docking studies of thiourea and urea derivatives. Bioorg Chem 2018;80:129–44.
- Zaborska W, Krajewska B, Kot M, et al. Quinone-induced inhibition of urease: elucidation of its mechanisms by probing thiol groups of the enzyme. Bioorg Chem 2007;35:233–42.
- Wu DW, Yu XD, Xie JH, et al. Inactivation of jack bean urease by scutellarin: elucidation of inhibitory efficacy, kinetics and mechanism. Fitoterapia 2013;91:60–7.
- Carter EL, Flugga N, Boer JL, et al. Interplay of metal ions and urease. Metallomics 2009;1:207–21.
- Soriano A, Hausinger RP. GTP-dependent activation of urease apoprotein in complex with the UreD, UreF, and UreG accessory proteins. Proc Natl Acad Sci USA 1999;96:11140–4.
- Yang XM, Li HY, Lai TP, et al. UreE-UreG complex facilitates nickel transfer and preactivates GTPase of UreG in Helicobacter pylori. J Biol Chem 2015;290:12474–85.
- Fong YH, Wong HC, Yuen MH, et al. Structure of UreG/UreF/UreH complex reveals how urease accessory proteins facilitate maturation of Helicobacter pylori urease. PLoS Biol 2013;11:e1001678.
- Gasper R, Meyer S, Gotthardt K, et al. It takes two to tango: regulation of G proteins by dimerization. Nat Rev Mol Cell Biol 2009;10:423–9.
- Ma F, Chen Y, Li J, et al. Screening test for anti-Helicobacter pylori activity of traditional Chinese herbal medicines. World J Gastroenterol 2010;16:5629–34.
- Li CL, Xie JH, Chen XY, et al. Comparison of Helicobacter pylori urease inhibition by Rhizoma Coptidis, cortex phellodendri and berberine: mechanisms of interaction with the sulfhydryl group. Planta Med 2016;82:305–11.
- Li B, Liu HR, Pan YQ, et al. Protective effects of total alkaloids from rhizoma Coptis chinensis on alcohol-induced gastric lesion in rats. China J Chin Mater Med 2006;31:51–4.
- Iizuka N, Oka M, Yamamoto K, et al. Identification of common or distinct genes related to antitumor activities of a medicinal herb and its major component by oligonucleotide microarray. Int J Cancer 2003;107:666–72.
- Tan LH, Li CL, Chen HB, et al. Epiberberine, a natural protoberberine alkaloid, inhibits urease of Helicobacter pylori and jack bean: susceptibility and mechanism. Eur J Pharm Sci 2017;110:77–86.
- Zhou JT, Li CL, Tan LH, et al. Inhibition of Helicobacter pylori and its associated urease by palmatine: investigation on the potential mechanism. PloS One 2017;12:e0168944.
- Jang MH, Kim HY, Kang KS, et al. Hydroxyl radical scavenging activities of isoquinoline alkaloids isolated from Coptis chinensis. Arch Pharm Res 2009;32:341–5.
- Yan D, Wei L, Xiao XH, et al. Microcalorimetric investigation of effect of berberine alkaloids from Coptis chinensis Franch on intestinal diagnostic flora growth. Sci Bull 2009; 54:369–73.
- Tanabe H, Suzuki H, Nagatsu A, et al. Selective inhibition of vascular smooth muscle cell proliferation by coptisine isolated from Coptis rhizoma, one of the crude drugs composing Kampo medicines Unsei-in. Phytomedicine 2006;13:334–42.
- Shin JS, Kim EI, Kai M, et al. Inhibition of dopamine biosynthesis by protoberberine alkaloids in PC12 cells. Neurochem Res 2000;25:363–8.
- Zhao HQ, Zhou SD, Zhang MM, et al. An in vitro AchE inhibition assay combined with UF-HPLC-ESI-Q-TOF/MS approach for screening and characterizing of AchE inhibitors from roots of Coptis chinensis Franch. J Pharm Biomed Anal 2016;120:235–40.
- Huang T, Xiao YB, Yi L, et al. Coptisine from Rhizoma Coptidis suppresses HCT-116 cells-related tumor growth in vitro and in vivo. Sci Rep 2017;7:38524.
- Hirano H, Osawa E, Yamaoka Y, et al. Gastric-mucous membrane protection activity of coptisine derivatives. Biol Pharm Bull 2001;24:1277–81.
- Tan LR, Inhibition efficacy of baicalin on jack bean urease [master's thesis]. Guangzhou: Guangzhou University of Chinese Medicine; 2013.
- Matsubara S, Shibata H, Ishikawa F, et al. Suppression of Helicobacter pylori-induced gastritis by green tea extract in Mongolian gerbils. Biochem Bioph Res 2003;310:715–9.
- Xu YF, Lian DW, Chen YQ, et al. In vitro and in vivo antibacterial activities of patchouli alcohol, a natural-occurring tricyclic sesquiterpene against Helicobacter pylori infection. Antimicrob Agents Chemother 2017;61:e00122–17.
- Mooney C, Munster DJ, Bagshaw PF, et al. Helicobacter pylori acid resistance. Lancet 1990;335:1232.
- Weatherburn M. Phenol-hypochlorite reaction for determination of ammonia. Anal Chem 1967; 39:971–4.
- Jadhav SG, Meshram RJ, Gond DS, et al. Inhibition of growth of Helicobacter pylori and its urease by coumarin derivatives: molecular docking analysis. J Pharm Res 2013;7:705–11.
- Morris GMGD, Halliday RS, Huey R, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998;19:1639–62.
- Baykov AA, Evtushenko OA, Avaeva SM. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal Biochem 1988;171:266–70.
- French GL. Bactericidal agents in the treatment of MRSA infections—the potential role of daptomycin. J Antimicrob Chemother 2006;58:1107–17.
- Dixon NE, Hinds JA, Fihelly AK, et al. Jack bean urease (EC 3.5.1.5). IV. The molecular size and the mechanism of inhibition by hydroxamic acids. Spectrophotometric titration of enzymes with reversible inhibitors. Can J Biochem 1980;58:1323–34.
- Breitenbach JM, Hausinger RP. Proteus mirabilis urease. Partial purification and inhibition by boric acid and boronic acids. Biochem J 1988;250:917–20.
- Mo ZZ, Wang XF, Zhang X, et al. Andrographolide sodium bisulphite-induced inactivation of urease: inhibitory potency, kinetics and mechanism. BMC Complement Altern Med 2015;15:238.
- Krajewska B. Hydrogen peroxide-induced inactivation of urease: mechanism, kinetics and inhibitory potency. J Mol Catal B Enzym 2011;68:262–9.
- Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol 1988;61:201–301.
- Yan D, Jin C, Xiao XH, et al. Antimicrobial properties of berberines alkaloids in Coptis chinensis Franch by microcalorimetry. J Biochem Biophys Methods 2008;70:845–9.
- Yan D, Jin C, Xiao XH, et al. Investigation of the effect of berberines alkaloids in Coptis chinensis Franch on Bacillus shigae growth by microcalorimetry. Sci China Chem 2007;50:638–42.
- Tanaka T, Metori K, Mineo S, et al. Inhibitory effects of berberine-type alkaloids on elastase. Planta Med 1993;59:200–2.
- Qu Q, Qu J, Han L, et al. Inhibitory effects of phytochemicals on metabolic capabilities of CYP2D6(*)1 and CYP2D6(*)10 using cell-based models in vitro. Acta Pharmacol Sin 2014;35:685–96.
- Yu D, Tao BB, Yang YY, et al. The IDO inhibitor coptisine ameliorates cognitive impairment in a mouse model of Alzheimer’s disease. J Alzheimers Dis 2015;43:291–302.
- Jung HA, Yoon NY, Bae HJ, et al. Inhibitory activities of the alkaloids from Coptidis Rhizoma against aldose reductase. Arch Pharm Res 2008;31:1405–12.
- Ro JS, Lee SS, Lee KS, et al. Inhibition of type A monoamine oxidase by coptisine in mouse brain. Life Sci 2001;70:639–45.
- Tanaka T, Metori K, Mineo S, et al. Studies on collagenase inhibitors. IV. Inhibitors of bacterial collagenase in Coptidis rhizoma. Yakugaku Zasshi 1991;111:538–41.
- Scott DR, Weeks D, Hong C, et al. The role of internal urease in acid resistance of Helicobacter pylori. Gastroenterology 1998;114:58–70.
- Ha NC, Oh ST, Sung JY, et al. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat Struct Biol 2001;8:505–9.
- Diaz-Sanchez AG, Alvarez-Parrilla E, Martinez-Martinez A, et al. Inhibition of urease by disulfiram, an FDA-approved thiol reagent used in humans. Molecules 2016;21:E1628.
- Mao WJ, Lv PC, Shi L, et al. Synthesis, molecular docking and biological evaluation of metronidazole derivatives as potent Helicobacter pylori urease inhibitors. Bioorg Med Chem 2009;17:7531–6.
- Xiao ZP, Peng ZY, Dong JJ, et al. Synthesis, molecular docking and kinetic properties of beta-hydroxy-beta-phenylpropionyl-hydroxamic acids as Helicobacter pylori urease inhibitors. Eur J Med Chem 2013;68:212–21.
- Xia W, Li H, Sze KH, et al. Structure of a nickel chaperone, HypA, from Helicobacter pylori reveals two distinct metal binding sites. J Am Chem Soc 2009;131:10031–40.
- Yang XM, Li HY, Cheng TF, et al. Nickel translocation between metallochaperones HypA and UreE in Helicobacter pylori. Metallomics 2014;6:1731–6.
- Leipe DD, Wolf YI, Koonin EV, et al. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol 2002;317:41–72.
- Hirano H, Tokuhira T, Yokoi T, et al. Gastric mucous membrane-protective principles of Coptidis Rhizoma. Nat Med 1997;51:516–18.
- He K, Ye XL, WU H, et al. The safety and anti-hypercholesterolemic effect of coptisine in Syrian golden hamsters. Lipids 2015;50:185–94.
- Yi J, Ye XL, Wang DZ, et al. Safety evaluation of main alkaloids from Rhizoma Coptidis. J Ethnopharmacol 2013;145:303–10.