
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In this work, design, synthesis, and screening of thiophene carboxamides 4–13 and 16–23 as dual vascular endothelial growth factor receptors (VEGFRs) and mitotic inhibitors was reported. All compounds were screened against two gastrointestinal solid cancer cells, HepG-2 and HCT-116 cell lines. The most active cytotoxic derivatives 5 and 21 displayed 2.3- and 1.7-fold higher cytotoxicity than Sorafenib against HepG-2 cells. Cell cycle and apoptosis analyses for compounds 5 and 21 showed cells accumulation in the sub-G1 phase, and cell cycle arrest at G2/M phase. The apoptotic inducing activities of compounds 5 and 21were correlated to the elevation of p53, increase in Bax/Bcl-2 ratio, and increase in caspase-3/7.Compounds 5 and 21 showed potent inhibition againstVEGFR-2 (IC50 = 0.59 and 1.29 μM) and β-tubulin polymerization (73% and 86% inhibition at their IC50 values).Molecular docking was performed with VEGFR-2 and tubulin binding sites to explain the displayed inhibitory activities.
Graphical Abstract
1. Introduction
Adenocarcinoma as hepatocellular carcinoma (HCC) and colorectal cancer are highly vascularized solid tumours characterized by angiogenesis signaling pathway abnormalitiesCitation1,Citation2. Vascular endothelial growth factor receptors (VEGFRs) are receptor tyrosine kinases (RTKs) that are implicated in angiogenesis. Reduction of angiogenesis via inhibition of the pro-angiogenic enzymes as VEGFR-2, have become the focus of a new wave of targeted chemotherapy for adenocarcinomaCitation3.
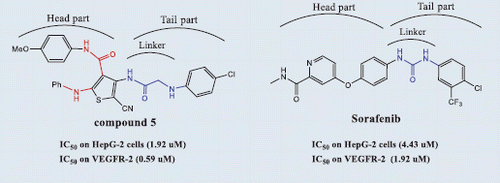
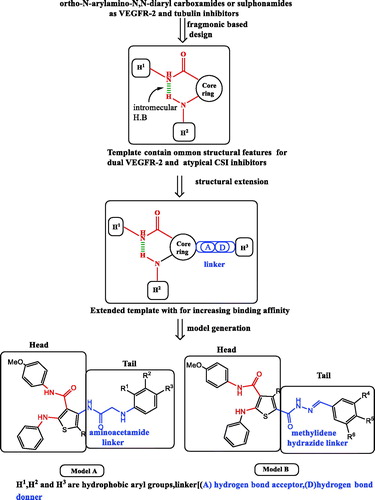
Despite of promising clinical outcomes of the angiokinase inhibitors as Sorafenib (Nexavar)™ and Rogerafenib (Stivarga)™ () in HCC and colon cancer chemotherapyCitation4, both tumours are still considered among the leading causes of mortality worldwideCitation1.,Citation2.,Citation4–6. Generally, RTK inhibitors are either ATP competitive or noncompetitive inhibitors according to the binding mode to kinase domainCitation7.,Citation8. ATP competitive inhibitors bind to ATP enzyme binding pocket, while noncompetitive inhibitors bind to ATP binding pocket, in addition to the allosteric binding site adjacent to the ATP pocketCitation9. Competitive inhibitors are structurally composed of orthogonal biaryl ring system with pharmacophoric featuresCitation10 including hydrogen bond (H–bond) acceptor–donor pair forming groups (A and D) that interact with the kinase “hinge” region, in addition to two hydrophobic groups (H1 and H2) around the region occupied by the ATP adenine ring. Vatalanib () is an example of competitive RTK inhibitors regarded as first generation VEGFR inhibitorsCitation11. that lack VEGFR selectivity specifically VEGFR-2 such accounts to pronounced side effectsCitation12. Noncompetitive inhibitors as Sorafenib represent the second generation of RTK inhibitors, are composed of two parts: (i) head part with similar structural features of ATP binding competitors and is responsible for potency and binding to the ATP binding siteCitation13. and (ii) tail part which binds to the allosteric site via H–bond forming linkers (amide or urea moieties) having a H–bond acceptor donor pair, and links the head part to another aryl group forming hydrophobic interactions (H3)Citation9,Citation13. Structural biology investigation performed on the complexes between vatalanib and the catalytic domain of the enzyme led to the discovery of AAL-993 (), that belongs structurally to the ortho-N-aryl-amino-N,N-diaryl carboxamidesCitation14. Several novel VEGFR kinase inhibitors () were identified as anti-cancer agents with close structural similarities to the aforementioned drug possessing different heterocyclic cores, such as Motesanib (ortho-N-aryl-2-amino-N′-arylpyridine-3-carboxamide)Citation15, OSI-930 (ortho-N-aryl-3-amnio-N′-arylthiophene-2-carboxamide)Citation16,Citation17. XL-844 (ortho-3-uriedothiophene-2-carboxamide) which was reported as a synthetic small-molecule with dual checkpoint and angiokinase inhibition activitiesCitation18, and CP-547,632 (ortho-5-ureidoisothiazole-4-carboxamide)Citation18. Discovery of the previous drugs proceeded a novel wave of selective VEGFR-2 kinase inhibitors as anti-angiogenic agents with low affinity against other RTKs, which are structurally based upon either ortho-N-arylamino-N,N′-diarylamide or ortho-ureido-N,N′-diarylamide pharmacophoric moieties. Earlier, the prototype RTK inhibitor (N-aryl-3-thienyl urea I)Citation19. () was reported as B-Raf kinase inhibitor and provided a route to the discovery of sorafenib. Unfortunately, although second generation VEGFR-2 inhibitors have the advantage of being selective enzyme inhibitors towards VEGF pathway other than malignant angiogenesis pathways, they face a major therapy problems as tumour cells can tolerate the drug induced hypoxia and hence acquire resistance to apoptosis inductionCitation20.,Citation21. Consequently, these drugs are used in high doses in advanced treatmentCitation22. causing severe drawbacks on patients’ lives quality. Thus, second generation VEGFR inhibitors are usually used in combination with other drugs targeting more than growth signaling pathways in order to promote apoptosisCitation22. A drawback of combination therapy is the increased risk of side effects of both drugsCitation22. As an alternative therapeutic approach, novel small molecule compounds with dual targets could successfully lead to the desired cytotoxic effect with minimal side effects leading to a better life quality for cancer patientsCitation23. Interestingly, combination chemotherapy of anti-angiogenesis agents with antimitotic drugs increased cytotoxicity to tumour cells without adding toxicity to normal cells, which led to reduced side effects associated with the chemotherapyCitation24. To discover pharmacophoric moieties that could be added to the core structure of anti-angiogenic agents, we studied colchicine binding site inhibitors, as the most commonly explored target for antimitotic drugs which are considered as one of the most successful targets for cancer chemotherapyCitation25.,Citation26. inhibiting cell division via suppression of microtubules growth, prevention of tubulin polymerization, blocking G2/M phase of the cell cycle and promoting apoptosisCitation27. Various drugs () were reported as potent antimitotic agents with common structural similarities such as ABT-751 (ortho-N-arylamino-N′-aryl-benzenesulphonamide)Citation28. a drug in phase II clinical trials that binds to the colchicine binding site for treatment of lung and prostate cancer, CA224 (N,N-alkylhetroaryl carboxamide)Citation29, Indibulin (N,N’-diarylglyoxyl-amide)Citation29. which was reported for treatment of solid tumours, and the natural product Tubulysin ACitation30. Colchicine binding site inhibitors (CSIs) are categorized into typical CSIs, as colchicines (), and atypical CSIs as EBT-751. The essential pharmacophoric features of typical CSIs are: biaryl ring system, methoxyphenyl fragment, and a restricted conformation moiety while atypical CSIs lacks at least one of these featuresCitation31.There are seven pharmacophoric points in colchicine binding site: three H–bond acceptors, one H–bond donor, two hydrophobic centers (H1, H2) and one planar group. The two hydrophobic and planar groups are responsible for the molecular skeleton and structural rigidity, while H–bond acceptor donor pair groups increase receptor binding interactionsCitation32. Surprisingly, although colchicine is a fused polycyclic compound, colchicine binding site is a folded architecture where, four points are in one plane, and three points are in a second plane. For this reason, complete planar anthracene and phenanthrene frameworks -analogs of colchicine- are inactiveCitation33. Hence, the design of the non-fused polycyclic flexible molecule is advantageous over fused planar polycyclic molecules. Since none of the known CSI structural models including colchicines occupy the seven pharmacophoric pointsCitation31, the design of novel hit molecules with increased binding affinity towards colchicine binding site is an interesting target in the field of medicinal chemistry research. Based on the history of drug discovery, we found that some drugs having common functional fragments often exhibit extended biological activitiesCitation34. Inspecting the structure of the mentioned antimitotic agents showed that they have similar constitutional characters to the mentioned VEGFR inhibitors where the N,N-diaryl carboxamide and ortho-amino-N,N′-diaryl sulfonamide are isosters to each other (). The biological activities of these drugs may be explained based upon their structure as it can form intramolecular H–bond between the ortho-disubstituted N-aryl amino-N-aryl carboxamide groups on the core ring. This pseudo fused ring system could simulate the adenine ring of ATP and the polycyclic ring skeleton of colchicine in interaction with binding sites. The observed structural similarities between VEGFR and tubulin polymerization inhibitors have stimulated our efforts to use the fragment-based lead generation approachCitation35. to design and synthesize dual VEGFR and β-tubulin polymerization inhibitors towards the road for discovery of potent third generation VEGFR inhibitors. Fragment-based lead generation approach is one of the leading methods in drug design and applied by identification of biological active fragments in drugs used in the market in order to generate novel lead structuresCitation35. Moreover, multi-targeted enzyme inhibitors are one of the hot topics in cancer drug discovery aiming to increase cytotoxicity by synergistic effect towards tumour cells and also to decrease cancer cells resistance to apoptosis inductionCitation36.,Citation37.. Combined with our previous studies on HCC and dual cytotoxic agentsCitation38.,Citation39., we attempted to combine anti-angiogenic and tubulin inhibition activities into several derivatives by using a common pharmacophoric moiety (ortho-N-arylamino-N,N-diaryl carboxamide) as a template to act as a dual angiokinase and tubulin polymerization inhibitor. To increase the binding of the novel molecules to both biological targets, the structure of the template is enriched with additional kinase and colchicine site interacting moieties. Structural extension of the common template resulted in two models (A and B), both of them have head and tail parts (). The head part (ortho-amino-N,N-diaryl carboxamide) is a common pharmacophore present to fulfill the basic requirement of VEGFR and colchicine binding site inhibitors. The tail part in both models is added to increase binding affinity to kinase and tubulin polymerization binding sites, and is formed from linker groups attached to aryl rings. The structure of tail parts has pharmacophoric similarities with ligands that bind to VEGFR and colchicine binding sites deduced from the known drugs Sorafenib and seven pharmacophoric features of CSI inhibitors. For instance, the 2-amino-acetamide group in model A is similar to the ureido group of sorafenib, in addition to the presence of methoxylated aryl ring and rigid configuration in model B to reach seven pharmacophoric points. Also, linkers used in both models differ in rigidity, where 2-amino-acetamide moiety is a flexible linker in model A, while methylidene hydrazide fragment is a restricted linker in model B. On the other hand, they have some similar structural features, such as the distance between head part and aryl groups in tail part and the capability of H–bond formation (acceptor–donor pair) that increase the interaction with binding sites. In Model A (N,N′-diaryl-2-amino-acetamide thiophenes 4–13), the isosteric 2-amino-acetamide group increases the probability to anchor colchicine and kinase binding sites by H-bond formation. In Model B (N-arylidene thiophene hydrazides 16–23), methylidene hydrazide constitute a rigid link to possess the model a restricted conformation property as that present in colchicine with the similar length and H–bond formation characters present in model A. The rigid configuration will help the model to possess the correct orientation to fit in both binding sites. Moreover, the aryl groups of the tail part were endowed with different electron withdrawing (halogens) or donating (hydroxyl or methoxy) groups to investigate the effect of substituents on the binding (). It is worthy to note that unsubstituted thiophene derivatives are metabolically unstable and toxic due to formation of irreversible bond interaction with cytochrome p450, therefore the head part in both models contain oxidation protecting groups (nitrile or carbamyl) as electron withdrawing substituents to increase the stability of thiophene ring against metabolic oxidationCitation40.,Citation41..
Figure 1. Chemical structures of the lead receptor tyrosine kinases (RTKs) inhibitors. ortho-amino-aryl carboxamide and ortho-uriedoaryl caboxamide pharmacophores showed in red color, urea pharmacophore as linker showed in blue color.
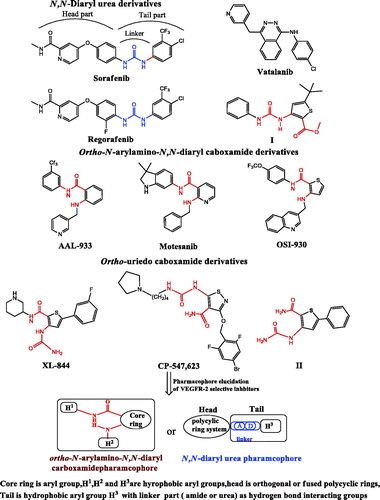
Figure 2. Chemical structures of some anti-mitotic drugs via tubulin polymerization inhibition. ortho-amino-aryl sulphoamide and diaryl caboxamide pharmacophores showed in red color.
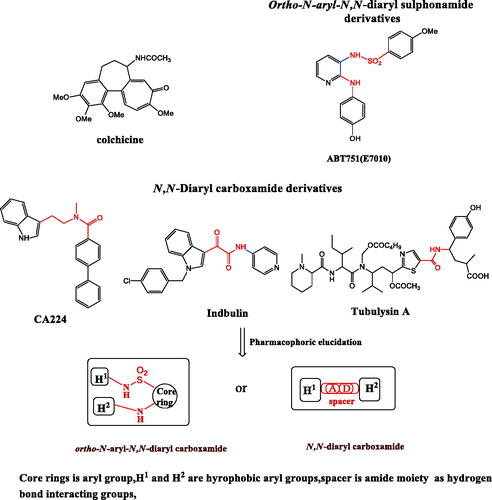
Figure 3. Diagrammatic sketch for molecular design strategy for elucidation of dual pharmacophoric template and structural extension of the template with the generation of the two models; Model A, substituted phenyl acetamides 4–13 and Model B substituted arylidene hydrazides 16–23. ortho-amino-aryl caboxamide pharmacophore showed in red color,isosteric groups(2-amino-acetamide and methylidene hydrazide) to urea pharmacophore as linker showed in blue color.
2. Experimental
2.1. Chemical synthesis
2.1.1. General
Melting points (oC, uncorrected) were determined in open capillaries on a Graffin melting point apparatus. Pre-coated silica gel plates (silica gel 0.25 mm, 60 G F 254; Merck, Germany) were used for thin layer chromatography, dichloromethane/methanol (9.5:0.5 ml) mixture was used as a developing solvent system.IR spectra were recorded as films on KBr discs using IR-470 Shimadzu spectrometer (Shimadzu, Tokyo, Japan). The NMR spectra were recorded by Varian Gemini-300BB 300 MHz FT-NMR spectrometers (Varian Inc., Palo Alto, CA). 1H and 13C spectra were run at 300 and 75 MHz, respectively, in deuterated dimethylsulfoxide (DMSO-d6). Chemical shifts (δH) are reported relative to TMS as internal standard. All coupling constant (J) values are given in hertz. Chemical shifts (δC) are reported relative to DMSO-d6 as internal standards. Elemental analysis was performed on Carlo Erba 1108 Elemental Analyzer (Heraeus, Hanau, Germany). Electron impact Mass Spectra (EIMS) were recorded on Hewlett Packard 5988 spectrometer, Micro analytical center, Cairo University, Cairo. All compounds were within ±0.4% of the theoretical values. All solvents and reagents were commercially available and used without further purification. Sieno-Mass-II microwave (2.45GMHz, 1000 W) synthesis workstation was used. 2-Cyano-N-(4-methoxyphenyl) acetamide1 was prepared as reportedCitation42.
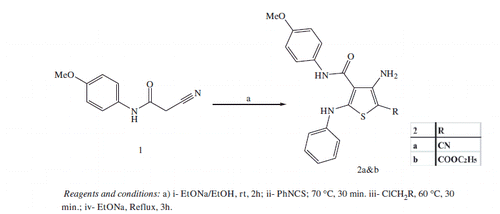
2.1.2. General procedure for synthesis of 4-amino-N-(4-methoxyphenyl)-5-substiuted-2-phenylaminothiophene-3-carboxamides (2a and b)
To a stirred solution of anilide1 (1.9 g, 0.01 mol) in absolute ethanol (20 ml), an ethanolic solution of sodium ethoxide (0.23 g sodium, 0.01 mol, 4 ml absolute ethanol) was added drop-wise and reaction mixture was stirred at room temperature. After 2 h, phenyl isothiocyanate (1.35 g, 0.01 mol) was added and the mixture was heated for 30 min at 70 °C then cooled to 25 °C. The α-chloromethylene derivative (0.01 mol) was then added, and the temperature raised to 60 °C for another 30 min. Sodium ethoxide (0.12 g sodium, 0.005 mol, 5 ml absolute ethanol) was then added and heating was continued under reflux temperature for additional 3 h.After cooling to room temperature, the reaction mixture was poured into ice cold water, the separated product was filtered, dried and recrystallized from ethanol to afford compounds 2a and 2b.
2.1.2.1. 4-Amino-5-cyano-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamide (2a)
Yield 62% (2.25 g); mp 291–293 °C; IR (KBr, υ cm−1): 3356–3224 (2NH, NH2), 2186 (C≡N), 1644 (C = O). 1H NMR (DMSO-d6): 3.67 (s, 3H, OCH3), 7.04–7.13 (m, 3H, Ar-H), 7.29-7.35 (m, 4H, Ar-H), 7.62 (d, J = 8.4 Hz, 2H, Ar-H), 9.74 (s, 1H, NH, D2O exchangeable), 9.89 (s, 1H, D2O exchangeable), 10.18 (s, 2H, NH2, D2O exchangeable). MS (m/z, %): 364 (M+, 6.60), 123 (100). Anal. Calcd. for C19H16N4O2S (364.42): C, 62.62; H, 4.43; N, 15.37. Found: C, 62.75; H, 4.37; N, 15.43.
2.1.2.2. Ethyl-3-amino-4-(4-methoxyphenylcarbamoyl)-5-phenylaminothiophene-2-carboxylate (2b)
Yield 73% (3 g); mp >300 °C; IR (KBr, υ cm−1): 3340–3262 (2NH, NH2), 1667–1625 (2C = O). 1H NMR (DMSO-d6): 1.21 (t, J = 6.9 Hz, 3H, OCH2CH3), 3.74 (s, 3H, OCH3), 4.14 (q, J = 6.9 Hz, 2H, OCH2CH3), 6.68 (s, 2H, NH2, D2O exchangeable), 7.09–7.12 (m, 3H, Ar-H), 7.30–7.39 (m, 4H, Ar-H), 7.50 (d,J = 8.4 Hz, 2H, Ar-H), 9.63 (s, 1H, NH, D2O exchangeable), 9.69 (s, 1H, NH, D2O exchangeable). MS (m/z, %): 412 (M + 1+., 9.65), 411 (M+., 18.70), 123 (100). Anal. Calcd. for C21H21N3O4S (411.47): C, 61.30; H, 5.14; N, 10.21. Found: C, 61.17; H, 5.26; N, 10.19.
2.1.3. Procedure for synthesis of 4-(2-chloro-acetamido)-5-cyano-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamide (3)
To a stirred solution of 4-aminothiophene-3-carboxamide derivative (2a, 0.55 g, 0.0015 mol) in n-butanol (5 ml), chloroacetyl chloride (0.17 g, 0.0015 mol) was added drop-wise with cooling at room temperature and the mixture was irradiated in the microwave oven at 150 °C for 5 min. After cooling to room temperature, the mixture was diluted with petroleum ether (5 ml). The solid formed was collected and treated with sodium carbonate solution (10%, 2 ml) then filtered, washed with water, dried and recrystallized from ethanol to afford white crystals of 4-(2-chloroacetamido)thiopnene-3-carboxamide derivative 3. Yield 59% (0.39 g); mp 242–244 °C; IR (KBr, υ cm−1): 3417–3234 (3NH), 2200 (C≡N), 1678–1628 (2C = O). 1H NMR (DMSO-d6): 3.63 (s, 3H, OCH3), 4.83 (s, 2H, CH2Cl), 7.05–7.16 (m, 2H, Ar-H), 7.24–7.50 (m, 3H, Ar-H), 7.59 (d, J = 8.1 Hz, 2H, Ar-H), 7.73 (d, J = 7.8 Hz, 2H, Ar-H), 10.56 (s, 1H, NH, D2O exchangeable), 10.81(s, 1H, NH, D2O exchangeable), 11.21 (s, 1H, NH, D2O exchangeable).MS (m/z, %): 442 (M + 2+., 8.27), 441 (M + 1+., 10.84), 440 (M+., 27.58), 267 (100). Anal. Calcd. for C21H17ClN4O3S (440.90): C, 57.21; H, 3.89; N, 12.71. Found: C, 57.39; H, 3.92; N, 12.68.
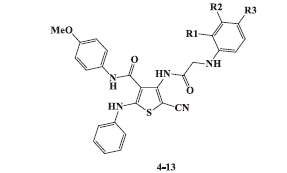
2.1.4. General procedure for synthesis of 5-cyano-N-(4-methoxyphenyl)-2-phenylamino-4-(2-arylamino-acetamido)thiophene-3-carboxamides (4-13)
To a stirred solution of 4-(2-chloroacetamido)thiophene-3-carboxamide 3 (0.44 g, 0.001 mol) in ethylene glycol (5 ml), the appropriate aromatic amine (0.002 mol) was added. The reaction mixture was irradiated in microwave oven at 120 °C for 15 min. After cooling to room temperature, the reaction mixture was diluted with petroleum ether (5 ml) and the precipitated solid was filtered, dried, and recrystallized from proper solvent to afford 4-(2-arylamino-acetamido)thiophene-3-carboxamides 4–13.
2.1.4.1. 5-Cyano-N-(4-methoxyphenyl)-2-phenylamino-4-(2-phenylamino-acetamido)thiophene-3-carboxamide (4)
Yield 76% (0.37 g); mp 233–236 °C(EtOH); IR (KBr, υ cm−1): 3425–3243 (4NH), 2210 (C≡N), 1665–1630 (2C = O). 1H NMR (DMSO-d6): δ 3.73 (s, 3H, OCH3), 4.82 (s, 2H, CH2), 7.07–7.13 (m,5H, Ar-H), 7.29–7.32 (m, 3H, Ar-H), 7.35–7.41 (m, 4H, Ar-H), 7.58 (d, J = 8.4 Hz, 2H, Ar-H), 9.58 (s, 1H, NH, D2O exchangeable),10.56 (s, 1H, NH, D2O exchangeable),10.81 (s, 1H, NH, D2O exchangeable), 11.26 (s, 1H, NH, D2O exchangeable). 13 C NMR (DMSO-d6): δ 54.9 (CH2), 55.6 (OCH3), 67.9, 97.8, 112.9 (C≡N), 119.0, 123.0, 123.5, 128.8, 129.2, 131.3, 136.4, 136.9, 138.5, 143.1, 147.6, 148.1, 156.1, 163.7, 165.8, 166.9. Anal. Calcd. for C27H23N5O3S (497.15): C, 65.17; H, 4.66; N, 14.08; Found: C,65.00; H, 4.40; N, 13.88.
2.1.4.2. 4-[2-(4-Chlorophenyl)aminoacetamido]-5-cyano-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamide (5)
Yield 72% (0.38 g); mp 210–212 °C(EtOH); IR (KBr, υ cm−1): 3410–3233 (4NH), 2200 (C≡N), 1678–1630 (2C = O). 1H NMR (DMSO-d6): δ 3.78 (s, 3H, OCH3), 4.81 (s, 2H, CH2), 7.19–7.24(m,2H, Ar-H), 7.25–7.28(m, 3H, Ar-H), 7.34–7.37 (m, 3H, Ar-H), 7.47–7.51 (m, 3H, Ar-H), 7.62 (d, J = 8.7 Hz, 2H, Ar-H), 8.63 (s, 1H, NH, D2O exchangeable), 9.21 (s, 1H, NH, D2O exchangeable), 10.62 (s, 1H, NH, D2O exchangeable), 11.22 (s, 1H, NH, D2O exchangeable).13C NMR (DMSO-d6): δ 54.8 (CH2), 55.9 (OCH3), 68.1, 98.1, 111.4 (C≡N), 115.1, 121.0, 123.0, 129.2, 131.3, 131.6, 136.4, 136.9, 137.9, 149.2, 151.4, 152.2, 156.2, 164.1, 165.8, 166.8. MS m/z (%):532(M + 1+, 18.45), 531 (M+., 21.55), 449 (100). Anal. Calcd. for C27H22ClN5O3S (531.01): C, 60.96; H, 4.17; N, 13.16; Found: C, 60.80; H, 4.30,; N, 13.18.
2.1.4.3. 4-[2-(4-Bromophenyl)amino-acetamido]-5-cyano-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamide (6)
Yield 55% (0.32 g); mp 217–219 °C(EtOH); IR (KBr, υ cm−1):3412–3225 (4NH), 2202 (C≡N), 1670–1645 (2C = O). 1H NMR (DMSO-d6): δ 3.87 (s, 3H, OCH3) , 4.83 (s, 2H, CH2), 7.08–7.13 (m,5H, Ar-H), 7.26–7.35 (m, 4H, Ar-H), 7.37–7.41 (m, 4H, Ar-H), 9.60 (s, 1H, NH, D2O exchangeable), 10.49 (s, 1H, NH, D2O exchangeable), 11.20 (s, 2H, 2NH, D2O exchangeable).13C NMR (DMSO-d6): δ 54.7 (CH2), 55.5 (OCH3), 67.8, 98.6, 111.2 (C≡N), 120.7, 120.8, 121.3, 126.5, 129.0, 131.4, 138.2, 138.3, 145.8, 148.8, 148.9, 152.1, 157.9, 164.3, 165.8, 166.5. MS m/z (%): 576 (M + 1+., 19.45), 575 (M+., 21.45), 448 (100). Anal. Calcd. for C27H22BrN5O3S (575.06): C, 56.25; H, 3.85; N, 12.15; Found: C, 56.10; H, 4.10; N, 12.28.
2.1.4.4. 5-Cyano-N-(4-methoxyphenyl)-2-phenylamino-4-[2-(4-methylphenyl)amino-acetamido]thiophene-3-carboxamide (7)
Yield 78% (0.40 g); mp 197–200 °C(DMF); IR (KBr, υ cm−1): 3425–3240 (4NH), 2200 (C≡N), 1675–1635 (2C = O). 1H NMR (DMSO-d6): δ 2.30 (s, 3H, CH3), 3.78(s, 3H, OCH3), 4.81 (s, 2H, CH2), 7.19–7.24 (m,4H, Ar-H), 7.25–7.28 (m, 3H, Ar-H), 7.34–7.37 (m, 2H, Ar-H), 7.47–7.51 (m, 2H, Ar-H), 7.62 (d, J = 8.7 Hz, 2H, Ar-H), 9.56 (s, 1H, NH, D2O exchangeable), 10.51 (s, 1H, NH, D2O exchangeable), 10.81 (s, 1H, NH, D2O exchangeable), 11.22 (s, 1H, NH, D2O exchangeable). 13 C NMR (DMSO-d6): δ 23.4 (CH3), 54.9 (CH2), 55.7 (OCH3), 68.3, 97.8, 111.2 (C≡N), 119.0, 122.9, 123.0, 129.1, 129.2, 131.3, 132.4, 136.1, 136.3, 149.1, 152.2, 156.2, 158.6, 164.1, 165.1, 166.7. Anal. Calcd. for C28H25N5O3S (511.17): C, 65.74; H, 4.93; N, 13.69; Found: C, 65.51; H, 4.80; N, 13.88.
2.1.4.5. 5-Cyano-N-(4-methoxyphenyl)-4-[2-(4-hydroxyphenyl)amino-acetamido]-2-phenylaminothiophene-3-carboxamide (8)
Yield 72% (0.37 g); mp 212-215 °C (DMF); IR (KBr, υ cm−1): 3435–3222 (OH, 4NH), 2204 (C≡N), 1672–1634 (2C = O). 1H NMR (DMSO-d6): δ 3.87 (s, 3H, OCH3), 4.83 (s, 2H, CH2), 7.08–7.11 (m, 2H, Ar-H), 7.12–7.15 (m, 3H, Ar-H), 7.29–7.32 (m, 3H, Ar-H), 7.3–7.36 (m, 3H, Ar-H), 7.37–7.43 (m, 2H, Ar-H), 9.60 (s, 1H, NH, D2O exchangeable), 10.19 (s, 1H, OH, D2O exchangeable), 10.53 (s, 1H, NH, D2O exchangeable), 10.81 (s, 1H, NH, D2O exchangeable), 11.21 (s, 1H, NH, D2O exchangeable). 13 C NMR (DMSO-d6): δ 54.9 (CH2), 55.4 (OCH3), 67.8, 98.4, 111.1 (C≡N), 119.0, 120.7, 120.8, 121.3, 126.5, 129.0, 131.3, 138.6, 145.8, 148.8, 148.9, 152.1, 157.9, 164.8, 165.3, 166.6. Anal. Calcd. for C27H23N5O4S (513.57): C, 63.14; H, 4.51; N, 13.64; Found: C, 62.94; H, 4.63; N, 13.59.
2.1.4.6. 5-Cyano-N-(4-methoxyphenyl)-4-[2-(4-methoxyphenyl)amino-acetamido]-2-phenylaminothiophene-3-carboxamide (9)
Yield 66% (0.35 g); mp 212–215 °C(DMF); IR (KBr, υ cm−1): 3410–3225 (4NH), 2198 (C≡N), 1676–1638 (2C = O). 1H NMR (DMSO-d6): δ 3.78 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 4.81 (s, 2H, CH2), 7.15–7.24 (m, 3H, Ar-H), 7.25–7.32 (m, 2H, Ar-H), 7.34–7.41 (m, 2H, Ar-H), 7.42–7.47 (m, 6H, Ar-H), 9.72 (s, 1H, NH, D2O exchangeable), 10.52 (s, 1H, NH, D2O exchangeable), 10.82 (s, 1H, NH, D2O exchangeable), 11.22 (s, 1H, NH, D2O exchangeable). 13 C NMR (DMSO-d6): δ 54.8 (CH2), 55.1 (OCH3), 55.4 (OCH3), 67.8, 98.4, 111.1 (C≡N), 120.6, 122.8, 122.9, 126.4, 129.2, 131.2, 133.9, 138.1, 142.7, 155.2, 155.7, 156.3, 164.4, 165.3, 166.7. Anal. Calcd. for C28H25N5O4S (527.16): C, 63.74; H, 4.78; N, 13.27; Found: C, 63.50; H, 4.70;N, 13.48.
2.1.4.7. 5-Cyano-N-(4-methoxyphenyl)-4-[2-(4-nitrophenyl) aminoacetamido]-2-phenylaminothiophene-3-carboxamide (10)
Yield 61% (0.33 g); mp 188–191 °C (EtOH); IR (KBr, υ cm−1): 3414–3230 (4NH), 2200 (C≡N), 1678–1635 (2C = O). 1H NMR (DMSO-d6): δ 3.79 (s, 3H, OCH3), 4.78 (s, 2H, CH2), 7.05–7.14 (m, 2H, Ar-H), 7.17–7.21 (m, 2H, Ar-H), 7.23–7.32 (m, 3H, Ar-H), 7.39--7.51 (m, 4H, Ar-H), 7.54–8.05 (m, 2H, Ar-H), 9.51 (s, 1H, NH, D2O exchangeable), 10.75 (s, 1H, NH, D2O exchangeable), 11.24 (s, 2H, 2NH, D2O exchangeable). 13 C NMR (DMSO-d6): δ 54.9 (CH2), 55.3 (OCH3), 67.8, 97.8, 111.0 (C≡N), 120.7, 121.3, 126.5, 129.0, 131.3, 131.4, 138.2, 138.6, 145.8, 148.8, 148.9, 152.1, 158.0, 164.6, 165.6, 166.9.MS (m/z, %): 543 (M + 1+., 18.15), 542 (M+., 30.46), 364 (100). Anal. Calcd. for C27H22N6O5S (542.14): C, 59.77; H, 4.09; N, 15.49; Found: C, 59.80; H, 4.30; N, 15.68.
2.1.4.8. 5-Cyano-N-(4-methoxyphenyl)-4-[2-(2-chlorophenyl) aminoacetamido]-2-phenylaminothiophene-3-carboxamide (11)
Yield, 66% (0.35 g); mp 156–158 °C(DMF); IR (KBr, υ cm−1): 3415–3234 (4NH), 2201 (C≡N), 1678–1635 (C = O). 1H NMR (DMSO-d6): δ 3.76 (s, 3H, OCH3) , 4.86 (s, 2H, CH2), 7.01–7.13 (m, 3H, Ar-H), 7.15–7.25 (m, 2H, Ar-H), 7.34–7.38 (m, 4H, Ar-H), 7.41–7.48 (m, 2H, Ar-H), 7.78 (d, J = 7.2 Hz, 2H, Ar-H), 8.62 (s, 1H, NH, D2O exchangeable), 9.21 (s, 1H, NH, D2O exchangeable), 10.18 (s, 1H, NH, D2O exchangeable), 11.34 (s, 1H, NH, D2O exchangeable). 13 C NMR (DMSO-d6): δ 54.8 (CH2), 55.5 (OCH3), 68.7, 99.1, 111.3 (C≡N), 120.7, 120.8, 121.3, 126.5, 129.0, 131.3, 131.4, 138.2, 138.6, 145.8, 148.8, 148.9, 152.1, 157.9, 164.0, 165.8, 166.1. Anal. Calcd. for C27H22ClN5O3S (532.01): C, 60.96; H, 4.17; N, 13.16; Found: C, 60.70; H, 4.20; N, 13.30.
2.1.4.9. 5-Cyano-N-(4-methoxyphenyl)-4-[2-(2-nitrophenyl)amino-acetamido]-2-phenylaminothiophene-3-carboxamide (12)
Yield 59% (0.32 g); mp 167–168 °C; IR (KBr, υ cm−1):3430–3224 (4NH), 2203 (C≡N), 1672–1639 (2C = O). 1H NMR (DMSO-d6): δ 3.84 (s, 3H, OCH3), 4.75 (s, 2H, CH2), 7.06–7.18 (m, 4H, Ar-H), 7.20–7.46 (m, 5H, Ar-H), 7.47–7.77 (m, 2H, Ar-H), 8.01–8.03 (m, 2H, Ar-H), 9.67 (s, 1H, NH, D2O exchangeable), 10.57 (s, 1H, NH, D2O exchangeable), 10.87 (s, 1H, NH, D2O exchangeable), 11.18 (s, 1H, NH, D2O exchangeable). 13 C NMR (DMSO-d6): δ 54.9 (CH2), 55.9 (OCH3), 68.2, 98.8, 111.9 (C≡N), 120.7, 120.8, 121.3, 126.5, 129.0, 131.3, 131.4, 138.2, 138.6, 145.9, 148.8, 148.9, 152.2, 157.9, 164.9, 165.4, 166.9. Anal. Calcd. for C27H22N6O5S (542.57): C, 59.77; H, 4.09; N, 15.49; Found: C, 59.50; H, 4.30; N, 15.28.
2.1.4.10. 5-Cyano-N-(4-methoxyphenyl)-4-[2-(3,4-dimethoxyphenyl) amino-acetamido]-2-phenylaminothiophene-3-carboxamide (13)
Yield, 52% (0.29 g); mp 167–168 °C; IR (KBr, υ cm−1):3425–3214 (4NH), 2209 (C≡N), 1672–1633 (2C = O). 1H NMR (DMSO-d6): δ 3.65 (s, 3H, OCH3), 3.71 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 4.83 (s, 2H, CH2), 7.04–7.07 (m, 2H, Ar-H), 7.18–7.23 (m, 3H, Ar-H), 7.33–7.40 (m, 3H, Ar-H), 7.45–7.47 (m, 4H, Ar-H), 9.59 (s, 1H, NH, D2O exchangeable),10.53 (s, 1H, NH, D2O exchangeable), 10.81 (s, 1H, NH, D2O exchangeable), 11.29 (s, 1H, NH, D2O exchangeable). 13 C NMR (DMSO-d6): δ 54.9 (CH2), 55.1 (OCH3), 55.9 (OCH3), 68.2, 98.8, 111.8 (C≡N), 115.1, 121.0, 123.0, 129.3, 131.4, 131.7, 136.4, 137.0, 138.0, 151.4, 152.2, 156.2, 164.4, 165.2, 166.4.Anal. Calcd. for C29H27N5O5S (557.62): C, 62.46; H, 4.88; N, 12.56; Found: C, 62.27; H, 5.03; N, 12.50.
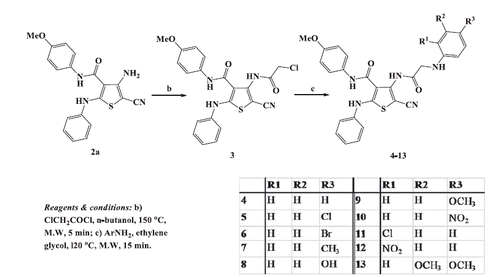
2.1.5. Procedure for synthesis of ethyl -3-acetamido-4-(4-methoxyphenylcarbamoyl)-5-phenylaminothiophene-2-carboxylate (14)
To a stirred solution of ethyl 3-amino-4-(4-methoxyphenylcarbamoyl)-5-phenylaminothiophene-2-carboxylate 2b(0.41 g, 0.001 mol) in n-butanol (5 ml), acetyl chloride (0.08 g, 0.001 mol) was added drop-wise with cooling and the reaction mixture was irradiated at 140 °C for 5 min in the microwave oven. After cooling to room temperature, the solid produced was collected and treated with sodium carbonate solution (10%, 20 ml) then filtered, washed with water, dried and recrystallized from the ethanol to afford crystals of ethyl 3-acetamido-4,5-disubstiutedthiophene-2-carboxylate 14. Yield 71% (0.32 g); mp 293–295 °C; IR (KBr, υ cm−1): 3429–3263 (3NH), 1682–1641 (3C = O); 1H NMR (DMSO-d6): δ 1.25 (t, J = 6.9 Hz, 3H, OCH2CH3), 2.26 (s, 3H, COCH3), 3.56 (s, 3H, OCH3), 4.23 (q, J = 6.9 Hz, 2H, OCH2CH3), 7.10–7.13 (m, 3H, Ar-H), 7.36–7.39 (m, 4H, Ar-H), 7.41–7.49 (m, 2H, Ar-H), 9.56 (s, 1H, NH, D2O exchangeable), 9.81 (s, 1H, NH, D2O exchangeable), 10.08 (s, 1H, NH, D2O exchangeable). 13 C NMR (DMSO-d6): δ 14.2 (OCH2CH3), 22.9 (COCH3), 55.9 (OCH3), 60.9 (OCH2CH3), 99.2, 114.8, 119.6, 123.0, 128.8, 131.3, 136.9, 138.5, 151.4, 152.2, 158.2, 160.4, 165.3, 166.7, 170.1. MS m/z: 454 (M + 1+., 8.76), 453 (M+., 13.23), 287 (100). Anal. Calcd. for C23H23N3O5S (453.51): C, 60.91; H, 5.11; N, 9.27. Found: C, 61.13; H, 4.98; N, 9.34.
2.1.6. Procedure for synthesis of 4-acetamido-5-hydrazinocarbonyl-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamide (15)
A mixture of ethyl 3-acetamido-4-(4-methoxyphenylcarbamoyl)-5-phenylaminothiophene-2-carboxylate14 (0.45 g, 0.001 mol) and hydrazine hydrate (99%, 0.001 mol) in n-butanol (5 ml) was heated at 150 °C for 5 min in the microwave oven. After cooling to room temperature, the separated solid was filtered, washed with hot ethanol, dried and recrystallized from acetic acid to yield hydrazide derivative 15. Yield, 84% (0.37g); mp> 300 °C; IR (KBr, υ cm−1): 3340–3275 (4NH, NH2), 1673–1632 (3C = O); 1H NMR (DMSO-d6): δ 2.27 (s, 3H, COCH3), 3.67 (s, 3H, OCH3) 7.11–7.21 (m, 2H, Ar-H), 7.39–7.45 (m, 3H, Ar-H), 7.48–7.54 (m, 2H, Ar-H), 7.84 (d, J = 8.4 Hz, 2H, Ar-H), 9.60 (s, 2H,NH2, D2O exchangeable), 9.64 (s, 1H,NH, D2O exchangeable),10.31(s, 1H,NH, D2O exchangeable), 11.40 (s, 1H, NH,D2O exchangeable). 13 C NMR (DMSO-d6): δ 22.8 (COCH3), 55.9 (OCH3), 99.2 114.8, 119.0, 122.9, 129.2, 131.3, 132.4, 136.0, 136.9, 149.1, 156.2, 158.6, 164.4, 165.3, 166.7.MS m/z: 440 (M + 1+., 0.49), 439 (M+., 2.75), 267 (100). Anal. Calcd. for C21H21N5O4S (439.49): C, 57.39; H, 4.82; N, 15.94. Found: C, 57.62; H, 5.01; N, 15.72.
2.1.7. General procedure for synthesis of 4-acetamido-5-(2-arylidenehydrazinocarbonyl)-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamides 16-23
To a well stirred solution of hydrazide 15 (0.44 g, 0.001 mol) in glacial acetic acid (5 ml), the appropriate aromatic aldehyde (0.001 mol) was added. The reaction mixture was irradiated at 100 °C in the microwave oven for 15 min and set aside to cool at room temperature. The separated solid was filtered, washed with hot ethanol and recrystallized from the proper solvent to afford 5-(2-arylidenehydrazinocarbonylthiophene-3-carboxamides 16-23.
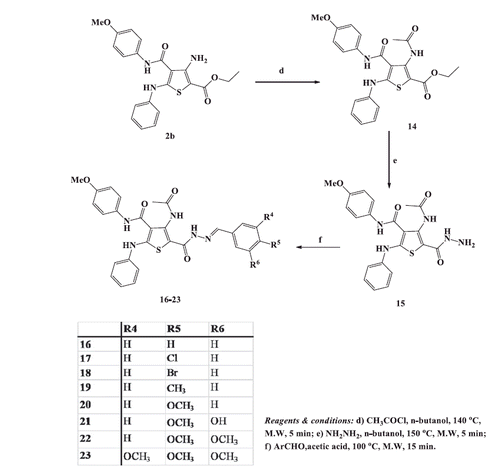
2.1.7.1. 4-Acetamido-5-(2-benzylidenehydrazinocarbonyl)-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamide (16)
Yield, 59% (0.31g); mp256–258 °C (EtOH); IR (KBr, υ cm−1); 3444–3212 (4NH), 1668–1630 (3C = O), 1587 (C = N); 1H NMR (DMSO-d6): δ 2.30 (s, 3H, COCH3), 3.62 (s, 3H, OCH3), 7.18–7.24 (m, 6H, Ar-H), 7.48–7.50 (m, 5H, Ar-H), 7.57–7.60 (m, 3H, Ar-H), 8.39 (s, 1H, CH=N), 8.64 (s, 1H, NH, D2O exchangeable), 9.76 (s, 1H, NH, D2O exchangeable), 11.43 (s, 1H, NH, D2O exchangeable), 11.47 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6): δ 22.9 (COCH3), 55.9 (OCH3), 99.2, 114.1, 120.7, 120.8, 121.3, 126.5, 129.0, 131.3, 131.4, 138.2, 138.6, 145.8, 148.8, 148.9, 152.1, 157.1 (CH=N), 164.0, 168.7, 168.8, 170.4. MS m/z: 528 (M + 1+., 18.38), 527 (M+., 22.62), 348 (100). Anal. Calcd. for C28H25N5O4S (527.59): C, 63.74, H, 4.78; N, 13.27; Found: C, 63.55; H, 4.58; N, 13.42.
2.1.7.2. 4-Acetamido-5-[2-(4-chlorobenzylidene)hydrazinocarbonyl]-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamide (17)
Yield, 61% (0.34g); mp234–244 °C (PrOH); IR (KBr, υ cm−1): 3338–3213 (4NH), 1650–1643 (3C = O), 1590 (C = N); 1H NMR (DMSO-d6): δ 2.32 (s, 3H, COCH3), 3.50 (s, 3H, OCH3), 7.07–7.14 (m, 3H, Ar-H), 7.28–7.36 (m, 2H, Ar-H), 7.38-7.48 (m, 6H, Ar-H), 7.59 (d, J = 8.1 Hz, 2H, Ar-H), 8.41 (s, 1H, CH=N), 8.63 (s, 1H, NH, D2O exchangeable), 9.76 (s, 1H, NH, D2O exchangeable), 9.84 (s, 1H, NH, D2O exchangeable), 10.10 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6): δ 22.0 (COCH3), 56.8 OCH3), 99.5, 114.2, 120.7, 120.8, 121.3, 126.5, 129.0, 131.3, 131.4, 138.2, 138.6, 145.8, 148.8, 148.9, 152.1, 157.9 (CH=N), 164.8, 168.1, 168.2, 170.1. MS m/z (%): 563 (M + 2+., 3.83), 561 (M+., 9.09), 541 (100). Anal. Calcd. for C28H24ClN5O4S (562.04): C, 59.84; H, 4.30; N, 12.46; Found: C, 59.72; H, 4.68; N, 12.59.
2.1.7.3. 4-Acetamido-5-[2-(4-bromobenzylidene)hydrazinocarbonyl]-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamide (18)
Yield, 84% (0.51 g); mp247-250 °C (EtOH); IR (KBr, υ cm−1): 3343–3210 (4NH), 1650–1634 (3C = O), 1590 (C = N); 1H NMR (DMSO-d6): δ 2.24 (s, 3H, COCH3), 3.62 (s, 3H, OCH3), 7.05–7.18 (m, 6H, Ar-H), 7.31–7.41 (m, 4H, Ar-H), 7.49–7.52 (m, 3H, Ar-H), 8.34 (s, 1H, CH=N), 8.59 (s, 1H, NH, D2O exchangeable), 9.49 (s, 1H, NH, D2O exchangeable) 9.57 (s, 1H, NH, D2O exchangeable), 11.28 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6): δ 23.2 (COCH3), 55.11 (OCH3), 99.7, 114.9, 120.7, 120.8, 121.3, 126.5, 129.0, 131.3, 131.4, 138.2, 138.6, 145.8, 148.8, 148.9, 152.1, 157.9 (CH=N), 164.6, 168.2, 168.3, 170.3. Anal. Calcd. for: C28H24BrN5O4S (606.49), C, 55.45; H, 3.99; N, 11.55; Found: C, 55.27; H, 3.90; N, 11.35.
2.1.7.4. 4-Acetamido-5-[2-(4-methylbenzylidene)hydrazinocarbonyl]-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamide (19)
Yield, 78% (0.42g); mp 180–183 °C(DMF); IR (KBr, υ cm−1): 3335–3218 (4NH), 1668–1633 (3C = O), 1593 (C = N); 1H NMR (DMSO-d6): δ 2.05 (s, 3H, CH3), 2.37 (s, 3H, COCH3), 3.68 (s, 3H, OCH3), 7.07–7.13 (m, 5H, Ar-H), 7.29–7.41 (m, 6H, Ar-H), 7.59 (d, J = 8.4 Hz, 2H, Ar-H), 8.41 (s, 1H, CH=N), 9.61 (s, 1H, NH, D2O exchangeable), 9.68 (s, 1H, NH, D2O exchangeable),9.86 (s, 1H, NH, D2O exchangeable), 11.28 (s, 1H, NH, D2O exchangeable), 13 C NMR (DMSO-d6): δ 22.9 (COCH3), 24.3 (CH3), 55.6 (OCH3), 99.1, 114.4, 120.7, 120.8, 121.3, 126.5, 129.0, 131.3, 131.4, 138.2, 138.6, 145.8, 148.8, 148.9, 152.1, 157.9 (CH=N), 164.4, 168.1, 168.2, 170.8.MS m/z: 542 (M + 1+., 17.34), 541 (M+., 24.17), 348 (100). Anal. Calcd. for C29H27N5O4S (541.62): C, 64.31; H, 5.02; N, 12.93; Found: C, 64.45; H, 5.11; N, 13.02.
2.1.7.5. 4-Acetamido-5-[2-(4-methoxybenzylidene)hydrazinocarbonyl] -N-(4-methoxyphenyl)-2-(phenylamino)thiophene-3-carboxamide (20)
Yield, 66% (0.37g); mp 259–661 °C (DMF); IR (KBr, υ cm−1): 3443–3225 (4NH), 1691–1640 (3C = O), 1592 (C = N); 1H NMR (DMSO-d6): δ 2.22 (s, 3H, COCH3), 3.78 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 7.07–7.13 (m, 3H, Ar-H), 7.25–7.27 (m, 2H, Ar-H), 7.36–7.41 (m, 4H, Ar-H), 7.57 (d, J = 8.4 Hz, 2H, Ar-H), 8.13 (d, J = 8.4 Hz, 2H, Ar-H), 8.39 (s, 1H, CH=N), 9.54 (s, 1H, NH, D2O exchangeable), 9.82 (s, 1H, NH, D2O exchangeable), 10.28 (s, 1H, NH, D2O exchangeable), 11.29 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6): δ 22.3 (COCH3), 54.2 (OCH3), 55.3 (OCH3), 99.6, 114.2, 121.0, 123.0, 129.2, 131.3, 131.6, 136.4, 136.9, 137.9, 151.4, 152.2, 156.2 (CH=N), 163.1, 164.6, 168.1, 168.2, 170.4. Anal. Calcd. for C29H27N5O5S (557.62): C, 62.46; H, 4.88; N, 12.56; Found: C, 62.53; H, 4.67; N, 12.72.
2.1.7.6. 4-Acetamido-5-[2-(3-hydroxy-4-methoxybenzylidene) hydrazinocarbonyl]-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamide (21)
Yield, 63% (0.36 g); mp292–295 °C (PrOH); IR (KBr, υ cm−1): 3440–3132 (OH, 4NH), 1667–1646 (3C = O), 1592 (C = N); 1H NMR (DMSO-d6): δ 2.29 (s, 3H, COCH3), 3.58 (s, 3H, OCH3), 3.61 (s, 3H, OCH3), 5.38 (s, 1H, OH, D2O exchangeable), 7.12–7.21 (m, 3H, Ar-H), 7.35–7.37 (m, 3H, Ar-H), 7.41–7.55 (m, 6H, Ar-H), 8.31 (s, 1H, CH=N), 8.61 (s, 1H, NH, D2O exchangeable), 9.64 (s, 1H, NH, D2O exchangeable), 9.72 (s, 1H, NH, D2O exchangeable), 11.23 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6): δ 22.5 (COCH3), 56.1 (OCH3), 56.2 (OCH3), 99.5, 119.0, 122.9, 123.0, 129.2, 129.3, 131.3, 132.4, 136.0, 136.2, 136.4, 136.9, 149.1, 152.3, 156.2 (CH=N), 158.7, 164.1, 168.4, 168.5, 170.2. MS m/z: 574 (M + 1+., 11.61), 573 (M+., 15.45), 307 (100). Anal. Calcd. for C29H27N5O6S (573.62): C, 60.72; H, 4.74; N, 12.21; Found: C, 60.86; H, 4.66; N, 12.48.
2.1.7.7. 4-Acetamido-5-[2-(3,4-dimethoxybenzylidene) hydrazinocarbonyl -N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamide (22)
Yield, 63% (0.37 g); mp233–235 °C (PrOH); IR (KBr, υ cm−1): 3411–3108 (4NH), 1667–1640 (3C = O), 1592 (C = N); 1H NMR (DMSO-d6): δ 2.24 (s, 3H, COCH3), 3.61 (s, 3H, OCH3), 3.66 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 7.10–7.23 (m, 5H, Ar-H), 7.34–7.44 (m, 7H, Ar-H), 8.39 (s, 1H, CH=N), 8.59 (s, 1H, NH, D2O exchangeable), 9.86 (s, 1H, NH, D2O exchangeable), 10.63 (s, 1H, NH, D2O exchangeable), 11.23 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6): δ 23.1 (COCH3), 55.8 (OCH3), 55.9 (OCH3), 56.0 (OCH3), 99.4, 114.0, 120.7, 120.8, 121.3, 126.5, 129.0, 131.3, 131.4, 138.2, 138.6, 145.9, 148.8, 148.9, 152.2, 157.9 (CH=N), 164.1, 168.3, 168.4, 170.1.MS m/z: 588 (M + 1+., 2.19), 587 (M+., 6.21), 347 (100). Anal. Calcd. for C30H29N5O6S (587.65): C, 61.32; H, 4.97; N, 12.92 Found: C, 60.98; H, 4.78; N, 12.70.
2.1.7.8. 4-Acetamido-N-(p-methoxyphenyl)-2-phenylamino-5-[2-(3,4,5-trimethoxybenzylidene)hydrazinocarbonyl]thiophene-3-carboxamide (23)
Yield, 61% (0.38 g); mp283–285 °C (PrOH); IR (KBr, υ cm−1): 3411–3108 (4NH), 1667–1646 (3C = O), 1592 (C = N); 1H NMR (DMSO-d6): δ 2.23 (s, 3H, COCH3), 3.76 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 7.16–7.23 (m, 6H, Ar-H), 7.32–7.53 (m, 5H, Ar-H), 8.39 (s, 1H, CH=N), 8.69 (s, 2H, 2NH, D2O exchangeable), 9.63 (s, 1H, NH, D2O exchangeable), 10.29 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6): δ 23.2 (COCH3), 55.7 (OCH3), 55.8 (OCH3), 55.9 (OCH3), 99.2, 107.1, 120.6, 122.8, 122.9, 126.4, 129.2, 131.2, 133.9, 138.1, 142.8, 155.2, 155.8, 156.4 (CH=N), 164.2, 168.6, 168.6, 170.6.MS m/z: 618 (M + 1+., 23.95), 617 (M+., 26.12), 502 (100). Anal. Calcd. for C31H31N5O7S (617.67): C, 60.28; H, 5.06; N, 11.34; Found: C, 60.33; H,5.18; N, 11.10.
2.2. Biological screening
2.2.1. Measurement of potential cytotoxic activity:
The cytotoxic activity of all the newly synthesized compounds was measured in vitro on HepG-2 and HCT-116 cell lines with Sulforhodamine-B stain (SRB) according to the reported procedureCitation43. Sorafenib was used as reference standard. Cells were seeded in 96-well microtiter plates at a concentration of 5 × 104–105 cells/well in a fresh medium and left to attach to the plates for 24 h before treatment with the tested compounds. Compounds were dissolved in dimethylsulfoxide (DMSO) and diluted with saline to the appropriate volume. After 24 h, cells were incubated with the appropriate concentration ranges of compounds (0.001, 0.01, 0.1, 1.0,10, and 100 µM), the wells were diluted to 200 μL with fresh medium and incubation was continued for 48 h. Control cells were treated with vehicle alone. Four wells were used for each drug concentration. After 48 h incubation, the cells were fixed with 50 μL cold 50% trichloroacetic acid for 1 h at 4 °C, washed 5 times with distilled water and then stained for 30 min. at room temperature with 50 μL 0.4% SRB dissolved in 1% acetic acid. The wells were then washed 4 times with 1% acetic acid. The plates were air-dried and the dye was solubilized with 100 μL/well of 10 mMtris base (PH 10.5) for 5 min on a shaker (Orbital shaker OS 20, Boeco, Germany) at 1600 rpm. The optical density (OD) of each well was measured spectrophotometrically at 564 nm with an enzyme-linked immunosorbent assay (ELISA) microplate reader (Meter tech. Σ 960, USA). The percentage of cell survival was calculated as follows: Survival fraction = OD (treated cells)/OD (control cells).The relation between surviving fraction and compound concentration was plotted and IC50 [the concentration required for 50% inhibition of cell viability] was calculated for each test compound. Experiment was repeated three times for each concentration.
2.2.2. DNA-flow cytometry cell cycle analysis for compounds 5 and 21 on HepG-2 cells:
HepG-2 cells were treated with compounds 5 and 21 at their IC50values for 24 h. After treatment, cells were washed twice with ice-cold phosphate buffer saline (PBS), collected by centrifugation, and fixed in ice-cold 70% (v/v) ethanol, washed with PBS, re-suspended with 0.1 mg/mL RNase, stained with 40 mg/mL propidium iodide (PI), and analyzed by flow cytometry using FACS Calibur (Becton Dickinson)Citation39. The cell cycle distributions were calculated using Cell-Quest software (Becton Dickinson). Exposure of HepG-2 cells to compounds5 and 21 resulted in an interference with the normal cell cycle distribution as indicated in the text.
2.2.3. In vitro ELISA immunoassay measurement of apoptotic markers for the effect for compounds 5 and 21 on HepG-2 cells (P53, bax, bcl-2, caspase-3/7)
2.2.3.1. Materials
The levels of Bax, Bcl-2, caspase-3 and -7 were assessed using ELISA immunoassay. Kits were purchased from the indicated suppliers. DRG® Human Bax. ELISA (EIA-4487) Marburg,Germany.Bcl-2ELISA Kit Invitrogen Corporation 1600 Faraday Avenue Carlsbad.Caspase-3, Invitrogen EIA kit Human (active) KHO1091 Invitrogen Corporation 1600 Faraday Avenue Carlsbad. Cloud clone caspase-7 (Active) eiakit, Cloud clone, Houston, USA. Procedures of the colorimetric kits were performed according to the manufacturer’sinstructions.
2.2.3.2. Methodology
The main principle of sandwich ELISA is the quantification of a specific protein through its containment in a sandwich of specific antibodies conjugated to the colorimetric TMB substrate, whose intensity is proportional to the protein quantity and is measured spectrophotometrically. The cell lysate was diluted 10 times, and 100 ml (50 mg protein) was added to the wells of separate micro titer plates for the ELISA kits that were precoated with primary antibodies specific to Bax, Bcl-2,caspase-3 or caspase-7. A secondary biotin-linked antibody specific to the protein captured by the primary antibody was added to bind the captured protein, forming a “sandwich” of specific antibodies around the desired protein in the cell lysate. The streptavidin-HRP complex was then used to bind the biotin-linked secondary antibody through its streptavidin portion. The HRP domain reacted with the added TMB substrate, forming a colored product that was measured at 450 nm by a plate reader (ChroMate-4300, FL,USA) after the reaction was terminated by the addition of stop solution.
2.2.4. In vitro β-tubulin polymerization inhibition assay for compounds 5 and 21
2.2.4.1. Materials
Enzyme linked immunosorbent assay (ELISA) was performed with human β-tubulin assay kit (SEB870HU) purchased from Cloud-Clone Corp. US. Procedures were performed according to manufacturer’sinstructionsCitation44.
2.2.4.2. Methodology
HepG-2 cell line was obtained from American Type Culture Collection, they were cultured using DMEM (Invitrogen/Life Technologies) supplemented with 10% FBS (Hyclone), 10 mg/mL of insulin (Sigma), and 1% penicillin–streptomycin. Plate cells (cells density 1.2–1.8 × 10,000 cells/well) in a volume of 100 ml complete growth medium and 100 ml of the tested compound at its IC50 per well in a 96-well plate for 18–24 h before the enzyme assay for β-TUB. The microtiter plate provided in this kit has been pre-coated with an antibody specific to β-TUB. Standards or samples are then added to the appropriate microtiter plate wells with a biotin-conjugated antibody specific to β-TUB. Next, Avidin conjugated to Horseradish Peroxidase (HRP) is added to each microplate well and incubated. After TMB substrate solution is added, only those wells that contain β-TUB, biotin-conjugated antibody and enzyme-conjugated Avidin will exhibit a change in color. The enzyme–substrate reaction is terminated by the addition of sulfuric acid solution and the color change is measured spectrophotometrically at a wavelength of50 nm ±10 nm. The concentration of β-TUB in the samples is then determined by comparing the O.D. of the samples to the standard curve. The data were compared with colchicine as standard β-TUB polymerization inhibitor. Each experiment was repeated three times.
2.2.5. In vitro vascular endothelial growth factor receptor-2(VEGFR-2) enzyme assay for
2.2.5.1. Materials
Enzyme linked immunosorbent assay (ELISA) for VEGFR-2 using human VEGF-R2/KDR ELISA kit [RBMS#2019R] according to manufacturer’s instructions.
2.2.5.2. Methodology
The samples were diluted with assay buffer 1:25 determine the number of microwell strips required. Microwell strips were washed twice with wash buffer.100 µL assay buffer was added in duplicate and to all standard wells, 100 µL of the prepared standard was pipetted into the first well and standard dilutions were created by transferring 100 µL from well to well. 100 µL of these standard dilutions were pipetted in the microwell strips, followed by addition of 100 µL assay buffer in duplicate to the blank wells and 50 µL assay buffer to sample wells. 50 µL of the prediluted samples were added in duplicates to designated sample wells. 50 µL of the prepared biotin-conjugate was added to all wells and then microwell strips were covered and incubated for 2 h at room temperature (18° to 25 °C). Microwell strips were washed six times with wash buffer and100 µL diluted Streptavidin-HRP was added to all wells. Microwell strips were covered again and incubated for 1 h at room temperature (18–25 °C). Microwell strips were washed six times with wash buffer and 100 µL of TMB substrate solution was added to all wells. Incubation of the microwell strips for about 30 min at room temperature (18–25 °C) followed by addition of 100 µL stop solution to all wells was done. Microwell reader was used to measure color intensity at 450 nm. The values of % activity versus a series of compound concentrations (0.01 µM, 0.1 µM, 1.0 µM, 10 µM, and 100 µM) was then plotted using non-linear regression analysis of sigmoidal dose-response curve. The IC50 values for compounds 5 and 21 against VEGFR-2 was determined by the concentration causing a half-maximal percent activity and the data were compared with Sorafenib as standard VEGFR-2 inhibitor. Each experiment was repeated three times.
2.2.6. Docking study
Molecular Operating Environment (MOE program 2008.10; Chemical Computing Group, Canada) was used to perform docking study. Crystal structures of VGEFR-2 complexed with sorafenib (PDB code 4ASD), and of tubulin (PDB ID: 1SA0) were downloaded from the protein data bank. The downloaded crystal structures were prepared for docking by adding the missing protons, deleting water molecules and all unnecessary co-crystallized ligands and metals using MOE software. Docking was performed using the default parameters of the software, and the best poses in terms of binding to the active site are discussed in text.
2.2.7. QSAR study
The synthesized compounds were divided into two sets, the training set composed of 15 compounds 4–11, 13–17, 19–20 and 22–23 that was used for a QSAR model development and their IC50 against HepG-2cells were converted into the logarithmic scale (pIC) and an external test subset using the compounds 12, 18 and 21 for validating the QSAR model was adopted. All the synthesized compounds were sketched in 2 D and minimized by the protocol “Prepared Ligands” in the Discovery Studio 2.5 software (Accelrys Inc., San Diego, CA, USA).The values of cytotoxic activity (IC50) were converted into the logarithmic scale pIC50 (pIC50= −log IC50) and then proceed to the QSAR analysis as the response variables. The obtained data set was further subdivided into the two subsets (Training and test set). Training set was composed of 15 compounds 4–11, 13–17, 19–20 and 22–23 and test set was three compounds12, 18 and 21. The training set was used to build a regression model, and the test set was used to external evaluation of the predictive ability of the model obtainedCitation45. In this study, several molecular descriptors representing thermodynamic, electronic, spatial, structural, thermodynamic, geometric, topological and quantum mechanical properties were calculated using “Calculate Molecular Properties” protocol of the Discovery Studio 2.5 (Table S8), where all the descriptor values for the molecules were considered as independent variables while, the inhibitory concentration results (pIC50) were taken as dependent variables.
3. Results and discussion
3.1. Chemistry
The work comprised the synthesis of two novel series of thiophene carboxamides; N,N-diaryl-2-amino-acetamido thiophenes 4–13 and arylidene thiophene hydrazides 16–23. Two Microwave-assisted organic synthesis (MWAOS) methodologies are known, which are open vessel microwave-assisted organic synthesis (OVMAOS) and pressurized microwave-assisted organic synthesis (PMAOS). OVMAOS have the advantage of being safer than PMAOS, and could be used in a large scale which is suitable for industrial chemistryCitation46.,Citation47.. The final target compounds were synthesized under OVMAOS methodology. All synthetic procedures adopted for preparation of key intermediates 3, 14, 15 and final target thiophenes 4–13 and 16–23 are illustrated in Schemes 1–3. Adopting Gewald methodCitation48, phenyl isothiocyanate was reacted with alkaline solution of 2-cyano-N-(4-methoxyphenyl)acetamide 1Citation42. in absolute ethanol, followed by treatment with α-halo active methylene compounds (chloro-acetonitrile or ethyl chloro-acetate) to yield thiophenes 2a and 2 b, respectively (Scheme 1). 1H NMR of thiophene derivatives 2a, b showed two D2O exchangeable signals at δ 9.63–9.74 and 9.69–9.89 ppm corresponding to two NH protons, and another exchangeable signal of two protons at δ 10.18 for compound 2a and δ 6.68 ppm for compound 2 b. Several synthetic trails for the preparation of compounds 2a and 2 b were done under MWAOS but all experimental trails failed. Literature survey revealed that aryl carboxamides could be prepared by reaction of the corresponding aryl amines with acid halides in polar solvents as dioxane for 24 h at room temperatureCitation49. In this study, acylation of 4-aminothiophene derivative 2a with chloro-acetyl chloride was performed in n-butanol as a high boiling solvent instead of ethanol and the reaction mixture was irradiated for 5 min at 150 °C to obtain the desired 4-(2-chloroacetamido)thiophene-3-carboxamide derivative 3 (Scheme 2). 1H NMR spectrum revealed the disappearance of the amine NH2 signal of the thiophene derivative 2a, and the appearance of a singlet signal at δ 4.83 of two protons corresponding to chloromethylene protons. In addition, mass spectrum of compound 3 showed two molecular ion peaks at m/z 442 and 440 with 3:1 ratio (Cl pattern). Conventional nucleophilic substitution reaction of alkyl chloride as the 2-chloro-acetamido derivatives with primary aromatic amines was reported in absolute ethanol and heating under reflux for long time (30 h)Citation50. In the current investigation, a solution of the alkyl halide 3 in ethylene glycol was irradiated for 15 min with different aromatic amines and afforded 4-(2-N-arylamino-acetamido)thiophene-3-carboxamides 4–13 (Scheme 2). 1H NMR spectra of the target compounds revealed the presence of two methylene protons and four exchangeable NH protons at δ 4.81–4.78 and δ 8.62–11.34 ppm, respectively. Moreover, 13 C NMR spectra of thiophenes 4–13 displayed a cyano group at δ 111.0–112.9 ppm. To synthesize the target thiophene hydrazides 16–23, amine group at C–3 of thiophene derivative 2 b must be protected. Masking of amine moiety via reaction with acyl halide as acetyl chloride is reported in dioxane as a reaction solventCitation49. In the present work, ethyl-3-amino-thiophene-2-carboxylate (2b) was treated with acetyl chloride to produce ethyl-3-acetamidothiophene-2-carboxylate derivative 14 (Scheme 3). The structure of intermediate 14 was confirmed by disappearance of the NH2 protons signal of the precursor 2 b, presence of signals corresponding to three NH protons at δ 9.56, 9.81, 10.08 ppm, and additional singlet signal of the acetyl protons at δ 2.26 in the 1H NMR spectrum of compound 14 (Scheme 3). The literature search disclosed that the ester group in ethyl thiophene-2-carboxylate derivatives could be converted into the corresponding acid hydrazides through hydrazinolysis of the parent ester in absolute ethanol and heating under reflux for 4 hCitation51. Herein, hydrazinolysis of the ester group in the ethylthiophene-2-carboxylate 14 using hydrazine hydrate in n-butanol was performed by heating at140 oC under OVMAOS conditions for 5 min which produced hydrazide 15(Scheme 3). Modifying the carboxylic acid hydrazides to the corresponding arylidene derivatives could be achieved by heating equimolar amounts of the acid hydrazide with the appropriate aromatic aldehydes for 2 h either in absolute ethanol only or with acetic acid as a catalystCitation52. In the present work, the arylidene thiophene hydrazides 16–23 were synthesized via reacting 4-acetamido-5-hydrazinocarbonyl-N-(4-methoxyphenyl)-2-phenylamino thiophene-3-carboxamide (15) with different aromatic aldehydes in acetic acid as a reaction solvent and acid catalyst at 100 °C for 15 min under OVMAOS conditions (Scheme 3). 1H NMR spectra of N-arylidene thiophene hydrazides 16–23 showed the presence of azomethine protons (N=CH) at δ 8.34–8.41 ppm, and confirmed the formation of the target small molecules.
Scheme 1. Synthesis of thiophene derivatives 2a and 2b.
Scheme 2. Synthesis of 5-Cyano-N-(4-methoxyphenyl)-4–(2-arylaminoacetamido)thiophene-3-carboxamides 4–13.
Scheme 3. Synthesis of 4-acetamido-5-(2-arylidenehydrazinocarbonyl)-N-(4-methoxyphenyl)-2-phenylaminothiophene-3-carboxamides 16–23.
3.2. In vitro screening of the synthesized compounds 4–13 and 16–23
3.2.1. In vitro cell growth inhibition of compounds 4–13 and 16–23 on HepG-2 and HCT-116 cancer cells
To assess cytotoxicity, liver hepatocellular carcinoma (HepG-2) and human colon carcinoma (HCT-116) cell lines were involved in this study, and Sulforhodamine-B stain (SRB) was used to assess cell viability according to the reported procedureCitation43. Sorafenib was also tested as a control compound. Sorafenib showed IC50 values of 4.43 and 3.22 µM against HepG-2 and HCT-116 cells, respectively.
3.2.1.1. In vitro cytotoxic activity of N,N′-diaryl-2-amino-acetamide thiophenes 4–13(model A) on HepG-2 and HCT-116 cells
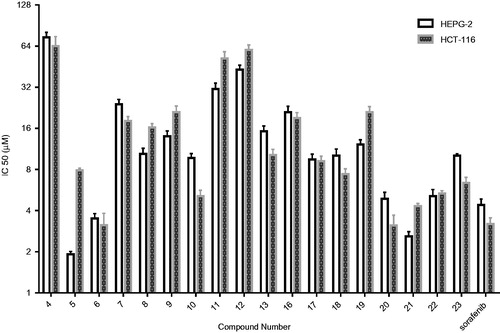
N,N′-Diaryl-2-aminoacetamide thiophene series(compounds 4–13) displayed cytotoxic activity against both HepG-2 and HCT-116 cell lines with varying IC50 values. The most potent thiophene derivatives were compounds 5, 6, and 10, which showed IC50 values of 1.92, 3.51, and 9.71 µM on HepG-2 cells, and 7.83, 3.13, and 5.12 µM on HCT-116 cells ( and ). All other analogs were less cytotoxic with IC50 values ranging from 10.42 to 74.32 µM on hepG-2, and 10.28 to 64.11 µM on HCT-116. Interestingly, compound 5 exhibited relatively higher cytotoxicity (IC50value of 1.92 µM) than the reference compound sorafenib (IC50value of 4.43 µM) with HepG-2 cells, while compound 6 exhibited the highest cytotoxicity (IC50value of 3.13 µM) with HCT-116 cells ( and ).
Figure 4. Dose response curves for the effect of the synthesized compounds 4–13 and 16–23 at different six concentrations (µM) on HepG-2 and HCT-116 cell lines for 24 h. (A)Effect of molecules 4–13 on Hep-G-2 cells. (B) Effect of molecules 16–23 on HepG-2 cells. (C)Effect of molecules 4–13 on HCT-116 cells. (D) Effect of molecules 16–23 on HCT-116 cells. Values represent the mean ± SEM for three experiments.
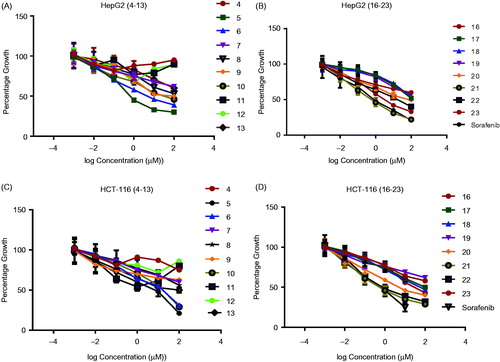
Table 1. IC50values of the 5-Cyano-N-(4-methoxyphenyl)-4–(2-N-(arylamino)acetamido)thiophene-3-carboxamide derivatives 4–13 against HepG-2 and HCT-116 cells.
3.2.1.2. Structural activity relationship (SAR) of cytotoxic activity of N,N′-diaryl-2-amino-acetamide thiophenes 4–13
Compounds 5 and 6 exhibited higher cytotoxicity than the rest of the compounds in the series, and compound 5 displayed cytotoxic effect with 2.31-fold higher than Sorafenib. Compound 6 was equipotent to Sorafenib. The results of the cytotoxicity screening experiments showed a correlation between the structural investigation of the compounds 4–13 with their cytotoxicity against HepG-2 and HCT-116 cells. Generally, substitution on the phenyl ring of the tail part contributed to cytotoxicity, while the unsubstituted derivative 4 was nearly not cytotoxic against HepG-2 and HCT-116 cells ( and ).The presence of an electron withdrawing group in the para position of the phenyl ring is essential for the observed cytotoxicity as in compounds 5, 6, and 10. Importantly, the presence of an electron donating groups on the para position in compounds 7, 8, 9, and 13 led to slight reduction of cytotoxicity compared to electron withdrawing groups containing compounds 5 and 6( and ). On the other hand, dramatic reduction in cytotoxicity was observed upon substituting the ring of the tail part on the ortho position as in (4-(2-(2-chlorophenylamino) acetamido)thiophene derivative 11 compared with(4-(2-(4-chlorophenylamino)acetamido) thiophene derivative 5 ( and ). It is worth mentioning that the most potent compounds 5 and 6 share a common pharmacophoric feature (a halogen in the para position) in the tail region with sorafenib, which might explain the high cytotoxicity observed in this experiment.
Figure 5. Graphical representation of IC50values (μM) of the synthesized compounds 4–13 and 16–23 against HepG-2 and HCT-116 cell lines.
3.2.1.3. In vitro cytotoxic activity of N-arylidene thiophene hydrazide derivatives 16–23(model B) over hepG-2 and HCT-116 cells
In this series, N-Arylidene thiophenehydrazides 20–23 showed good cytotoxicity against both cell lines with IC50values in the range of 2.61–10.82 μM with HepG-2 cell line, and 3.12–6.46 μM with HCT-116 cell line ( and ).
Table 2. IC50values of 4-acetamido-5-(2-arylidene-hydrazinocarbonyl)-N-(4-methoxyphenyl)thiophene-3-carboxamide derivatives 16–23 against HepG-2 and HCT-116 cells.
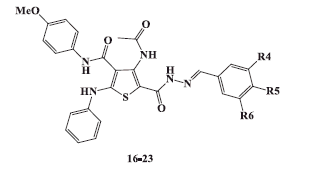
3.2.1.4. Structural activity relationship (SAR) of cytotoxic activity of N-arylidene thiophene hydrazides 16–23
N-(3-Hydroxy-4-methoxybenzylidene) thiophene hydrazide 21 was the most potent cytotoxic agent (IC50value of 2.61 µM) with about 1.7-fold higher cytotoxicity compared to Sorafenib (IC50value of 4.43 µM).N-(4-methoxybenzylidene) thiophene hydrazide 20 was the most active (IC50value of 3.12 µM) and almost equipotent to the reference compound .Remarkably, compound 21 possessed more cytotoxic effect within the series on HepG-2, while compound 20 exhibited the most potent cytotoxicity on HCT-116 cells ( and ).Within the N-arylidene thiophene hydrazideseries16–23,investigation of the electronic nature of the arylidene moieties in the tail part showed that hydrazides 20 and 21 that contain one methoxy group on arylidene groups exhibited higher cytotoxicity than compounds 17–18 which contain a halogen on the same position. The increased cytotoxicity might be due to the structural similarities of arylidene moieties of compounds 20–23 with the potent anti-mitotic compounds as colchicine and CA-4( and ).Analysis of the structural variations between the N,N′-diaryl-2-aminoacetamide thiophenes (model A) and arylidene thiophene hydrazide (model B) demonstrated that the presence of electron withdrawing substituents on the aryl rings of the tail part in N,N′-diaryl-2-amino-acetamide thiophenes (5,6, and 10) is essential for cytotoxic activity. On the other hand, an arylidene group bearing electron donating group in the tail part of model B (compounds 20–23) seems to be also crucial for the observed good cytotoxicity. Collectively, this experiment revealed the presence of a significant correlation between the linker (spacer) moiety and the substitution pattern on the aryl ring in the tail part. Furthermore, the electronic properties of the substituents on the aryl ring exert an additional effect on cytotoxic activity. Finally, the cytotoxicity results for the synthesized compounds showed higher cytotoxic sensitivity over HepG-2 cancer cells than HCT-116 cells.
3.2.2. In vitro DNA-flow cytometry and detection of apoptosis for compounds 5 and 21 on HepG-2 cells
The most cytotoxic thiophene derivatives 5 and 21 were selected to further evaluate their effect on the cell cycle progression and induction of apoptosis. HepG-2 cells were treated with each derivative at its IC50 values for 24 h, and then the DNA content was analyzed by flow cytometry using FACS Calibur (Becton Dickinson). A control experiment with no treatment was also included. Flow cytometric analysis revealed that treatment with compound 5 and 21 resulted in apoptosis as denoted by the increase in the apoptotic sub-G1 cell population (8.23% and 6.07%, respectively), compared with the control (0.10%) ( and Table S3). In addition, a G2/M arrest was also observed upon treatment with compounds 5 or 21, with about 2- and 1.9-fold increase compared to the control ( and Table S3).Accumulation of cells in the G0 phase and cell cycle arrest at G2/M phase indicates the possible role of compounds 5 and 21 in apoptosis induction and cell death. In conclusion, compounds 5 and 21 showed pre-apoptotic activity and cell growth inhibition through cell cycle arrest at G2/M phase. N-aryl-2-N-(4-chlorophenyl) amino-acetamide 5 showed higher apoptotic inducing activity than N-(3-hydroxy-4-methoxybenzylidene) thiophene hydrazide 21.
Figure 6. (I) Flow cytometric analysis of the most potent compounds 5 and 21 on cell cycle distribution of HepG-2 cancer cells (24 h). (A) Control HepG-2 cells. (B) Cells treated with compound 5 (2.61 µM) 24 h showing most of the cells arrested in G2/M phase and apoptosis. (C) Cells treated with compound 21 (1.92 µM) for 24 h showing cell cycle arrest at G2/M phase. II) Data expressed as mean (n = 4 experiments) ± SEM and statistical comparisons were carried out using one-way analysis of variance (ANOVA) followed by Tukey multiple comparisons.
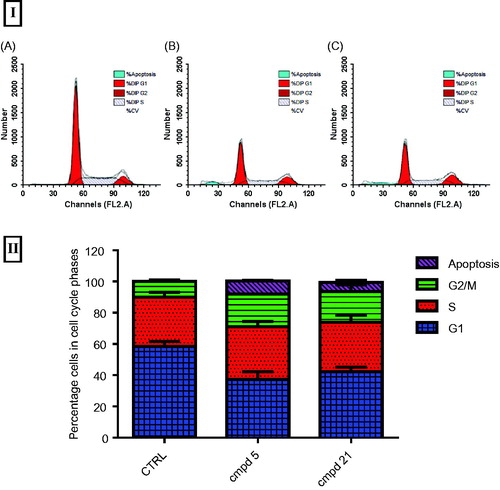
Table 3. IC50values (μM) of thiophene-3-carboxamides5, 6, 10, 17, 20–23, sorafenib, and colchicinein VEGFR-2 enzyme assay, and β-tubulin polymerization inhibition, along with the IC50values with HepG-2 cell line.
3.2.3. In vitro apoptosis identification by FITC-annexin V-staining
To further assess the tumour suppression property of the compounds, and to confirm that the observed sub-G1 cells accumulation was due to apoptosis, HepG-2 cells treated with 5 and 21for 24 h were analyzed with PI and FITC-annexin V stain for apoptosis determination ( and Table S4). Both early apoptosis (Q4 quadrant in ) and late apoptosis (Q2 quadrant in ) stages in HepG-2 cells increased after treatment with compounds 5 and 21 ().Interestingly, the late apoptotic cells percentage showed an increase after treatment with compounds 5 and 21 from 0.34% in control to 7.85% (23-fold increase) and 7.2% (21-fold increase). In addition, early apoptosis was significantly increased from 0.72% in the no treatment control to 5.47% (8-fold increase) and 4.29% (6-fold increase) upon treatment with 5 and 21, respectively. Furthermore, a profile of early apoptotic cell population was also observed for the tested compounds 5 and 21. The results in this experiment are in line with the previous experiment, and confirm that these compounds exhibit their cytotoxic effect and pro-apoptotic activity.
Figure 7. FITC-annexin-V and PI staining of HepG-2 cells after treatment with compounds 5 and 21 at their IC50 values. (A) Dot plots showing: dead cells and cells debris (Q1, top left quadrant), late apoptosis cells (Q2, top right quadrant), viable cells (Q3, bottom left quadrant), and early apoptosis cells (Q4, bottom right quadrant). (B) Graphical representation of the early and late apoptotic cells percentage upon treatment with 5, 21or no treatment control.
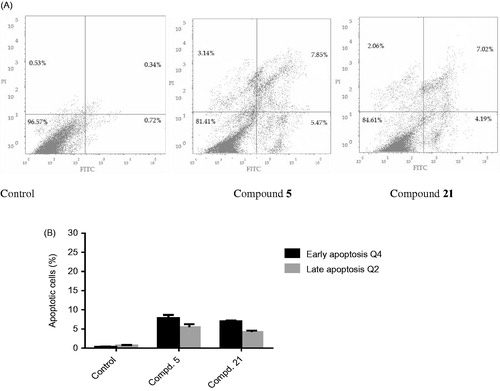
Table 4. Estimated pIC50 data of the training set analogs against HepG-2 cell line according to QSAR Equation.
3.2.4. In vitro measurement of the concentration of the apoptotic marker proteins (p53, bax, bcl-2, caspase-3/7) for compounds 5 and 21in HepG-2 cells
To further investigate the apoptosis inducing activity of N,N′-diaryl-2-amino-acetamide 5 and N-arylidene hydrazide 21,the levels of apoptosis-induction biomarkers, p53, Bax,Bcl-2, caspase-3/7 proteins were measured after 24 h treatment of HepG-2 cells with compound 5 or compound 21 at their IC50 values, and were then compared with a no treatment control ( and Table S5).The tumour suppression gene, p53, is a transcription factor that blocks cell cycle at G1 phase and induces apoptosis via mitochondrial pathway through several proteins such as Bax and Bcl-2 via loss of cell contents and restriction of cell growth. In this experiment, treatment of HepG-2 cells with compounds 5 and 21 led to significant increase in p53 protein level, with about 3.4- and 1.7-fold increase, respectively, compared with the control ( and Table S5).This result suggests that compounds 5 and 21 caused an elevation in the concentration of p53, which might have led to the observed cell growth inhibition and apoptosis induction. Among the resistance mechanisms of tumour cells to apoptosis is the increase in the expression of anti-apoptotic proteins, such as Bcl-2, and the decrease of the expression of pro-apoptotic proteins, such as BaxCitation53.Treatment of HepG-2 cells with compounds 5 and 21 resulted in a significant increase in the level of Bax by 6.6- and 11.8-fold, respectively compared to the control ( and Table S4). On the other hand, levels of Bcl-2 decreased by 2.2- and 2.5-fold, respectively compared to the control ( and Table S5). Finally, the tested compounds 5 and 21 showed significant higher Bax/Bcl-2 ratio than the control, which is consistent with the results of the DNA-flow cytometry that revealed the ability of compounds 5 and 21 to induce apoptosis in HepG-2 cells ().
Figure 8. In vitro ELISA immunoassay measurement of the p53,Bcl-2, Bax, caspase-3 and caspase-7 concentration in HepG-2cells after treatment for 24 h with thiophene-3-carboxamides 5 and 21 at their IC50. (A) effect of compounds 5 and 21 on p53 concentration (B) effect of compounds 5 and 21 on Bcl-2 concentration (C) effect of compounds 5 and 21 on Bax concentration (D)effect of compound 5 and 21 on Bax/Bcl-2 ratio (E)effect of compounds 5 and 21 on caspase-3 concentration (F)effect of compounds 5 and 21 on caspase-7 concentration. Data were expressed as mean (n= experiments) ± SEM and statistical comparisons were carried out using one-way analysis of variance (ANOVA) followed by Tukey multiple comparisons test at p < .05.
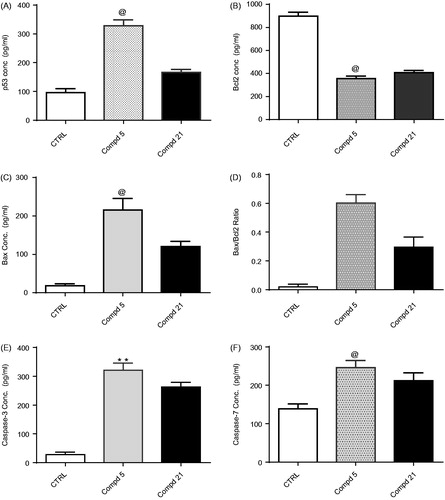
Table 5. External validation for the established QSAR model utilizing potent (21), moderate (18) and inactive (12) anticancer active agents.
Activation ofcaspase-3/7 is the final effective step in apoptosis induction leading to DNA fragmentation and cell deathCitation54.Moreover, it was suggested that the caspase proteins family -especially caspase-7-are potentially involved in the regulation at M phaseCitation55. Given that compounds 5 and 21 significantly elevated Bax/Bcl-2 ratio, we were encouraged to subsequently evaluate the effect of these derivatives on the levels of active caspase-3/7 upon treatment of HepG-2cells. As expected, compound 5 increased the level of caspase-3/7 by about 11.2- and 6.9-fold,respectively compared to the control, while compound 21 increased the level of caspase-3/7 by 9.2- and 5.9-fold,respectively compared to the control().In conclusion, compound 5 showed a pro-apoptotic effect higher than compound 21, through increasing p53 protein level, Bax/Bcl-2 ratio, and levels of the terminal caspase-3/7, which supported the results obtained from cell cycle analysis().
3.2.5. In vitro screening against vascular endothelial growth factor receptor-2 (VEGFR-2) enzyme for N,N′-diaryl-2-amino-acetamides 5, 6, 10 and N-arylidene thiophene hydrazides 17 and 20–23
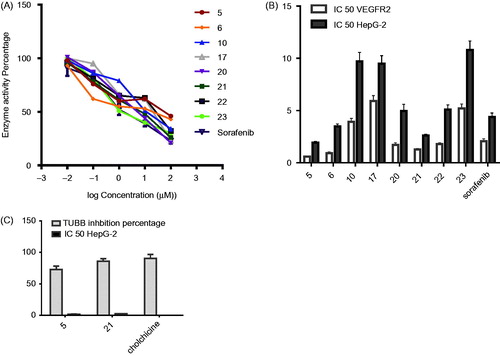
To assess the success of the approach used to design the pharmacophores developed in this work as VEGFR inhibitor, we extended the biological investigation to determine VEGFR-2 enzyme inhibitory activity of the analogs. The IC50 values of the most promising cytotoxic derivatives in each library (N,N′-diaryl-2-amino-acetamides 5, 6, and 10 from model A, and N-arylidene thiophene hydrazides17, 20–23 from model B) were determined. Sorafenib, as a reference VEGFR-2 inhibitor was also tested as positive control ( and ). The N,N′-diarylamino-acetamide derivatives 5, 6, and 10 showed IC50values in the range of 0.59–3.92 μM,which is nearly equipotent to standard drug Sorafenib (IC50value of 2.07 μM). Significantly, compounds 5 and 6 showed IC50value of 0.59 and 0.93 μM, which are potent than Sorafenib. The results point to the likelihood that the good inhibitory activity displayed by the 5-cyano-N-(4-methoxyphenyl)-4-(2-(arylamino) acetamido) thiophene-3-carboxamide derivatives 5, 6, and 10 might be due to the structural similarity in molecular skeleton to the second generation ortho-uriedo-aryl carboxamide VEGFR-2 inhibitors, such as XL-880 and CP-547-623, in addition to structural similarities with tail part of Sorafenib ( and ).In parallel, N-arylidene thiophene hydrazides 17 and 20–23 that belong to pharmacophoric model B showed good inhibitory activities, with the lowest activity observed in N-(4-chlorobenzylidene)thiophene hydrazide 17 and N-(3,4,5-trimethoxybenzylidene)thiophene hydrazide 23. It’s worth mentioning that compound 23 bears a bulky tri-methoxyphenyl moiety, which could explain its lower inhibitory activity among the series. The most potent analog in compounds 16–23 was compound 21, which displayed a slightly lower IC50value (1.29 µM) than the reference compound Sorafenib (IC50 value of 2.07 µM) ().
Figure 9. (A) Dose response curve for compounds 5, 6, 10, 17, 20–23 and Sorafenib for IC50 determination of VEGFR-2 enzyme.Values represent the mean ± SE for four experiments. B) Graphical representation of IC50 for compounds 5, 6, 10, 17, 20-23 and Sorafenib on VEGFR-2 enzyme, HepG-2 cells. (C) Graphical representation of IC50 for compounds 5, 21 on HepG-2 cells and β-tubulin inhibition percentage of compounds 5 and 21 compared with colchicine. Values represent the mean ± SE for two experiments.
3.2.6. In vitro β-tubulin polymerization inhibition assay (TUB) for N,N′-diaryl-2-aminoacetamide 5 and N-arylidene hydrazide 21 on HepG-2 cells
To investigate the growth inhibition showed by the synthesized compounds 5 and 21 and the observed cell cycle arrest at G2/M phase, further enzyme assay for β-tubulin polymerization inhibition percentage on HepG-2 was carried out at their IC50 values. The inhibitory activity in this experiment is given as the percentage of β-tubulin polymerization inhibition. The obtained results showed that compounds 5 and 21 produced promising tubulin polymerization suppression compared with colchicine ( and ).Compounds 5 and 21 showed 73% and 86% β-tubulin polymerization inhibition, respectively. Compound 21 was nearly equipotent to colchicine, while compound 5 exhibited lower inhibition percentage, compared with the reference anti-mitotic drug, Colchicine (). The observed inhibitory activity of compound 21 might be due to close structural similarities to CSI inhibitors (). On the other hand, the obtained tubulin polymerization inhibition for N-aryl-2-N-(4-chlorophenyl) amino-acetamide 5 suggested that it is one of atypical CSI agents. In summary, the good inhibitory activity against β-tubulin polymerization for compounds 5 and 21 might explain the cell cycle block at G2/M phase exhibited by 5 and 21 in the previous experiments and supports the hypothesized design of the pharmacophoric models mentioned in this work.
3.3. Molecular docking studies
3.3.1. Molecular docking in the active site of VEGFR-2 enzyme
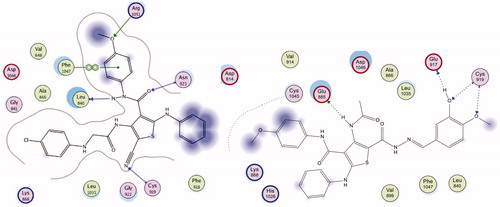
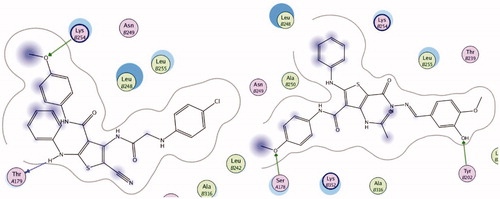
Docking study was done to assess the binding affinities of the most active thiophenes 5 and 21 in VEGFR-2 active site. The crystal structure of VEGFR-2 (PDB ID:4ASD) enzyme co-crystallized with sorafenib was used in this study. Compounds 5 and 21 showed promising binding affinity with VGEFR-2, as both inhibitors occupied the active site of the enzyme. The co-crystallized ligand, sorafenib formed four H-bonding interactions with Glu885, Cys919, and Asp1046 amino acid residues in theVEGFR-2 active site (Figure S58). Compound 5 formed four hydrogen bonding interaction with Leu840, Cys919, Asn923, and Arg1051, in addition to arene-arene interaction with Phe1047, total 5 binding interactions with the active site of VEGFR-2 (). On the other hand, 21 showed four hydrogen bonding interactions with Glu885, Glu927, and Cys919 ().This study shows that the binding interactions of derivatives 5 and 21 to the active site of VEGFR-2 are similar to Sorafenib, which explains the low IC50 values observed with VEGFR-2 in this work ().
Figure 10. Docking poses displaying 2D interactions of compounds 5 (left) and 21 (right) with VEGFR-2 active site.
3.3.2. Molecular docking in tubulin colchicine binding site
To evaluate the binding of analogs 5 and 21 to colchicine binding site of tubulin, a docking study was carried out with the colchicine binding site in the x-ray crystal structure of tubulin (PDB ID: 1SA0). Both compounds 5 and 21 occupied colchicine binding sit of the enzyme. Compounds 5 showed good binding interactions with two hydrogen bonds with ThrA179 and LysB254 (). In addition, 21 bound to the active site with two hydrogen bonds with SerA178 and TyrB202 (). Aromatic rings in both derivatives displayed favorable hydrophobic interactions with different amino acids lining the active site and the active site entrance (Leu245, Lys251, Lys 341, Leu 242, Val 307, Ile 359 and Leu 252). The results of this docking study are in agreement with the β-tubulin polymerization inhibition assay (), where both studies suggest that compounds 5 and 21 bind to the colchicine binding site of β-tubulin, and can act as β-tubulin polymerization inhibitors ( and ).
Figure 11. Docking poses displaying 2D interactions of compounds 5 (left) and 21 (right) with tubulin colchicine binding site.
3.4. Quantitative structure activity relationship (QSAR)
To validate the results of cytotoxicity against HepG-2 cancer cells, QASR model was generated. The synthesized compounds were divided into 2 sets, the training set is composed of 15 compounds (4–11, 13–17, 19–20 and 22–23). The training set was used to develop the QSAR model in which their IC50 against HepG-2cells were converted into the logarithmic scale (pIC), while compounds 12, 18 and 21 were used in the external test subset to validate the developed QSAR model.
3.4.1. Development of QSAR model
Several molecular descriptors were calculated for each compound employing a calculated molecular properties module. The 3 D structures of the training set compounds were imported into the Discovery Studio program to calculate various molecular descriptors including energies of highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO) for each of the synthesized compoundCitation39. First, Genetic function approximation (GFA) which is a useful statistical analysis tool to correlate biological activity or property with chemical characteristics of molecules was employed to search for the best possible QSAR regression equation, then Multiple linear regression (MLR) analysis were employed to optimize QSAR modelCitation39.The MLRTemp Model equation bearing the relevant descriptors shows our best performing QSAR models is given as the following equation:
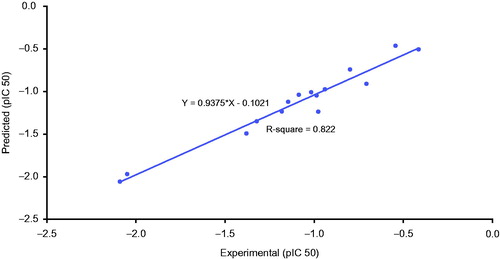
According to the equation, the QSAR model was represented graphically by scattering plots of the experimental cytotoxic activity pIC50 versus the predicted activity values pIC50 for the training set compounds against HepG-2 cells as ().
Figure 12. Plot of predicted versus experimental pIC50 values of the training set compounds and results against HepG-2 human tumor cell line according toQSARequation.
3.4.2. Validation of QSAR model
QSAR model was validated employing leave one-out cross-validation by setting the folds to a number much larger than the number of samples, r2(squared correlation coefficient value) and r2prediction (predictive squared correlation coefficient value), residuals between the predicted and experimental activity of the training set (), (N = 15, r2 = 0.822, r2ad = 0.745, r2pred = 0.708), where N, represents number of compounds in the training set, r2 (adj) is r2 adjusted for the number of terms in the model, r2 (pred) is the prediction r2, equivalent to q2 from a leave one-outcross-validation.
3.4.3. External validation of QSAR
In addition to validation of QSAR model by employing leave one-out cross-validation, r2adj and r2pred, external validation of the determined QSAR equations was performed utilizing three of compounds exhibiting potent (21), moderate (18) and inactive (12) anticancer properties. The observed pIC50 activities and those provided by QSAR study and residuals between the predicted and experimental activity of the test set are shown in . It should be noted that the predicted cytotoxic activity by QSAR model was very close to the experimentally observed values. In conclusion, descriptors which played important role in developing QSAR model were jurs-RPCS. Jurs-RPCS indicated that the relative hydrophobic characters of the whole molecule are important for the observed activity. Other descriptors used were Radius of gyration (Rad of Gyration), which is a geometrical descriptor and measure of the size of the molecule. Another important descriptor was the principal moment of inertia (PMI), which is a physical property that is related to the rotational dynamics of a module that combine shape and electronic information to characterize moleculesCitation56.QSAR model suggested that the cytotoxic activity of the two models A and B was affected by electronic effect of the substituents and shape of the model as previously explained in the cytotoxicity results.
4. Conclusions
In this study, a library of small synthetic molecules sharing a common ortho-aminothiophene carboxamide moiety was designed as dual anti-angiogenesis agents and antimitotic agents. The therapeutic rationale behind the design of compounds with dual biological targets was to decrease the acquired resistance of tumour cells towards hypoxia induced by these molecules and hence improved apoptosis induction by the anti-angiogenic drugs. The ortho-aminothiophene carboxamides 4–13 and 16–23 were synthesized under microwave assisted synthesis protocol in good yield as prerequisite for sustainable development of organic synthesis. The N,N′-diaryl-2-amino-acetamide thiophenes 4–13 and N-arylidene thiophene hydrazides 16–23 were evaluated for their in vitro anti-proliferative activity against gastrointestinal solid cancer cell lines(HepG-2 and HCT-116).The most cytotoxic compounds with both cancer cells were 5 and 21. Compound 5 showed higher cytotoxic activity onHepG-2cells than Sorafenib with 2.3-fold, while the restricted conformation hydrazide derivative 21 displayed 1.7-fold higher cytotoxicity compared to Sorafenib. Moreover, compounds 5 and 21 showed pro-apoptotic activity by inducting significant increase in the percentage of apoptotic cells (8.23% and 6.07%, respectively) in DNA-flow cytometry compared to control (0.10%), indicating potent apoptosis-inducing cancer cell death. The apoptosis mechanistic pathway was confirmed by a significant elevation in Bax/Bcl-2 ratio with 30- and 15-fold after treatment of HepG-2 cells with compounds 5 and 21compared to the control. In addition, significant increase in the protein level concentrations of caspase-3/-7 was also observed. The most potent cytotoxic compound 5 showed promising dual inhibitory activity againstVEGFR-2 enzyme with IC50 value 0.59 µM which was 4-fold more potent than Sorafenib (IC50 = 2.07 μM), and against β-tubulin polymerization (72% polymerization inhibition at 1.92 μM).Molecular docking of compounds 5 and 21 at the binding site of VEGFR-2 and colchicine tubulin binding site was done to explain the exhibited inhibitory activity against both targets. Finally, quantitative structure-activity relationship (QSAR) studies delivered equation of five 3 D descriptors with a good correlation between experimental and predicted IC50values (R2 = 0.822).This QSAR model provides as an effective technique for understanding the observed pharmacological properties. The compounds reported in this work represent potential cytotoxic agents that could help to develop more effective anticancer agents against gastrointestinal tumours.
supplmentary_data.pdf
Download PDF (4.2 MB)Acknowledgement
The authors are filled with respect to the memory of Dr. Ibrahim Abouelish, founder of Sekem Corporation. The authors are grateful to all members of the department of Cancer Biology, National Cancer Institute, Cairo, Egypt, for carrying out the cytotoxicity testing. The authors thank Dr. Esam Rashwan, Head of the confirmatory diagnostic unit VACSERA, Egypt, for carrying out VEGFR-2 inhibition assays.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Lanza G, Messerini L, Gafà R, et al. Colorectal tumors: the histology report. Dig Liver Dis 2011;43:S344–S55.
- Gao JJ, Shi ZY, Xia J, et al. Sorafenib-based combined molecule targeting in treatment of hepatocellular carcinoma. WJG 2015;21:12059–70.
- Zhang X, Raghavan S, Ihnat M, et al. The design and discovery of water soluble 4-substituted-2,6-dimethylfuro[2,3-d]pyrimidines as multitargeted receptor tyrosine kinase inhibitors and microtubule targeting antitumor agents. Bioorg Med Chem 2014;22:3753–72.
- Von Felden J, Schulze K, Gil-Ibanez I, et al. First- and second-line targeted systemic therapy in hepatocellular carcinoma—an update on patient selection and response evaluation. Diagnostics 2016;6:44.
- Arnold M, Sierra MS, Laversanne M, et al. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2016;0:1–9.
- Frenette C, Gish R. Targeted systemic therapies for hepatocellular carcinoma: clinical perspectives, challenges and implications. WJG 2012; 18:498–506.
- Zwick E, Bange J, Ullrich A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr-Relat Cancer 2001;8:161–73.
- Wu J, Ji J, Weng B, et al. Discovery of novel non-ATP competitive FGFR1 inhibitors and evaluation of their anti-tumor activity in non-small cell lung cancer in vitro and in vivo. Oncotarget 2014;5:4543–53.
- Garuti L, Roberti M, Bottegoni G. Non-ATP competitive protein kinase inhibitors. Curr Med Chem 2010;17:2804–21.
- Chen X, Lu T, Lu S, et al. Structure-based and shape-complemented pharmacophore modeling for the discovery of novel checkpoint kinase1 inhibitors. J Mol Model 2010;16:1195–204.
- Farsangi MH. Small-molecule inhibitors of the receptor tyrosine kinases: promising tools for targeted cancer therapies. Int. J. Mol. Sci 2014;15:13768–801.
- Chen Y, Fu L. Mechanisms of acquired resistance to tyrosine kinase inhibitors. Acta Pharmaceutica Sinica B 2011;1:197–207.
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol 2006; 2:358–64.
- Avendaño C, Menéndez JC. Drugs that inhibit signalling pathways for tumor cell growth and proliferation. Med Chem Anticancer Drugs 2008;251–305.
- Blumenschein GR, Reckamp K, Stephenson GJ, et al. Phase Ib Study of motesanib, an oral angiogenesis inhibitor, in combination with carboplatin/paclitaxel and/or panitumumab for the treatment of advanced non–small cell lung cancer. Clin Cancer Res 2010;16:279–90.
- Garton AJ, Crew APA, Franklin M, et al. OSI-930: a novel selective inhibitor of kit and kinase insert domain receptor tyrosine kinases with antitumor activity in mouse xenograft models. Cancer Res 2006;66:1015–24.
- Yap TA, Arkenau HT, Camidge DR, et al. First-in-human phase I trial of two schedules of OSI-930, a novel multikinase inhibitor, incorporating translational proof-of-mechanism studies. Clin Cancer Res 2013;19:909–19.
- Riesterer O, Matsumoto F, Wang L, et al. A novel Chk inhibitor, XL-844, increases human cancer cell radiosensitivity through promotion of mitotic catastrophe. Invest New Drugs 2011;29:514–22.
- Lowinger TB, Riedl B, Dumas J, et al. Design and discovery of small molecules targeting Raf-1 kinase. Curr Pharm Des 2002;8:2269–78.
- El-Aarag BYA, Kasai T, Zahran MAH, et al. In vitro anti-proliferative and anti-angiogenic activities of thalidomide dithiocarbamate analogs. Int Immunopharmacol 2014;21:283–92.
- Barnes TA, O’Kane GM, Vincent MD, et al. Third-generation tyrosine kinase inhibitors targeting epidermal growth factor receptor mutations in non-small cell lung cancer. Front Oncol 2017;7:113.
- Hoelder S, Clarke PA, Workman P. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol Oncol 2012;6:155–76.
- Lu M, Kong X, Wang H, et al. A novel microRNAs expression signature for hepatocellular carcinoma diagnosis and prognosis. Oncotarget 2017;8:8775–84.
- Nerini IF, Cesca M, Bizzaro F, et al. Combination therapy in cancer: effects of angiogenesis inhibitors on drug pharmacokinetics and pharmacodynamics. Chin J Cancer 2016;35:61
- Bates D, Eastman A. Microtubule destabilising agents: far more than just antimitotic anticancer drugs. Br J Clin Pharmacol 2017;83:255–68.
- Lu Y, Chen J, Xiao M, et al. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res 2012;29:2943–71.
- Mikstacka R, Stefański T, Różański J. Tubulin-interactive stilbene derivatives as anticancer agents. Cell Mol Biol Lett 2013;18:368–97.
- Mauer AM, Cohen EE, Ma PC, et al. A Phase II study of ABT-751 in patients with advanced non-small cell lung cancer. J Thorac Oncol 2008;3:631–6.
- Hu MJ, Zhang B, Yang HK, et al. Design, synthesis and molecular docking studies of novel indole–pyrimidine hybrids as tubulin polymerization inhibitors. Chem Biol Drug Des 2015;86:1491–500.
- Staben LR, Yu S, Chen J, et al. Stabilizing a tubulysin antibody–drug conjugate to enable activity against multidrug-resistant tumors. ACS MedChem Lett 2017;8:1037–41.
- Nguyen TL, McGrath C, Hermone AR, et al. A common pharmacophore for a diverse set of colchicine site inhibitors using a structure-based approach. J Med Chem 2005;48:6107–16.
- Zefirova ON, Diikov AG, Zyk NV, et al. Ligands of the colchicine site of tubulin:a common pharmacophore and new structural classes. Russ Chem Bull Int Ed 2007;56:680–8.
- Botta M, Forli S, Magnani M, et al. Molecular modeling approaches to study the binding mode on tubulin of microtubule destabilizing and stabilizing agents. Top Curr Chem 2009;286:279–328.
- Fortin S, Bérubé G. Advances in the development of hybrid anticancer drugs. Expert Opin Drug Discov 2013;8:1029–47.
- Albert JS, Blomberg N, Breeze AL, et al. An integrated approach to fragment-based lead generation:philosophy, strategy and case studies from AstraZeneca’s drug discovery programmes. Curr Top Med Chem 2007;7:1600–29.
- Chekler ELP, Kiselyov AS, Ouyang X, et al. Discovery of dual VEGFR-2 and tubulin inhibitors with in vivo efficacy. ACS Med Chem Lett 2010;1:488–92.
- Wilson C, Ye X, Pham T, et al. AXL inhibition sensitizes mesenchymal cancer cells to anti-mitotic drugs. Cancer Res 2014;74:5878–90.
- Elshaier YAMM, Shaaban MA, Abd El Hamid MK, et al. Design and synthesis of pyrazolo[3,4-d]pyrimidines: Nitric oxide releasing compounds targeting hepatocellular carcinoma. Bioorg Med Chem 2017;25:2956–70.
- Abdelhaleem EF, Abdelhameid MK, Kassab AE, et al. Design and synthesis of thienopyrimidine urea derivativeswithpotential cytotoxic and pro-apoptotic activity against breast cancer cell line MCF-7. Eur J Med Chem 2018;143:1807–25.
- Hu Y, Yang S, Shilliday FB, et al. Novel metabolic bioactivation mechanism for a series of anti-inflammatory agents (2,5-diaminothiophene derivatives) mediated by cytochrome P450 enzymes. Drug MetabDispos 2010;38:1522–31.
- Medower C, Wen L, Johnson WW. Cytochrome P450 oxidation of the thiophene-containing anticancer drug 3-[(quinolin-4-ylmethyl)-amino]-thiophene-2-carboxylic acid (4-trifluoromethoxy-phenyl)-amide to an electrophilic intermediate. Chem Res Toxicol 2008;21:1570–77.
- Fadda AA, Bondock S, Rabie R, et al. Cyanoacetamide derivatives as sythons in heterocyclic synthesis. Turk J Chem 2008;32:259–86.
- Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107–22.
- El-Meligie S, Taher AT, Kamal AM, et al. Design, synthesis and cytotoxic activity of certain novel chalcone analogous compounds. Eur J Med Chem 2017;126:52–60.
- Irfan M, Aneja B, Yadava U, et al. Synthesis, QSAR and anticandidal evaluation of 1,2,3-triazoles derived from naturally bioactive scaffolds. Eur J Med Chem 2015;93:246–54.
- Ravichandran S, Karthikeyan E. Microwave synthesis-a potential tool for green chemistry. Int J Chemtech Res 2011;3:466–70.
- Stefanidis G and Stankiewicz AJ. Energy sources for green chemistry Alternative Energy Sources for Green Chemistry,1st ed. London:The Royal Society of Chemistry; 2016:1–3.
- Gewald K, Hain U, Schmidt M. Substituierte 3,4-diamino-thieno[2,3-b]pyrrole. J Prakt Chem 1986;328:459–64.
- Raslan MA, Sayed SM. Studies with Heterocyclic -Enaminoniriles: a simple route for the synthesis of polyfunctionally substituted thiophene, Imidazo[1,2:1,6]pyrimido[5,4-b]thiophene and Thieno[3,2-d]pyrimidine. J Chin Chem Soc 2003;50:909.
- Santilli AA, Kim DH, Wanser SV. Synthesis of pyrimido[4,5-e][1,4]oxazepin-5-ones. J Heterocyclic Chem 1972;9:309–453.
- Ho YW, Yao WH. Synthesis and characterization of new pyrimidine based 1,3,4-oxa(thia)diazoles, 1,2,4-triazoles and 4-thiazolidinones. Dyes and Pigments 2009;2009:281.
- Mabkhoot YN. Synthesis and chemical characterisation of new bis-thieno [2,3-b]thiophene derivatives. Molecules 2010;15:3329–27.
- Tolba MF, Esmat A, Al-Abd AM, et al. Caffeic acid phenethyl ester synergistically enhances docetaxel and paclitaxel cytotoxicity in prostate cancer cells. Int Union Biochem Mol Biol 65 2013;65:716–29.
- Hassan M, Watari H, AbuAlmaaty A, et al. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int 2014;2014:150845.
- Brentnall M, Rodriguez-Menocal L, De Guevara RL, et al. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol 2013;14:32.
- Roy K, Kar S, Narayan Das R. Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment. 1st ed. 2015;70–4.