
Abstract
Bile acids have been shown to inhibit human (h) carbonic anhydrases (CA, EC 4.2.1.1) along the gastrointestinal tract, including hCA II. The elucidation of the hormonal inhibition mechanism of the bile acid cholate to hCA II was provided in 2014 by X-ray crystallography. Herein, we extend the inhibition study to a wealth of steroids against four relevant hCA isoforms. Steroids displaying pendants and functional groups of the carboxylate, phenolic or sulfonate types appended at the tetracyclic ring were shown to inhibit the cytosolic CA II and the tumor-associated, transmembrane CA IX in a medium micromolar range (38.9–89.9 µM). Docking studies displayed the different chemotypes CA inhibition mechanisms. Molecular dynamics (MD) gave insights on the stability over time of hyocholic acid binding to CA II.
Introduction
Steroids encompass a great variety of structurally related compounds that are widely distributed in the animal and plant kingdomCitation1. The common chemical feature among the diverse classes is constituted by a perhydrocyclopentanophenanthrene nucleus. Steroids include crucial compounds for life as cholesterol, bile acids, and sex hormones that play several physiological responses mediated by both genomic and non-genomic actionsCitation2. A variety of natural and semi-synthetic steroids are used in therapy as anti-inflammatory, immunosuppressive, anabolic, and contraceptive agents, as well as for the prevention of coronary disease, and for the management of diabesity and declared AIDS. Remarkably, steroids are a wealthy source of therapeutic agents for specific forms of cancerCitation3: indeed, they act as aromatase and sulfatase modulators against breast cancer, and as 5α-reductase and CYP17 inhibitors to treat benign prostatic hyperplasia and advanced prostate cancer, respectively. Additionally, semi-synthetic steroidal derivatives are very relevant for the discovery of chemical probes for exploring molecular mechanisms of action of understudied biological targets and pathwaysCitation4–6. In this paper, we aim to determine the activity of a set of major naturally-occurring steroids ( and ) as carbonic anhydrase inhibitors (CA, EC 4.2.1.1).
Figure 1. Structures of bile acids 1–10.
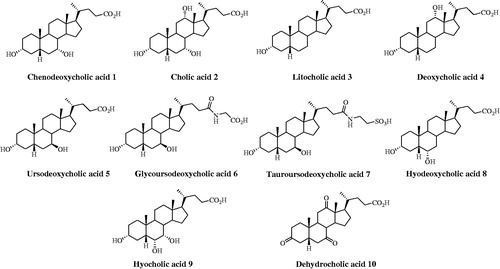
Figure 2. Structures of steroids 11–22.
Carbonic anhydrases consist in a superfamily of zinc enzymes which catalyze the reversible hydration of CO2 into HCO3 and protons by a metal hydroxide nucleophilic mechanismCitation7,Citation8. Seven genetically distinct CA families (α-, β-, γ-, δ-, ζ-. η- and θ-CAs.) are known to dateCitation7–10 .The 15 different α-CA isoforms isolated in humans (h) feature catalytic activity, sub-cellular localization and organ/tissue distribution. Among the catalytically active isoforms, some are cytosolic (CA I, CA II, CA III, CA VII and CA XIII), others are membrane bound (CA IV, CA IX, CA XII, CA XIV and CA XV), two of them are mitochondrial (CA VA and CA VB), and one isozyme is secreted in saliva (CA VI)Citation7. A variety of human patho-physiological processes shows abnormal levels or activities of these enzymes, and this makes CA isozymes valuable targets for many pharmacological applications such as antiglaucoma drugs, diuretics, antiobesity, anticonvulsant and/or antitumor agents/diagnostic toolsCitation7. Several hCAs along the gastrointestinal tract, including hCA II, have been shown to be inhibited by the 5β-steroids bile acidsCitation11, the primary end-products of cholesterol catabolism, resulting in a damage of the gastric mucosa. The damage produced mainly by primary bile acids and their conjugates has been related with gastric mucosal CAs inhibition in rats and humansCitation12. The structural evidence of the hormonal inhibition mechanism of the bile acid cholate to hCA II has been given in 2014Citation13. The carboxylate was found to bind to the zinc ion in a bivalent manner displacing the zinc-bound solvent molecule.
Herein, we extend the inhibition study to a wealth of steroids against 4 relevant hCA isoforms. Beyond understanding the interference of the CA activity by steroids, the knowledge of the CA inhibition profiles of several such derivatives could be of interest and drive the design of new non-sulfonamide-like compounds, that can be easily transported across the cellular membrane.
Experimental section
Steroids
Chenodeoxycholic acid (1), ursodeoxycholic acid (5), hyodeoxycholic acid (8), hyocholic acid (9), coprostan-3-ol (12), trans-dehydroandrosterone (15), progesterone (17), 11α-hydroprogesterone (18), α-estradiol (19), and diosgenin (22) were purchased from Sigma-Aldrich. Cholic acid (2), lithocholic acid (3), deoxycholic acid (4), cholesterol (11), testosterone (13), and oestron (20) were purchased from Fluka. Dehydrocholic acid (10) and hydrocortisone (21) were purchased from Janssen Chimica and BDH Chemicals, respectively. Glyco (6) and tauroursodeoxycholic acid (7) were prepared as previously reportedCitation14,Citation15. Purity of tested compounds 1–22 was >95%.
Carbonic anhydrase inhibition
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activityCitation16. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.4) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation17–19, and represent the mean from at least three different determinations. All CA isofoms were recombinant ones obtained in-house as reported earlierCitation20,Citation21.
Computational studies
4E3H26 and 4PXX13 crystal structures was prepared according to the Protein Preparation module in Maestro – Schrödinger suite, assigning bond orders, adding hydrogens, deleting water molecules, and optimizing H-bonding networksCitation22. Finally, energy minimization with a root mean square deviation (RMSD) value of 0.30 was applied using an Optimized Potentials for Liquid Simulation (OPLS_2005) force field. 3 D ligand structures were prepared by MaestroCitation22a and evaluated for their ionization states at pH 7.4 ± 0.5 with EpikCitation22b OPLS-2005 force field in MacromodelCitation22e was used for energy minimization for a maximum number of 2500 conjugate gradient iteration and setting a convergence criterion of 0.05 kcal mol−1 Å−1. The docking grid was generated using GlideCitation22f with default settings, with the center located on the center of mass of the cocrystallized ligand. Ligands were docked employing the standard precision mode (SP) retaining the best five poses of each molecule as output. The top ranked binding pose of each compound was then analyzed in terms of coordination, hydrogen bond interactions and hydrophobic contacts.
The best scored binding pose of 9 to the CA II active site was submitted to a MD simulation using DesmondCitation23 and the OPLS2005 force field. Specifically, the system was solvated in an orthorhombic box using TIP4PEW water molecules, extended 15 Å away from any protein atom. Then, it was neutralized adding a concentration of 0.15 M chlorine and sodium ions. The simulation protocol included a starting relaxation step and a final production phase of 10 ns. In particular, the relaxation step comprised the following: (a) a stage of 100 ps at 10 K retaining the harmonic restraints on the solute heavy atoms (force constant of 50.0 kcal mol−1 Å−2) using the NPT ensemble with Brownian dynamics; (b) a stage of 12 ps at 10 K with harmonic restraints on the solute heavy atoms (force constant of 50.0 kcal mol−1 Å−2), using the NVT ensemble and Berendsen thermostat; (c) a stage of 12 ps at 10 K and 1 atm, retaining the harmonic restraints and using the NPT ensemble and Berendsen thermostat and barostat; (f) a stage of 12 ps at 300 K and 1 atm, retaining the harmonic restraints and using the NPT ensemble and Berendsen thermostat and barostat; (g) a final 24 ps stage at 300 K and 1 atm without harmonic restraints, using the NPT Berendsen thermostat and barostat. The final production phase of MD was run using a canonical the NPT Berendsen ensemble at temperature 300 K. During the MD simulation, a time step of 2 fs was used while constraining the bond lengths of hydrogen atoms with the M-SHAKE algorithm. The atomic coordinates of the system were saved every 50 ps along the MD trajectory. The occupancy of intermolecular hydrogen bonds and hydrophobic contacts was calculated along the production phase of the MD simulation with the Simulation Interaction Diagram tools implemented in Maestro. MD snapshots were clustered with the script Cheminformatics – Clustering of Conformers from Schrodinger, using the average linkage clustering method based on the RMSD matrix between the conformers Cartesian coordinates – non hydrogen atoms only.
Results and discussion
Biological activity
Among the twelve catalytically active hCAs, the isoforms chosen for our studies involved the cytosolic hCA I and II (involved in a host of physiologic processes)Citation7, the membrane-bound hCA IV (involved in glaucoma, retinitis pigmentosa, stroke and rheumatoid arthritis)Citation24,Citation25 and the tumor-associated hCA IX (abundant in hypoxic tumors and recently validated as antitumor target)Citation26,Citation27.
Inhibition data of steroids 1–22 against hCA I, II, IV and IX were measured by a stopped flow CO2 hydrase assay and are shown in Citation16. Acetazolamide, a clinically used sulfonamide inhibitor, was used as standard.
Table 1. Inhibition data of human CA isoforms hCA I, II, IV and IX with compounds reported here and the standard sulfonamide inhibitor acetazolamide (AAZ) by a stopped flow CO2 hydrase assay.
The data of show that a basic requirement to address a though weak inhibitory efficacy to steroidal derivatives is the presence of a functional moiety that plays the role of zinc-binding or anchoring group to the metal-coordination center, i.e. carboxylates, sulfonates and phenols. Unlike this latter, it should be stressed that analog derivatives bearing OH moieties of the aliphatic type do not exhibit any inhibitory efficacy.
Nevertheless, the cytosolic hCA I and the membrane-bound hCA IV are inhibited by none of the assayed derivatives below 100 µM, except steroids 5, 9 and 19 which feebly affect hCA I activity with inhibition constants (KIs) of 95.9, 83.3, 87.8 µM, respectively.
The ubiquitous hCA II and tumor-associated hCA IX were comparably inhibited by carboxylic acids 1–6, 8–10, sulfonic acid 7 and phenols 19, 20 in the micromolar range spanning between 38.9 and 89.9 µM. Tauroursodeoxycholic acid (7) stands out as the most efficient hCA IX inhibitor, being instead its action the least efficient against hCA II. Repositioning of the alcoholic moieties mainly located at the outer edge of the molecular structures has been shown to slightly alter the weak inhibition profiles. The lengthening of the carboxyalkyl chain of ursodeoxycholic acid (5) by a glycine unit as in 6 does not affect the derivatives efficacy against both considered isoforms. Reduction of the enone system at ring A of testosterone (13) to 5α-steroids androstanolone (16) and androsterone (14) does not interfere with the hCA II and IX inhibitory efficacy.
Computational studies
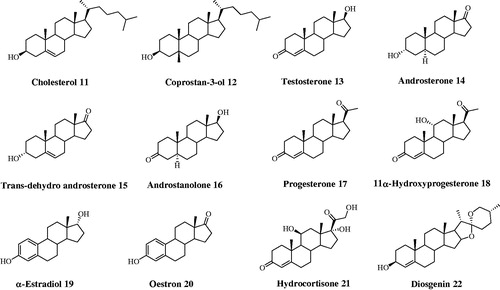
According to the binding mechanism of the different class of CAI (phenols, sulfonates, and carboxylates) herein studiedCitation28–35, docking simulations were carried out on representative steroids with hCA II, namely 7, 9, 19. Sulfonate (7) and compounds bearing a phenol group (19, 20) act as zinc-bound nucleophile anchoring group, whereas carboxylates (1–6, 8–10) bind directly to the catalytic Zn ion (zinc binders). It was found that the phenolic OH of α-estradiol (19) is H-bonded to the zinc-bound hydroxide ion, that is in turn stabilized by three other H-bonds, acting as donor to Thr199 and Thr200 side chain OH and as acceptor with the Thr199 backbone NH. Noteworthy, the phenolic portion of α-estradiol anchors to the nucleophile locating more externally than cocrystalized simple ligands such as hydroquinone (PDB code 4E3H), owing to the steric hindrance lined by the tetrahydrophenantrenic core. This latter is involved in π-π and π-alkyl interactions with Leu198, Thr200, Phe131, Val121, His94, Gln92, Asn67, Asn62, and Leu60 side chain. Furthermore, the docked pose features an H-bond between the alcoholic moiety of 19 and the Asn67 side chain carbonyl group. The terminal sulfonate group of tauroursodeoxycholic acid (7) anchors to the pseudo-tetracoordinated zinc-bound water molecule by a five H-bonds network involving the ligand, the nucleophile and the enzyme (Thr199 and Thr200). Further H-bonds stabilize the docked pose. The carboxy amidic moiety acts as acceptor by the amidic NH2 of Gln92, the alcoholic function in C7 position acts as donor to Ile91 and Phe70 backbone carboxy and NH group respectively and the C3-OH donates a H-bonds to Glu69 side chain. Like 19, the tetrahydrophenantrenic ring of 7 was involved in Van der Waals interactions with Val143, Val207, Trp209, Leu198, Thr199, Thr200, His94, His119, Gln92, Phe131, Val121, Ile91, Glu69, and Arg58 residues ().
Figure 3. Dockings of (A) α-estradiol (19) and (B) tauroursodeoxycholic acid (7) within hCA II. (C) Superposed docked hyocholic acid (9) (blue) and cholic acid (2) (yellow) X-ray solved orientation within hCA II.
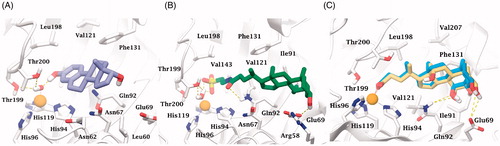
According to the X-ray solved structure (PDB code 4PXX) of cholic acid (2) in complex with hCA IICitation13, the carboxylic group of the docked hyocholic acid (9) directly coordinates the zinc ion in a bidentate manner, and showing a very good agreement with the positioning of the crystallographic complex ligand. Thorough analogies between cholic acid (2) crystallography and hyocholic acid (9) docking with hCA II were found. As in the X-ray solved structure, the coordination is further sustained by a H-bond occurring between the backbone NH of Thr199 and the carboxylate group. The alcoholic function in C7 accepts an H-bond from Gln92 side chain NH2, and further stabilization of the pose comes from hydrophobic interactions taking place between the tetrahydrophenantrenic core and His94, His96, His119, Thr199, Thr200, Leu198, Pro201, Val121, Gln92, Phe131 and Ile91 residues. Unlike cholic acid (2), the hydroxy group at C3 position of 9 is not in H-bond contact with Glu69.
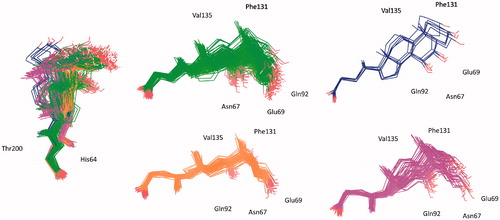
The statistical stability of the docked pose of hyocholic acid (9) as well as the stability of the H-bonds network involving the hydroxy moieties has been studied by analyzing the ligand conformation and the positional changes upon 10 ns molecular dynamics (MD) simulation of 9 docked to hCA II, as starting point. The blue line in shows how the protein C-alphas evolve over the 10 ns MD period and it indicates that the system is equilibrated. The green line, representing the aligned-ligand RMSD (only heavy atoms), gives insights onto how compound 9 is stable with respect to the enzyme binding pocket. The bidentate coordination is stably maintained (). The H-bond that is established either directly or mediated by a water molecule between the C7-OH and the Gln92 side chain NH2 displayed a 60% stability over 10 ns, whereas analog interaction taking place between the C3-OH and Glu69 side chain is maintained for 4 ns (40% of the overall dynamics simulation time) (). The inspection of the MD trajectory points out interesting conformational transitions taking place in between 3 and 7 ns of the overall 10 ns time scale. Cluster analysis may help in identifying the relevant internal fluctuations which occur within the ligand structure during the simulation (). Four main conformer families were found which substantially shares the zinc binding mode and differ for the orientation of the methyl group on C21 (orientation A and B according to the side - His64 or Thr200 - towards which the methyl group points to) and the hydrophobic or hydrophilic portion of hCA II occupied by the polycyclic scaffold (). The percentage (about 60%) of time spent by 9 in the docking found conformation testifies the stability of the pose.
Figure 4. Analysis of the MD simulation of 9 docked to hCA II. (A) Coordination and H-bonds occupancies within 10 ns MD for 9 - hCA II complex. (B) Rmsd representation of the heavy atoms of the receptor and the ligand from the starting model structure during the simulation.
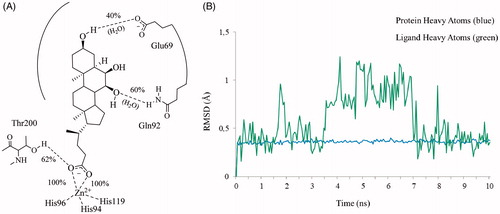
Figure 5. Superposed representative orientations of the four identified clusters within superposed protein backbones of 200 frames of MD.
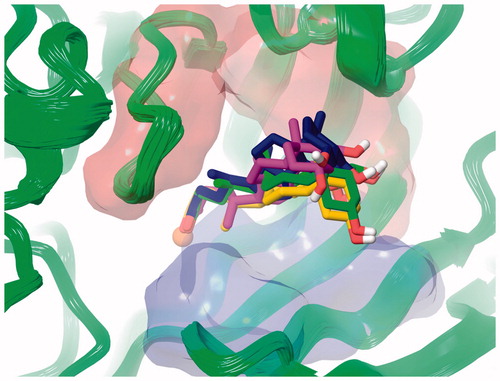
Figure 6. Conformer families of 9 identified over the 10 ns MD period.
Conclusions
Bile acids are known to inhibit hCAs along the gastrointestinal tract, including hCA IICitation11. The structural elucidation of the hormonal inhibition mechanism of the bile acid cholate to hCA II was furnished in 2014. The present study reports inhibition studies of physio-pathologically relevant CAs with a set of selected steroids. Steroids displaying pendants and functional groups of the carboxylate, phenolic or sulfonate types appended at the tetracyclic ring were shown to inhibit the cytosolic CA II and the tumor-associated, transmembrane CA IX in a medium micromolar range. Since the aforementioned functional groups are known to exert a CA inhibitory action by different binding mechanism, computational studies on representative derivatives were undertaken. A 10 ns MD gave insights on the stability of hyocholic acid (9) binding to CA II. Beyond understanding the interference of the CA activity by steroids, the knowledge of the CA inhibition profiles of such derivatives could be of interest and drive the design of novel steroidal non-sulfonamide-like CA inhibitors.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Zeelen FJ. Medicinal chemistry of steroids. In: Timmerman H, ed. Pharmacochemistry Library. Amsterdam: Elsevier; 1997:1–357.
- Falkenstein E, Tillmann HC, Christ M, et al. Multiple actions of steroid hormones–a focus on rapid, nongenomic effects. Pharmacol Rev 2000;52:513–56.
- Salvador JA, Carvalho JF, Neves MA, et al. Anticancer steroids: linking natural and semi-synthetic compounds. Nat Prod Rep 2013;30:324–74.
- Bucci M. Plant development: get lit on steroids. Nature Chem Biol 2017;13:569.
- Hanson JR. Steroids: partial synthesis in medicinal chemistry. Nat Prod Rep 2007;24:1342–9.
- Djerassi C. A steroid autobiography-Carl Djerassi. Steroids 1984;43:351–61.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discovery 2008;7:168–81.
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Lane TW, Saito MA, George GN, et al. Biochemistry: a cadmium enzyme from a marine diatom. Nature 2005;435:42.
- Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum-the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96.
- Milov DE, Jou WS, Shireman RB, et al. The effect of bile salts on carbonic anhydrase. Hepatology 1992;15:288–96.
- Kivilaakso E. Inhibition of gastric mucosal carbonic anhydrase by taurocholic acid and other ulcerogenic agents. Am J Surg 1982;144:554–7.
- Boone CD, Tu C, McKenna R. Structural elucidation of the hormonal inhibition mechanism of the bile acid cholate on human carbonic anhydrase II. Acta Crystallogr D Biol Crystallogr 2014;70:1758–63.
- Mostarda S, Filipponi P, Sardella R, et al. Glucuronidation of bile acids under flow conditions: design of experiments and Koenigs-Knorr reaction optimization. Org Biomol Chem 2014;12:9592–600.
- Mostarda S, Passeri D, Carotti A, et al. Synthesis, physicochemical properties, and biological activity of bile acids 3-glucuronides: Novel insights into bile acid signalling and detoxification. Eur J Med Chem 2018;144:349–58.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and CJ Biol Chem 1971;246:2561–73.
- Nocentini A, Cadoni R, Del Prete S, et al. Benzoxaboroles as Efficient Inhibitors of the β-Carbonic Anhydrases from Pathogenic Fungi: Activity and Modeling Study. ACS Med Chem Lett 2017;8:1194–8.
- Vullo D, Del Prete S, Nocentini A, et al. Dithiocarbamates effectively inhibit the β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa. Bioorg Med Chem 2017;25:1260–5.
- Nocentini A, Bua S, Lomelino CL, et al. Discovery of New Sulfonamide Carbonic Anhydrase IX Inhibitors Incorporating Nitrogenous Bases. ACS Med Chem Lett 2017;8:1314–9.
- Entezari Heravi Y, Bua S, Nocentini A, et al. Inhibition of Malassezia globosa carbonic anhydrase with phenols. Bioorg Med Chem 2017;25:2577–82.
- Ibrahim HS, Allam HA, Mahmoud WR, et al. Dual-tail arylsulfone-based benzenesulfonamides differently match the hydrophobic and hydrophilic halves of human carbonic anhydrases active sites: Selective inhibitors for the tumor-associated hCA IX isoform. Eur J Med Chem 2018;152:1–9.
- Schrodinger Suite Release 2016-1, Schrodinger, LLC, New York, NY, 2016: a) Maestro v.10.5; b) Epik v.3.5; c) Impact v.7.0; d) Prime v.4.3; e) Macromodel v.11.1; f) Glide v.7.0. Available from: https://www.schrodinger.com [last accessed 24 Jul 2018].
- Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2016. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2016. Available from: https://www.schrodinger.com. [last accessed 15 Jul, 2018].
- Tang Y, Xu H, Du X, et al. Gene expression in blood changes rapidly in neutrophils and monocytes after ischemic stroke in humans: a microarray study. J Cereb Blood Flow Metab 2006;26:1089–102.
- Liu C, Wei Y, Wang J, et al. Carbonic anhydrases III and IV autoantibodies in rheumatoid arthritis, systemic lupus erythematosus, diabetes, hypertensive renal disease, and heart failure. Clin Dev Immunol 2012;2012:1.
- a) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77; b) Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev 2018; in press. doi:10.1002/med.21497.
- a) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88; b) Capasso C, Supuran CT. An overview of the alpha-beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Ch 2015;30:325–32; c) Supuran CT. Carbon-versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Ch 2018;33:485–95.
- a) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60; b) Supuran C.T., Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- Simonsson I, Jonsson BH, Lindskog S. Phenol, a competitive inhibitor of CO2 hydration catalyzed by carbonic anhydrase. Biochem Biophys Res Commun 1982;108:1406–12.
- a) Karioti A, Carta F, Supuran CT. Phenols and Polyphenols as Carbonic Anhydrase Inhibitors. Molecules 2016;21:E1649; b) Abbate F, Winum JY, Potter BV, Casini A, Montero JL, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with EMATE, a dual inhibitor of carbonic anhydrases and steroid sulfatase. Bioorg Med Chem Lett 2004;14:231–4.
- a) Nocentini A, Moi D, Balboni G, et al. Discovery of thiazolin-4-one-based aromatic sulfamates as a new class of carbonic anhydrase isoforms I, II, IV, and IX inhibitors. Bioorg Chem 2018;77:293–99; b) Casey JR, Morgan PE, Vullo D, Scozzafava A, Mastrolorenzo A, Supuran CT. Carbonic anhydrase inhibitors. Design of selective, membrane-impermeant inhibitors targeting the human tumor-associated isozyme IX. J Med Chem 2004;47:2337–47.
- a) Nocentini A, Bua S, Del Prete S, et al. Natural Polyphenols Selectively Inhibit β-Carbonic Anhydrase from the Dandruff-Producing Fungus Malassezia globosa: Activity and Modeling Studies. ChemMedChem 2018;13:816–23; b) Menchise V, De Simone G, Alterio V, et al. Carbonic anhydrase inhibitors: stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J Med Chem. 2005;48:5721–27; c) Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors—part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents in rabbits. Eur J Med Chem 1998;33:247–54; d) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8; e) Şentürk M, Gülçin İ, Beydemir Ş, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77: 494-99; f) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47.
- a) Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56(1):293–300; b) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–38; c) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3; d) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94; e) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11; f) Alper Türkoğlu E, Şentürk M, Supuran CT, Ekinci D. Carbonic anhydrase inhibitory properties of some uracil derivatives. J Enzyme Inhib Med Chem 2017;32:74–7; g) Soydan E, Güler A, Bıyık S, et al. Carbonic anhydrase from Apis mellifera: purification and inhibition by pesticides. J Enzyme Inhib Med Chem 2017;32:47–50
- a) Lomelino CL, Supuran CT, McKenna R. Non-Classical Inhibition of Carbonic Anhydrase. Int J Mol Sci 2016;17:E1150; b) Nishimori I, Minakuchi T, Morimoto K, et al. Carbonic anhydrase inhibitors: DNA cloning and inhibition studies of the alpha-carbonic anhydrase from Helicobacter pylori, a new target for developing sulfonamide and sulfamate gastric drugs. J Med Chem 2006;49:2117–26; c) Supuran CT. Carbonic anhydrase inhibitors: an editorial. Expert Opin Ther Pat 2013;23:677–9.
- Martin DP, Cohen SM. Nucleophile recognition as an alternative inhibition mode for benzoic acid based carbonic anhydrase inhibitors. Chem Commun (Camb) 2012;48:5259–61.