
Abstract
The treatment of neuropathic pain is one of the urgent unmet medical needs and T-type calcium channels are promising therapeutic targets for neuropathic pain. Several potent T-type channel inhibitors showed promising in vivo efficacy in neuropathic pain animal models and are being investigated in clinical trials. Herein we report development of novel pyrrolidine-based T-type calcium channel inhibitors by pharmacophore mapping and structural hybridisation followed by evaluation of their Cav3.1 and Cav3.2 channel inhibitory activities. Among potent inhibitors against both Cav3.1 and Cav3.2 channels, a promising compound 20n based on in vitro ADME properties displayed satisfactory plasma and brain exposure in rats according to in vivo pharmacokinetic studies. We further demonstrated that 20n effectively improved the symptoms of neuropathic pain in both SNL and STZ neuropathic pain animal models, suggesting modulation of T-type calcium channels can be a promising therapeutic strategy for the treatment of neuropathic pain.
Introduction
Neuropathic pain is chronic pain originated from lesions or diseases of the peripheral or central somatosensory nervous systemCitation1. It is usually associated with unpleasant feeling such as a burning, abnormal crawling, or electric shock-like sensations and is further characterised by evoked pain after stimuli such as hypersensitivity and summation. Current therapeutic options for neuropathic pain include anti-convulsants such as sodium and calcium channel blockers, anti-depressants, opioids, NSAIDs, cannabinoids, topical agents, and combinations of these drugsCitation2,Citation3. However, administration of these drugs only reduce 30–50% of pain in approximately 50% of neuropathic pain patientsCitation4,Citation5. This low efficacy issue can be attributed to the substantial heterogeneity of pain mechanisms, and requests precise elucidation of underlying mechanisms for different types of neuropathic pain in order to improve both diagnosis and prescription. Thus, identification of potential biomarkers for each type of neuropathic pain and subsequent development of chemical probes that can modulate the biomarkers will provide exciting opportunities to better understand the heterogeneity of neuropathic pain and produce efficacious pain drugsCitation6,Citation7.
One of the encouraging strategies to treat neuropathic pain is modulation of intracellular calcium levelsCitation8,Citation9 either directly by controlling voltage-gated calcium channels or indirectly by regulating receptors such as nicotinic acetylcholine receptorsCitation10,Citation11. Among several voltage-gated calcium channels, T-type calcium channels have received great attention as one of the promising molecular targets for neuropathic painCitation12,Citation13. These are low voltage-activated (LVA) channels composed of α1 subunit single pore, and three isoforms of T-type calcium channels have been identifiedCitation14: Cav3.1 (α1G)Citation15, Cav3.2 (α1H)Citation16, Cav3.3 (α1I)Citation17. In contrast to high voltage-activated (HVA) channels, T-type calcium channels are activated even after a slight depolarisation of the cell membrane, functioning at near-resting membrane potentialsCitation18. Due to this unique voltage sensitivity, T-type calcium channels can regulate neuronal excitability and oscillatory behaviourCitation19.
Among T-type calcium channels, Cav3.2 is the major isoform expressed in primary sensory afferent neurons and spinal cordCitation20, and it has been strongly implicated that Cav3.2 channel contributes to the development of neuropathic painCitation12. For example, in a neuropathic pain rat model derived by a chronic constriction injury (CCI), the expression of Cav3.2 channel is highly up-regulated, and delivery of Cav3.2-specific antisense oligonucleotide generated robust pain-relieving effectsCitation20. Furthermore, in a STZ-induced neuropathic pain model, the level of Cav3.2 expression is also increased and specific in vivo knock-down of Cav3.2 effectively alleviated mechanical and thermal hypersensitivityCitation21. In addition, Cav3.1 channels are mainly expressed in dorsal horn neuronsCitation22, and its significance in neuropathic pain is also well reported. It was demonstrated that after spinal nerve ligation (SNL) CaV3.1 knock-out mice showed a higher threshold to mechanical stimuli than their wild-type litter mates, and thermal hypersensitivity was also substantially decreasedCitation23. Furthermore, the mechanical hypersensitivity was markedly attenuated after induction of trigeminal neuropathy in the CaV3.1 knock-out mice as compared to wild type miceCitation24.
Based on these observations, it was believed that T-type calcium channel inhibitors would provide effective treatment options for neuropathic painCitation13, leading to development of a variety of T-type calcium channel blockers, such as MibefradilCitation25, ethosuximideCitation26, and (3β,5α,17β)-17-hydroxyestrane-3-carbonitrile (ECN)Citation27. In fact, treatment of Mibefradil or ethosuximide in the rat CCI model relieved behavioural symptoms of neuropathic painCitation28. It was shown that ECN also alleviated mechanical and thermal hypersensitivity in rats with neuropathic painCitation29 and in diabetic ob/ob miceCitation30. Although these early T-type channel inhibitors showed great promise for the treatment of neuropathic pain, however, they suffered relatively low selectivity against other ion channels, especially voltage-gated sodium channels in neurons raising adverse side effect issuesCitation25,Citation31,Citation32. Moreover, Mibefradil was withdrawn just 1 year after the FDA approval due to drug–drug interactionsCitation33.
Thus, development of selective T-type channel inhibitors is highly desired to treat neuropathic pain with minimal side effects. Recently, selective T-type channel inhibitors such as R-(-)-efonidipineCitation34, TTA-P2Citation35, ML-218Citation36, ABT-639Citation37, Z-944Citation38, and benzodiazepine- and dihydropyrazole-based Actelion inhibitorsCitation39,Citation40 have been developed, and some of them presented robust analgesic effects in several neuropathic pain modelsCitation41,Citation42, illustrating selective T-type calcium channel inhibitors hold strong potential to be effective treatment options for neuropathic pain.
Herein we describe our ongoing efforts to develop selective T-type channel inhibitors, identifying pyrrolidine derivatives as potent T-type channel inhibitors after evaluation of Cav3.1 (α1G) and Cav3.2 (α1H) T-type calcium channel blocking activities with fluorescence-based FDSS6000 assayCitation43 and whole-cell patch clamp recordingsCitation44. Pharmacokinetic parameters of a promising compound 20n and its in vivo analgesic efficacy in two different neuropathic pain models are investigated.
Materials and methods
Chemistry
Commercially available reagents and solvents were used without further purification. Air- and moisture-sensitive reactions were performed under a positive pressure of nitrogen. All reactions were monitored by analytical thin layer chromatography (TLC) using glass pates pre-coated with silica gel from Merck (0.25 mm, 60 Å pore-size) impregnated with a fluorescent indicator (254 nm). Visualisation of TLC was carried out by ultraviolet light (λ = 254 and 365 nm). Flash column chromatography was performed on Merck silica gel 60 (70–230 mesh). 1H NMR were recorded on Bruker AVANCE II 400 (400 MHz), 300 (300 MHz) NMR spectrometers at 23 °C. Proton chemical shifts are expressed in parts per million (ppm, δ scale) and are referenced to residual protium in the NMR solvent (CDCl3, δ 7.26; DMSO-d6, δ 2.50). Peak splitting patterns are presented as follows: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet. Coupling constants (J) are recorded in hertz (Hz) values. 13C NMR spectra were recorded on Bruker AVANCE II 400 (100 MHz), 300 (75 MHz) NMR spectrometers at 23 °C. Carbon chemical shifts are expressed in parts per million (ppm, δ scale) and are referenced to the carbon resonances of the NMR solvent (CDCl3, δ 77.0; DMSO-d6, δ 39.50). High-resolution mass spectra (HRMS) were recorded on a LTQ Orbitrap (Thermo Electron Corporation). Purity was measured using Waters e2695/2489 HPLC instrument (1 ml/min flow rate; H2O/CH3CN 90:10 → 0:100 in 15 min; 10 μL of injection volume; λ = 254, 280 nm).
Synthesis of (S)-5-isobutyl-1-phenyl-N-(pyrrolidin-3-ylmethyl)-1H-pyrazole-3-carboxamide (15)
(R)-tert-Butyl 3-((5-isobutyl-1-phenyl-1H-pyrazole-3-carboxamido)methyl)pyrrolidine-1-carboxylate 14 (2.93 g, 6.87 mmol) in anhydrous DCM (15 ml) was treated with trifluoroacetic acid (10 ml) and stirred at room temperature for 2 h. The reaction mixture was evaporated and neutralised by adding an aqueous solution of saturated sodium bicarbonate. The aqueous layer was extracted with DCM, dried over sodium sulfate and concentrated in vacuo. The desired compound (2.22 g) was obtained. Yield 99%; 1H NMR (400 MHz, CDCl3) δ: 7.54–7.40 (m, 5H), 6.75 (s, 1H), 3.45 (d, J = 7.2 Hz, 2H), 3.30–3.24 (br, 2H), 3.17–3.14 (m, 1H), 3.00 (dd, J = 11.6, 6.4 Hz, 1H), 2.63–2.56 (m, 1H), 2.51 (d, J = 7.2 Hz, 2H), 2.10–2.05 (m, 1H), 1.88–1.81 (m, 1H), 1.73–1.68 (m, 1H), 0.87 (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.89, 146.04, 145.22, 139.16, 129.27, 128.84, 125.84, 106.26, 48.29, 44.70, 40.67, 38.65, 34.93, 28.19, 22.20; HPLC purity 100%; HR-MS m/z [M + H]+ (ESI+) calcd. for C19H27N4O = 327.2179, found 327.2180.
Synthesis of (R)-N-((1-(3,3-dimethylbutyl)pyrrolidin-3-yl)methyl)-5-isobutyl-1-phenyl-1H-pyrazole-3-carboxamide (16)
(S)-1-methyl-3-propyl-N-(pyrrolidin-3-ylmethyl)-1H-pyrazole-5-carboxamide 15 (100 mg, 0.40 mmol) and 3,3-dimethylbutanal (50 μL, 0.40 mmol) were dissolved in anhydrous DCM. Molecular sieves were added to the mixture. The mixture was stirred at room temperature for 2 h. NaBH(OAc)3 (254 mg, 1.20 mmol) was added to the mixture. The reaction was stirred at room temperature for 10 h. It was quenched with an aqueous solution of saturated sodium bicarbonate, water and extracted with DCM. The residue was dried over magnesium sulfate and concentrated in vacuo. The residue was purified via column chromatography on silica gel (DCM/MeOH =10:1) to obtain the desired compound (16 mg). Yield: 12%; 1H NMR (300 MHz, CDCl3) δ: 7.52–7.47 (m, 3H) 7.43–7.40 (m, 2H) 6.75 (s, 1H) 3.50–3.44 (m, 2H) 2.80–2.52 (m, 7H) 2.51 (d, J = 7.2 Hz, 2H) 2.14 (m, 1H) 1.84 (m, 1H) 1.69 (m, 1H) 1.51 (m, 2H) 0.89 (s, 9H) 0.86 (d, J = 6.78 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ: 162.91, 146.15, 145.29, 139.27, 129.32, 128.83, 125.90, 106.51, 57.09, 53.21, 52.54, 41.66, 39.25, 37.78, 35.09, 29.76, 29.18, 28.32, 27.58, 22.32.
General procedure for the preparation of compounds 20a-p
The pyrrolidine 15 and potassium carbonate in acetonitrile was stirred at room temperature for 30 min. The tosylate 19a–p in acetonitrile was added dropwise and refluxed for 24 h. The reaction mixture was cooled to room temperature, quenched with water and extracted with ethyl acetate. It was dried over sodium sulfate and concentrated in vacuo. The residue was purified via column chromatography on silica gel, and the desired compound was obtained.
(R)-5-Isobutyl-1-phenyl-N-((1-(4-(trifluoromethyl)phenethyl)pyrrolidin-3-yl)meth-yl)-1H-pyrazole-3-carboxamide (20a)
Yield 72%; 1H NMR (300 MHz, CDCl3) δ: 7.52–7.40 (m, 8H) 7.28–7.24 (m, 2H) 6.78 (s, 1H) 3.48–3.43 (m, 2H) 2.88–2.83 (m, 2H) 2.76–2.65 (m, 4H) 2.56–2.47 (m, 5H) 2.08–2.02 (m, 1H) 1.90–1.81 (m, 1H) 1.63–1.57 (m, 1H) 0.88 (d, J = 6.60 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ: 162.98 146.09 145.37 141.22 139.26 129.60 129.34 129.11 128.88 125.90 125.77 125.67 122.27 106.4857.15 56.65 53.53 41.65 37.93 35.08 32.60 28.32 27.61 22.32.
(R)-5-Isobutyl-1-phenyl-N-((1-(3-(trifluoromethyl)phenethyl)pyrrolidin-3-yl)methy-l)-1H-pyrazole-3-carboxamide (20b)
Yield 35%; 1H NMR (300 MHz, CDCl3) δ: 7.51–7.30 (m, 9H), 6.79 (s, 1H), 3.47–3.38 (m, 2H), 2.87–2.82 (m, 2H), 2.78–2.65 (m, 4H), 2.58–2.49 (m, 5H), 2.08 (s, 1H), 1.9–1.78 (m, 1H), 1.64–1.53 (m, 1H), 0.89 (d, J = 6.6 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 162.55 146.85 145.07 141.24 139.45 132.08 130.74 130.43 129.25 128.72 128.70 125.96 125.57 125.31 125.28 122.91 122.88 58.38 57.54 53.82 43.90 37.06 35.18 35.12 28.37 28.32 22.34.
(R)-5-Isobutyl-1-phenyl-N-((1-(2-(trifluoromethyl)phenethyl)pyrrolidin-3-yl)methy-l)-1H-pyrazole-3-carboxamide (20c)
Yield 27%; 1H NMR (300 MHz, CDCl3) δ: 7.58–7.24 (m, 9H), 6.75 (s, 1H), 3.46–3.43 (m, 2H), 2.97–2.94 (m, 2H), 2.78–2.46 (m, 9H), 2.18–1.99 (m, 1H), 1.88–1.78 (m, 1H), 1.63–1.55 (m, 1H), 0.87 (d, J = 6.6 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ: 162.54 146.82 145.08 139.43 138.89 131.72 131.59 129.27 128.80 128.69 128.41 126.37 126.13 125.97 125.89 125.82 125.74 122.74 106.53 58.24 57.74 53.68 43.85 37.14 35.12 32.10 28.41 28.33 22.35.
(R)-N-((1-(3-fluorophenethyl)pyrrolidin-3-yl)methyl)-5-isobutyl-1-phenyl-1H-pyrazole-3-carboxamide (20d)
Yield 70%; 1H NMR (400 MHz, CDCl3) δ: 7.49–7.38 (m, 6H), 7.20–7.15 (m, 1H), 6.90–6.81 (m, 3H), 6.76 (s, 1H), 3.47–3.39 (m, 2H), 2.79–2.61 (m, 6H), 2.54–2.45 (m, 5H), 2.04–1.98 (m, 1H), 1.86–1.79 (m, 1H), 1.61–1.54 (m, 1H), 0.85 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.78 (d, JC-F = 243.7 Hz), 162.50, 146.86, 144.97, 142.97 (d, JC-F = 7.2 Hz), 139.43, 129.63 (d, JC-F = 8.2 Hz), 129.21, 128.63, 125.88, 124.23 (d, JC-F = 2.7 Hz), 115.39 (d, JC-F = 20.8 Hz), 112.76 (d, JC-F = 20.9 Hz), 106.50, 58.34, 57.49, 53.82, 43.92, 37.01, 35.28, 35.07, 28.34, 28.26, 22.29; HR-MS m/z [M + H]+ (ESI+) calcd. for C27H34FN4O = 449.2711, found 449.2707.
(R)-N-((1-(3,4-difluorophenethyl)pyrrolidin-3-yl)methyl)-5-isobutyl-1-phenyl-1H-p-yrazole-3-carboxamide (20e)
Yield 75%; 1H NMR (400 MHz, CDCl3) δ: 7.47–7.37 (m, 6H), 7.01–6.94 (m, 1H), 6.92–6.87 (m, 1H), 6.81–6.79 (br m, 1H), 6.75 (s, 1H), 3.43–3.38 (m, 2H), 2.73–2.58 (m, 6H), 2.51–2.43 (m, 5H), 2.04–1.95 (m, 1H), 1.85–1.78 (m, 1H), 1.59–1.51 (m, 1H), 0.84 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.49, 150.05 (d, JC-F = 245.6 Hz), 149.92 (d, JC-F = 245.7 Hz), 148.79 (d, JC-F = 244.1 Hz), 148.66 (d, JC-F = 244.1 Hz), 146.84, 144.98, 139.42, 137.41 (d, JC-F = 4.2 Hz), 137.35 (d, JC-F = 4.0 Hz), 129.19, 128.62, 125.87, 124.39 (d, JC-F = 3.5 Hz), 124.33 (d, JC-F = 3.5 Hz), 117.23 (d, JC-F = 16.7 Hz), 116.80 (d, JC-F = 16.8 Hz), 106.47, 58.28, 57.43, 53.76, 43.88, 36.99, 35.04, 34.64, 28.29, 28.25, 22.26; HR-MS m/z [M + H]+ (ESI+) calcd. for C27H33F2N4O = 467.2616, found 467.2612.
(R)-N-((1-(3,5-difluorophenethyl)pyrrolidin-3-yl)methyl)-5-isobutyl-1-phenyl-1H-pyrazole-3-carboxamide (20f)
Yield 70%; 1H NMR (400 MHz, CDCl3) δ: 7.51–7.37 (m, 6H), 6.75 (s, 1H), 6.64–6.55 (m, 3H), 3.46–3.39 (m, 2H), 2.76–2.59 (m, 6H), 2.51–2.46 (m, 5H), 2.03–1.97 (m, 1H), 1.85–1.78 (m, 1H), 1.58–1.54 (m, 1H), 0.84 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 164.17, 162.94 (d, JC-F = 245.9 Hz), 162.81 (d, JC-F = 246.2 Hz), 146.83, 145.03, 144.29 (d, JC-F = 18.0 Hz), 144.29, 139.43, 129.23, 128.68, 125.91, 111.42 (d, JC-F = 6.4 Hz), 111.24 (d, JC-F = 6.2 Hz), 106.49, 101.51 (d, JC-F = 25.3 Hz), 101.25 (d, JC-F = 25.1 Hz), 58.30, 56.98, 53.75, 43.88, 37.00, 35.20, 35.07, 28.32, 28.28, 22.30; HR-MS m/z [M + H]+ (ESI+) calcd. for C27H33F2N4O = 467.2616, found 467.2613.
(R)-N-((1-(2,3-difluorophenethyl)pyrrolidin-3-yl)methyl)-5-isobutyl-1-phenyl-1H-pyrazole-3-carboxamide (20g)
Yield 76%; 1H NMR (400 MHz, CDCl3) δ: 7.46–7.35 (m, 6H), 6.94–6.84 (m, 3H), 6.72 (s, 1H), 3.43–3.35 (m, 2H), 2.82–2.78 (m, 2H), 2.70–2.59 (m, 4H), 2.52–2.43 (m, 5H), 2.00–1.95 (m, 1H), 1.83–1.76 (m, 1H), 1.56–1.52 (m, 1H), 0.82 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.45, 150.53 (d, JC-F = 245.7 Hz), 150.40 (d, JC-F = 245.6 Hz), 148.98 (d, JC-F = 244.5 Hz), 148.86 (d, JC-F = 244.6 Hz), 146.82, 139.40, 129.77, 129.64, 129.15, 128.56, 125.83, 125.44 (d, JC-F = 3.6 Hz), 125.40 (d, JC-F = 3.5 Hz), 123.70 (d, JC-F = 6.8 Hz), 123.66 (d, JC-F = 6.9 Hz), 114.84 (d, JC-F = 16.9 Hz), 106.45, 58.20, 55.94, 53.68, 43.85, 37.04, 35.03, 28.50, 28.30, 28.23, 22.24; HR-MS m/z [M + H]+ (ESI+) calcd. for C27H33F2N4O = 467.2616, found 467.2609.
(R)-N-((1-(3-chloro-5-fluorophenethyl)pyrrolidin-3-yl)methyl)-5-isobutyl-1-phenyl-1H-pyrazole-3-carboxamide (20h)
Yield 71%; 1H NMR (400 MHz, CDCl3) δ: 7.55 (t, J = 5.5 Hz, 1H), 7.50–7.39 (m, 5H), 6.90–6.87 (m, 2H), 6.77 (s, 1H), 6.73 (d, J = 9.2 Hz, 1H), 3.49–3.37 (m, 2H), 2.76–2.60 (m, 6H), 2.52–2.46 (m, 5H), 2.06–1.98 (m, 1H), 1.89–1.79 (m, 1H), 1.62–1.54 (m, 1H), 0.86 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.56 (d, JC-F = 247.2 Hz), 162.51, 146.88, 144.99, 144.21 (d, JC-F = 8.2 Hz), 139.45, 134.53 (d, JC-F = 10.8 Hz), 129.21, 128.66, 125.90, 124.62 (d, JC-F = 2.8 Hz), 113.99 (d, JC-F = 21.2 Hz), 113.76 (d, JC-F = 25.3 Hz), 106.48, 58.38, 57.00, 53.76, 43.96, 36.96, 35.08, 28.34, 28.28, 22.29; HR-MS m/z [M + H]+ (ESI+) calcd. for C27H33ClFN4O = 483.2321, found 483.2316.
(R)-N-((1-(4-chloro-3-fluorophenethyl)pyrrolidin-3-yl)methyl)-5-isobutyl-1-phenyl-1H-pyrazole-3-carboxamide (20i)
Yield 77%; 1H NMR (400 MHz, CDCl3) δ: 7.50 (t, J = 5.5 Hz, 1H), 7.45–7.35 (m, 5H), 7.18 (t, J = 7.9 Hz, 1H), 6.85 (dd, J = 10.1, 1.7 Hz, 1H), 6.79 (d, J = 8.0 Hz, 1H), 6.73 (s, 1H), 3.43–3.34 (m, 2H), 2.72–2.56 (m, 6H), 2.48–2.42 (m, 5H), 2.01–1.92 (m, 1H), 1.83–1.76 (m, 1H), 1.57–1.49 (m, 1H), 0.82 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.47, 157.76 (d, JC-F = 246.7 Hz), 146.83, 144.93, 141.50, 141.44, 139.41, 130.12, 129.17, 128.60, 125.83, 125.04 (d, JC-F = 3.3 Hz), 118.08 (d, JC-F = 17.5 Hz), 116.63 (d, JC-F = 20.4 Hz), 106.46, 58.27, 57.14, 53.74, 43.89, 36.96, 35.03, 34.77, 28.28, 28.23, 22.25; HR-MS m/z [M + H]+ (ESI+) calcd. for C27H33ClFN4O = 483.2321, found 483.2316.
(R)-N-((1-(3,4-dichlorophenethyl)pyrrolidin-3-yl)methyl)-5-isobutyl-1-phenyl-1H-pyrazole-3-carboxamide (20j)
Yield 77%; 1H NMR (400 MHz, CDCl3) δ: 7.50 (t, J = 5.5 Hz, 1H) 7.47–7.36 (m, 5H), 7.25 (d, J = 8.2 Hz, 1H), 7.17 (d, J = 1.9. Hz, 1H), 6.92 (dd, J = 8.2, 2.0 Hz, 1H), 6.74 (s, 1H), 3.45–3.35 (m, 2H), 2.71–2.56 (m, 6H), 2.49–2.43 (m, 5H), 2.02–1.94 (m, 1H), 1.84–1.77 (m, 1H), 1.59–1.51 (m, 1H), 0.83 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.49, 146.85, 144.95, 140.73, 139.42, 131.99, 130.47, 130.10, 129.76, 129.19, 128.62, 128.09, 125.86, 106.47, 58.31, 57.20, 53.76, 43.92, 36.95, 35.05, 34.59, 28.30, 28.25, 22.28; HR-MS m/z [M + H]+ (ESI+) calcd. for C27H33Cl2N4O = 499.2025, found 499.2019.
(R)-N-((1-(2,4-bis(trifluoromethyl)phenethyl)pyrrolidin-3-yl)methyl)-5-isobutyl-1-phenyl-1H-pyrazole-3-carboxamide (20k)
Yield 40%; 1H NMR (400 MHz, CDCl3) δ: 7.87 (s, 1H), 7.69 (d, J = 8.1 Hz, 1H), 7.52–7.40 (m, 7H), 6.79 (s, 1H), 3.53–3.41 (m, 2H), 3.04 (t, J = 7.9 Hz, 2H), 2.79–2.67 (m, 4H), 2.60–2.49 (m, 5H), 2.11–2.02 (m, 1H), 1.89–1.83 (m, 1H), 1.66–1.58 (m, 1H), 0.88 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.52, 146.83, 145.09, 143.42, 139.43, 132.30, 129.37 (q, JC-F = 30.0 Hz), 129.22, 128.73 (q, JC-F = 33.3 Hz), 128.65, 128.38, 125.94, 123.72 (q, JC-F = 272.4 Hz, CF3), 123.50 (q, JC-F = 270.4 Hz, CF3), 123.10, 123.06, 123.00, 106.52, 58.20, 57.03, 53.63, 43.85, 37.14, 35.08, 32.00, 28.36, 22.29; HR-MS m/z [M + H]+ (ESI+) calcd. for C29H33F6N4O = 567.2553, found 567.2549.
(R)-N-((1-(3-fluoro-4-(trifluoromethoxy)phenethyl)pyrrolidin-3-yl)methyl)-5-isobutyl-1-phenyl-1H-pyrazole-3-carboxamide (20l)
Yield 62%; 1H NMR (400 MHz, CDCl3) δ: 7.51–7.38 (m, 6H), 7.15 (td, J = 8.1, 0.9 Hz, 1H), 6.95 (dd, J = 11.0, 1.9 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 6.76 (s, 1H), 3.47–3.38 (m, 2H), 2.79–2.61 (m, 6H), 2.53–2.47 (m, 5H), 2.05–2.00 (m, 1H), 1.86–1.80 (m, 1H), 1.60–1.56 (m, 1H), 0.86 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.54, 154.23 (d, JC-F = 250.4 Hz), 146.80, 145.07, 141.37 (d, JC-F = 6.4 Hz), 139.44, 134.58 (d, JC-F = 1.9 Hz), 134.46 (d, JC-F = 2.1 Hz), 129.22, 128.67, 125.92, 125.83, 124.66, 124.60, 124.53 (d, JC-F = 6.9 Hz), 123.41, 123.32, 120.48 (q, JC-F = 256.6 Hz, CF3), 117.25 (d, JC-F = 18.1 Hz), 115.41 (d, JC-F = 20.1 Hz), 112.89 (d, JC-F = 19.4 Hz), 106.49, 58.14, 57.38, 57.08, 53.69, 43.74, 43.63, 37.10, 35.08, 34.91, 34.62, 28.28, 22.28.
(R)-5-isobutyl-1-phenyl-N-((1-(4-(trifluoromethoxy)-3-(trifluoromethyl)phenethyl)-pyrrolidin-3-yl)methyl)-1H-pyrazole-3-carboxamide (20m)
Yield 68%; 1H NMR (400 MHz, CDCl3) δ: 7.52 (t, J = 5.4 Hz, 1H), 7.48–7.38 (m, 6H), 7.34 (dd, J = 8.5, 1.8 Hz, 1H), 7.26 (d, J = 8.2 Hz, 1H), 6.76 (s, 1H), 3.49–3.37 (m, 2H), 2.82 (t, J = 7.9 Hz, 2H), 2.76–2.62 (m, 4H), 2.53–2.47 (m, 5H), 2.05–1.98 (m, 1H), 1.86–1.79 (m, 1H), 1.62–1.56 (m, 1H), 0.85 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.53, 146.83, 145.03, 144.71, 139.46, 139.41, 133.46, 129.18, 128.63, 127.47 (q, JC-F = 4.7 Hz), 125.87, 122.71 (q, JC-F = 31.5 Hz), 122.49 (q, JC-F = 271.3 Hz, OCF3), 120.89, 120.88, 120.26 (q, JC-F = 257.5 Hz, CF3), 106.47, 58.28, 57.10, 53.71, 43.91, 36.99, 35.03, 34.62, 28.29, 28.26, 22.23; HR-MS m/z [M + H]+ (ESI+) calcd. for C29H33F6N4O2 = 583.2502, found 583.2497.
(R)-5-Isobutyl-N-((1-(4-methyl-3-(trifluoromethyl)phenethyl)pyrrolidin-3-yl)methyl)-1-phenyl-1H-pyrazole-3-carboxamide (20n)
Yield 89%; 1H NMR (400 MHz, CDCl3) δ: 7.49–7.37 (m, 7H), 7.20–7.14 (m, 2H), 6.77 (s, 1H), 3.48–3.40 (m, 2H), 2.81–2.61 (m, 6H), 2.56–2.47 (m, 5H), 2.42 (d, J = 1.6 Hz, 3H), 2.05–2.01 (m, 1H), 1.87–1.81 (m, 1H), 1.61–1.56 (m, 1H), 0.87 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.50, 146.84, 144.97, 139.42, 138.00, 134.04, 131.84, 131.77, 129.28, 129.18, 128.63 (q, JC-F = 29.3 Hz), 128.61, 125.87, 125.83, 125.77, 125.72, 124.59 (q, JC-F = 272.0 Hz, CF3), 106.47, 58.33, 57.59, 53.76, 43.85, 37.03, 35.06, 34.81, 28.31, 22.26, 18.81; HR-MS m/z [M + H]+ (ESI+) calcd. for C29H36F3N4O = 513.2835, found 513.2826.
(R)-5-Isobutyl-N-((1-(2-methyl-5-(trifluoromethyl)phenethyl)pyrrolidin-3-yl)methyl)-1-phenyl-1H-pyrazole-3-carboxamide (20o)
Yield 69%; 1H NMR (400 MHz, CDCl3) δ: 7.49–7.39 (m, 6H), 7.34 (s, 2H), 7.20 (d, J = 8.4 Hz, 1H), 6.77 (s, 1H), 3.52–3.40 (m, 2H), 2.89–2.76 (m, 4H), 2.67–2.61 (m, 3H), 2.58–2.55 (m, 2H), 2.51 (d, J = 7.2 Hz, 2H), 2.32 (s, 3H), 2.11–2.03 (m, 1H), 1.89–1.78 (m, 1H), 1.67–1.59 (m, 1H), 0.86 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.57, 146.76, 145.05, 140.30, 139.39, 138.92, 130.46, 129.20, 128.66, 128.25 (q, JC-F = 31.6 Hz), 125.87, 125.75, 125.68, 125.64, 124.35 (q, JC-F = 271.8 Hz, CF3), 122.96, 122.92, 106.49, 58.22, 56.17, 53.76, 43.64, 37.14, 35.06, 32.44, 28.27, 22.27, 19.30; HR-MS m/z [M + H]+ (ESI+) calcd. for C29H36F3N4O = 513.2835, found 513.2829.
(R)-5-Isobutyl-N-((1-(5-methyl-2-(trifluoromethyl)phenethyl)pyrrolidin-3-yl)methyl)-1-phenyl-1H-pyrazole-3-carboxamide (20p)
Yield 68%; 1H NMR (400 MHz, CDCl3) δ: 7.49–7.37 (m, 7H), 7.08–7.05 (m, 2H), 6.76 (s, 1H), 3.50–3.40 (m, 2H), 2.95–2.91 (m, 2H), 2.76–2.46 (m, 9H), 2.34 (s, 3H), 2.08–2.00 (m, 2H), 1.89–1.79 (m, 1H), 1.63–1.55 (m, 1H), 0.87 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 162.50, 146.84, 144.98, 141.91, 139.42, 138.60, 132.24, 129.18, 128.59, 126.77, 125.87, 125.81 (q, JC-F = 29.5 Hz), 125.80, 125.75, 124.77 (q, JC-F = 271.7 Hz, CF3), 106.49, 58.24, 57.75, 53.62, 43.80, 37.12, 35.08, 32.00, 28.27, 22.28, 21.16.
Biological assay methods
FDSS6000 assay. HEK 293 cells which express both stable CaV3.1 and CaV3.2 were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum, penicillin (100 μg/mL), streptomycin (100 μg/mL), geneticin (500 μg/mL), and puromycin (1 μg/mL) at 37 °C in a humid atmosphere of 5% CO2 and 95% air. Cells were seeded into 96-well black wall clear bottom plates at a density of 40,000 cells/well and were used on the next day for FDSS6000 assay. Cells were incubated for 60 min at room temperature with 5 μM fluo3/AM and 0.001% Pluronic F-127 in a HEPES-buffered solution composed of 115 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 20 mM HEPES and 13.8 mM glucose (pH 7.4). During fluorescence-based FDSS6000 assay, CaV3.1 and CaV3.2 channels were activated using a high concentration of 70 mM KCl in 10 mM CaCl2 contained a HEPES-buffered solution, and the increase in [Ca2+]i by KCl-induced depolarisation was detected. Throughout the entire procedure, cells were washed in a BIO-TEK 96-well washer. All data were collected and analysed using FDSS6000 and related software (Hamamatsu, Japan).
Whole-cell patch-clamp method
Cav3.1: Cav3.1 channel was stably expressed in HEK293 cells. The cells used for T-type calcium channel activity assay were cultured on a coverslip coated with poly-L-lysine (0.5 mg/mL) whenever sub-cultured, and their calcium channel activity was recorded 2–7 days after the cultivation. Current of the T-type calcium channel at a single cell level was measured according to an electrophysiological whole-cell patch-clamp method using EPC-9 amplifier (HEKA, Germany). At this time, a cell exterior solution composed of 140 mM NaCl, 2 mM CaCl2, and 10 mM HEPES (pH 7.4) and a cell interior solution composed of KCl 130 mM, HEPES 10 mM, EGTA 11 mM, and MgATP 5 mM (pH 7.4) were employed. Inward current caused by the T-type calcium channel activation which occurred when the cells were converted into a whole-cell recording mode by stabbing a microglass electrode having 3–4 MΩ resistance which was filled with the cell interior solution into a single cell and depolarised at –30 mV (50 ms duration period) every 10 s with fixing membrane potential to –100 mV was measured according to a T-type calcium channel protocol activated at low current. Cav3.2: current of the calcium channel expressed from the Xenopus unfertilised oocytes was measured according to a two-electrode voltage clamp method. Current was measured in 10 mM Ba2+ solution composed of 10 mM Ba(OH)2, 90 mM NaOH, 1 mM KCl, 0.1 mM EDTA, and 5 mM HEPES, and pH was adjusted to 7.4 with methanesulfonic acid. At this time, voltage clamp and current measurements were regulated with an amplifier for unfertilised oocytes (Model OC-725C, Warner Instrument Corp., LLC., Hamden, CT, USA), analog signals were converted into digital signals using Digidata 2000 A (Analog-Digital converter, Axon Instrument), and acquisition, storage, and analysis of all data were recorded in Pentium IV computer via pCLAMP8. The data were mainly collected at 5 KHz and filtered at l KHz (Model 902 filter; Frequency devices located at Haverhill, MA, USA). The generation of T-type current was occurred by imposing test electric potential of –20 mV every 15 s on the unfertilised oocytes whose potential was fixed at –90 mV, and a blocking percentage was calculated by comparing the potentials before and after the drug treatment.
In vivo assay
Behavioural tests were conducted for rats at 1 day prior to surgery and 14 days after surgery. After the postoperative behavioural test, the animals were treated orally with 100 mg/kg compound 20n or gabapentin. The tests were reevaluated at 1 h, 3 h, and 5 h after administration. Mechanical allodynia: Testing was performed according to methods described in the literatureCitation45. Rats were acclimated in a transparent plastic boxes permitting freedom of movement with a wire mesh floor to allow access to the planter surface of the hind paws. A von Frey filament (Stoelting, Wood Dale, IL) was applied 5 times (once every 3–4 s) to hind paw. Von Frey filaments were used to assess the 50% mechanical threshold for paw withdrawal. The 50% withdrawal threshold was determined by using the up-down method and formula given by DixonCitation46: 50% threshold =10(X + kd)/104, where X is the value of the final von Frey hair used (in log units), k is the tabular value for the pattern of positive/negative responses modified from the same literature, and d is the mean difference between stimuli in log units (0.17). Cold allodynia: To quantify cold sensitivity of the paw, brisk paw withdrawal in response to acetone application was measured as reported previouslyCitation47. The rat was placed under a transparent plastic box on a metal mesh floor and acetone was applied to the plantar surface of the hind paw. To do this, an acetone bubble was formed at the end of a small piece of polyethylene tubing which was connected to a syringe. The bubble was then gently touched to the heel. The acetone quickly spread over the proximal half of the plantar surface of the hind paw. The acetone was applied 5 times to the hind paw at 2 min interval. The frequency of paw withdrawal was expressed as a percentage [(no. of trials accompanied by brisk foot withdrawal/total no. of trials) × l00]. The results of behavioural tests are expressed as a %MPE. For example, paw withdrawal thresholds were converted to %MPE by the following formula, using a cutoff of 15 g (the threshold for normal rats) to define maximum possible effect: (post drug threshold-baseline threshold)/(cutoff-baseline threshold) × 100. %MPE values near 100 indicate normal mechanical thresholds (i.e. at or near 15 g), whereas values near 0 indicate allodynia. The result of cold allodynia was also expressed as %MPE.
Results and discussion
Inhibitor design
Many promising T-type channel inhibitors have been developed based on a piperidine moiety such as Penfluridol (1)Citation48, TTA-P2 (2) and Z-944 (3) (). We also synthesised T-type channel inhibitors based on a piperidine moiety such as compound 4 which showed approximately 60% inhibition of Cav3.1 at 10 μMCitation49. However, pyrrolidine, a close homolog of piperidine, has not been frequently introduced so far. Therefore, in an effort to develop novel potent T-type channel inhibitors, we decided to synthesise pyrrolidine-based inhibitors. Our initial molecular structure was constructed via a structure-hybridisation between the template of TTA-P2 or Z-944 and 5-isobutyl-1-phenyl-1H-pyrazole moiety that consistently showed robust activity in our previous T-type inhibitors ()Citation50,Citation51. Furthermore, this compound series accommodating hydrophobic R group are well mapped with 3D ligand-based pharmacophore that was produced by common feature hypothesis generation approach (HipHop) implemented in CATALYST program for the rational design of T-type calcium channel inhibitors ()Citation52.
Figure 1. Previous T-type calcium channel inhibitors based on the piperidine scaffold.
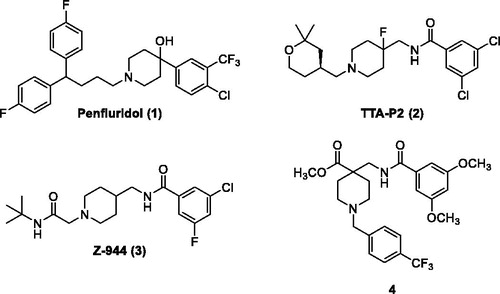
Figure 2. Design of new pyrrolidine-based T-type calcium channel inhibitors via a structure-hybridisation strategy.
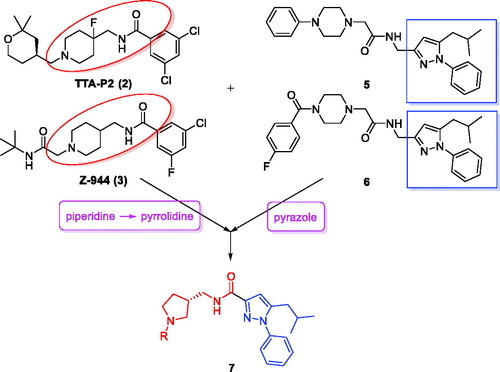
Figure 3. Left: Reference pharmacophore model. Right: pharmacophore mapping results of 20n. Green: hydrogen bond acceptor, cyan: hydrophobic, red: positive ionisable.
Synthesis of inhibitors
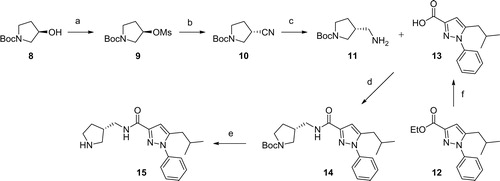
We envisioned that the designed compounds would be obtained by an amide formation between (N-tert-butoxycarbonyl-pyrrolidin-3-yl)methanamines 11 and 5-isobutyl-1-phenyl-1H-pyrazole-3-carboxylic acid 13 followed by derivatisation of the free pyrrolidine 15. The synthesis of the pyrroldine part commenced with mesylation of commercially available (R)-N-(tert-butoxycarbonyl)-3-hydroxypyrrolidine 8 (Scheme 1). Subsequent SN2 reaction with sodium cyanide followed by reduction with lithium aluminum hydride provided (R)-(N-(tert-butoxycarbonyl)-pyrrolidin-3-yl)methanamine 11 in good yield. 5-isobutyl-1-phenyl-1H-pyrazole-3-carboxylic acid 13 was produced by hydrolysis of the known ester 12Citation53 with 1 N NaOH. Next, carbonyldiimidazole-mediated amide formation smoothly provided the desired amide 14 in 90% yield, and the Boc group deprotection with TFA furnished the free amine 15 as a common synthetic intermediate.
Scheme 1. Reagents and conditions: (a) MsCl, TEA, DCM, rt, 100%; (b) NaCN, DMF, 80 °C, 84%; (c) 1.0 M LiAlH4 in THF, AlCl3, THF, 0 °C to rt, 56%; (d) 1,1d) % 56%lHaCN, DMFMFb) NaCN,rt, 90%; (e) TFA, DCM, rt, 94%; (f) 1 N NaOH, EtOH, rt, 82%.
Preparation of N-alkylated pyrrolidines (20a–p) was started from synthesis of tosylates (19a–p): a variety of homobenzylic alcohol 18a–p were either purchased or produced by reduction of commercially available homobenzylic acid 17d, 17f, and 17h–p with lithium aluminum hydride (Scheme 2). After tosylation of 18a–p, SN2 reaction with 15 underwent smoothly with potassium carbonate furnishing desired compounds 20a–p in good yields. Additionally, N-(3,3-dimethybutyl)pyrrolidine 16 was obtained by reductive amination reaction with 3,3-dimethylbutanal.
Scheme 2. Reagents and conditions: (a) 1.0 M LiAlH4 in THF, THF, 0 °C to rt, 39–97%; (b) p-toluenesulfonyl chloride, TEA, DCM, 0 °C to rt, 12–87%; (c) K2CO3, acetonitrile, reflux, 17–89%; (d) 3,3-dimethylbutanal, NaBH(OAc)3, DCM, rt, 10–29%.
Biological activity
In vitro T-type calcium channel inhibitory activity
The in vitro T-type calcium channel inhibitory activity of synthesised pyrrolidine compounds were evaluated against CaV3.1 and CaV3.2 at two different concentrations, 10 μM and 1 μM, respectively (). We stably expressed CaV3.1 and CaV3.2 in HEK293 cells and measured the concentration of intracellular Ca2+ with fluo3/AM after the activation of CaV3.1 and CaV3.2 channels by 70 mM of KCl in 10 mM of CaCl2. Then, we presume that the degree of the fluorescence decrease read by FDSS6000 after co-incubation with synthesised compounds can reflect the inhibitory activity of each pyrrolidine compoundCitation43. First, the free pyrrolidine 15 was subjected to this FDSS6000-based fluorescent assay, but the compound was not able to inhibit both T-type channels more than 20% at 10 μM (18.60% inhibition against CaV3.1 and 10.72% inhibition against CaV3.2). Guided by the pharmacophore mapping results we decided to put a hydrophobic group to the free pyrrolidine to increase the potency. N-Alkylation of pyrrolidine dramatically increased inhibitory activities as exemplified with 16 and 20a which inhibited the activation of CaV3.1 and CaV3.2 channels more than 47% at 10 μM. Since 20a was more potent than 16, we focused on the synthesis of N-phenethylpyrrolidine compounds. A variety of substituents such as methyl-, fluoro-, chloro-, trifluoromethyl- and trifluoromethoxy groups were introduced to the phenyl ring to investigate the substituent effects on the activity. Most compounds showed great inhibitory activities more than 65% against both channels at 10 μM, and 10 compounds (20b–h, 20 m, 20n, 20p) inhibited greater than 80% of activation of at least one of either CaV3.1 or CaV3.2 channels. Furthermore, IC50 values of selected compounds were precisely measured by the whole-cell patch-clamp assayCitation44, and compounds displaying high inhibition percentage at 10 μM also showed great IC50 values against both T-type calcium channels (IC50 (CaV3.1) 2.14–6.11 μM, IC50 (CaV3.2) 2.88–8.14 μM) strongly implicating that these pyrrolidine compounds possess great therapeutic potential for neuropathic pain.
Table 1. Inhibitory activities of pyrrolidine-based inhibitors 16 and 20a–p against CaV3.1 and CaV3.2 T-type calcium channels.
In vitro ADME properties
In order to further evaluate the potential of these pyrrolidine compounds as promising therapeutic agents, their respective physicochemical properties were evaluated by assessing CYP450 inhibitory activities at 10 μM and metabolic stability in human liver microsome (). Most compounds barely inhibited CYP1A2 enzyme, but several compounds such as 20k showed moderate to strong inhibition of other tested CYP enzymes. Also, we determined remaining compound percentage at 30 min after incubation of compounds in human liver microsome, but unfortunately the metabolic stability of synthesised pyrrolidines appeared not quite high. Nevertheless, among N-phenethylpyrrolidine compounds, 20n seemed most stable in human liver microsome, approximately 15% of 20n left intact after 30 min. As a next step, IC50 values of selected compounds against hERG channels were evaluated employing the same whole-cell patch-clamp assay (). Blockage of hERG channels induces long QT syndrome leading to potentially fatal cardiac arrhythmiaCitation54. Often, t-type calcium channel inhibitors suffer from low selectivity against hERG channels which we should investigate in developing t-type channel blockers to avoid potential cardiac side effects. The IC50 value of Mibefradil against hERG channels was reported as approximately 1.34 μMCitation49. It was observed that most of our compounds showed lower IC50 values than Mibefradil requiring further improvement of selectivity against hERG channels. Fortunately, better selectivity against hERG channels than Mibefradil was obtained with compounds 16, 20k and 20n, among which 20n exhibited IC50 value of 8.13 μM representing safest compounds in this series of compounds. Comprehensive evaluation of current pyrrolidine compound series based on T-type channel inhibitory activity and in vitro ADME properties led us to determine 20n as a promising candidate for the next in vivo evaluation.
Table 2. Results of in vitro ADME assay (CYP inhibition, microsomal stability, hERG inhibition) of pyrrolidine-based inhibitors 16 and 20a–p.
In vivo pharmacokinetic study
First, in vivo pharmacokinetic studies were carried out with Sprague-Dawley (SD) male rats (). Total eight rats were equally divided into two groups and 10 mg/kg of 20n was administered to each group but in two different routes, orally and intravenously. Then, pharmacokinetic parameters of each administration route were calculated from compound concentrations in the blood. It turned out moderate Cmax value (0.20 μg/mL) was observed after the oral administration. Nevertheless, because the clearance rate (29.7 ml/min/kg) was relatively low, total AUC0-∞ values after both oral and intravenous dosing were 339.17 and 61.89 μg min/mL sufficient enough to exert therapeutic effects in vivo. Of particular importance is the observation that brain-to-plasma ratio at 2 h was 8.78 after intravenous administration, strongly suggesting that once exposed to the plasma, 20n is highly likely to cross the tight blood-brain barrier efficiently and well penetrated into the brain. Despite low oral bioavailability presumably caused by poor metabolic stability, 20n can be employed to develop promising candidates for the treatment of neuropathic pain.
Table 3. Pharmacokinetic parameters of 20n after intravenous and oral administration (10 mg/kg) to SD male rats.
In vivo efficacy studies with two neuropathic pain animal models
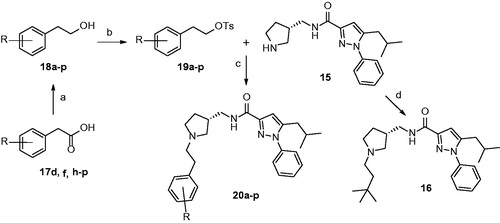
In order to further demonstrate the potential of 20n as a therapeutic agent to treat neuropathic pain, we tested in vivo efficacy of 20n with two different animal models of neuropathic pain: the SNL modelCitation55 and the streptozotocin (STZ)-induced diabetic neuropathy modelCitation56. The SNL model is generated by ligating one (L5) or two (L5 and L6) segmental spinal nerves of the rat. In addition, streptozotocin is known to destroy pancreatic beta cells via DNA alkylation followed by activation of apoptosis resulting in diabetes. Both models result in behavioural symptoms such as increased sensitivity to non-noxious mechanical or cold stimuli and heat hypersensitivity, and have been widely used as animal model systems to investigate neuropathic pain. In the SNL model, we first observed that the paw withdrawal threshold to mechanical stimuli was significantly decreased and also the frequency of the paw withdrawal to cold stimuli was substantially increased confirming that neuropathic pain was induced 2 weeks after the surgery (). Then, 20n was orally administered to rats (n = 4) with 100 mg/kg. The current drug for neuropathic pain, gabapentin (100 mg/kg) was also dosed orally to another rat group (n = 4) side by side to directly compare pain-relieving effects with 20n. The mechanical threshold was evaluated by von Frey filaments. It was found that 20n was able to restore mechanical hypersensitivity close to the normal level and showed superior analgesic effects as compared to gabapentin at 3 h after the treatment. Furthermore, 20n significantly lowered the paw withdrawal response rate to cold stimuli as well, and the analgesic effects of 20n were again similar to that of gabapentin.
Figure 4. Analgesic effects to reduce hypersensitivity to non-noxious mechanical stimuli (A and B) and cold stimuli (C and D) after oral administration of gabapentin (○, 100 mg/kg, n = 4) and 20n (•, 100 mg/kg, n = 4) to the SNL model. Experimental time expressed as D for days after neuropathic injury (N) and h for hours after gabapentin or 20n administration, *p < .05 (gabapentin), *p < .05 (20n) versus pre-administration value (paired t-test), ♣p < .05 gabapentin versus 20n (unpaired t-test).
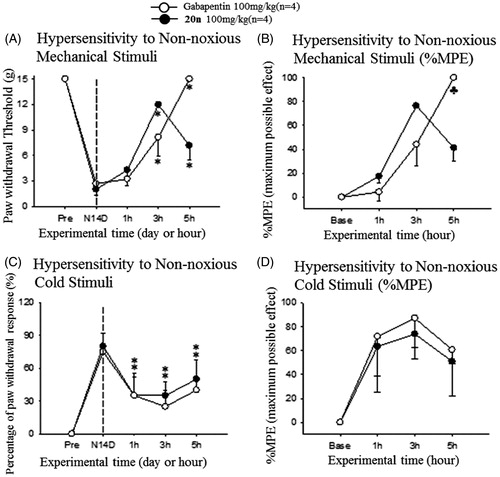
After observing treatment of 20n relieved neuropathic pain response in the SNL model, we decided to further prove that the ability of 20n to reduce neuropathy can be general and are not confined to one specific neuropathic model. The streptozotocin (STZ)-induced diabetic neuropathy model for diabetic peripheral neuropathy (DPN) was selected. We confirmed that intraperitoneal injection of 65 mg/kg of streptozotocin to rats reduced both the paw withdrawal threshold to non-noxious mechanical stimuli and the paw withdrawal latency to heat stimuli at 2 weeks after the surgery (). Subsequently, 100 mg/kg of 20n and gabapentin was orally dosed to seven and six rats, respectively. Once again, 20n increased the paw withdrawal threshold similarly to gabapentin, and delayed the paw withdrawal latency comparable to gabapentin suggesting 20n is also effective in alleviating heat hypersensitivity.
Figure 5. Analgesic effects to reduce hypersensitivity to non-noxious mechanical stimuli (A and B) and heat hypersensitivity (C and D) after oral administration of gabapentin (○, 100 mg/kg, n = 6) and 20n (•, 100 mg/kg, n = 7) to the STZ model. Experimental time expressed as D for days after neuropathic injury (N) and h for hours after gabapentin or 20n administration, *p < .05 (gabapentin), *p < .05 (20n) versus pre-administration value (paired t-test), ♣p < .05 gabapentin versus 20n unpaired t-test).
Conclusion
In conclusion, we have developed pyrrolidine-based T-type calcium channel inhibitors by pharmacophore mapping and structural hybridisation followed by evaluation of their Cav3.1 and Cav3.2 channel inhibitory activities with FDSS and whole-cell patch-clamp assay. Most pyrrolidine compounds potently inhibited activation of both Cav3.1 and Cav3.2 T-type channels showing IC50 values between 2.14 and 8.14 μM. Profiling of in vitro ADME properties revealed that among this series 20n has great selectivity against hERG channel and high plasma exposure in the subsequent in vivo pharmacokinetic study. Furthermore, it was notable that 20n can be potentially well penetrated into the brain according to the excellent brain to plasma compound concentration ratio at 2 h after intravenous administration. Finally, 20n exhibited comparable analgesic effects to gabapentin in the both SNL and STZ neuropathic pain animal models, implicating potent T-type calcium channel inhibitor 20n can be developed as promising candidates to treat neuropathic pain. Further optimisation of 20n in regard to physicochemical properties like metabolic stability is currently in progress.
Supplemental Material
Download PDF (1.2 MB)Disclosure statement
The authors report no declarations of interest.
Additional information
Funding
Related Research Data
References
- O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med 2009;122:S22–S32.
- Muthuraman A, Singh N, Jaggi AS, Ramesh M. Drug therapy of neuropathic pain: current developments and future perspectives. Curr Drug Targets 2014;15:210–53.
- Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol 2010;9:807–19.
- Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 2015;14:162–73.
- Farrar JT, Young JP, Jr, LaMoreaux L, et al. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. Pain 2001;94:149–58.
- Finnerup NB, Jensen TS. Mechanisms of disease: mechanism-based classification of neuropathic pain-a critical analysis. Nat Clin Pract Neurol 2006;2:107–15.
- Colloca L, Ludman T, Bouhassira D, et al. Neuropathic pain. Nat Rev Dis Primers 2017;3:17002.
- Bourinet E, Altier C, Hildebrand ME, et al. Calcium-permeable ion channels in pain signaling. Physiol Rev 2014;94:81–140.
- Di Cesare Mannelli L, Zanardelli M, Landini I, et al. Effect of the SOD mimetic MnL4 on in vitro and in vivo oxaliplatin toxicity: possible aid in chemotherapy induced neuropathy. Free Radic Biol Med 2016;93:67–76.
- Di Cesare Mannelli L, Cinci L, Micheli L, et al. α-conotoxin RgIA protects against the development of nerve injury-induced chronic pain and prevents both neuronal and glial derangement. Pain 2014;155:1986–95.
- Pacini A, Micheli L, Maresca M, et al. The α9α10 nicotinic receptor antagonist α-conotoxin RgIA prevents neuropathic pain induced by oxaliplatin treatment. Exp Neurol 2016;282:37–48.
- Todorovic SM, Jevtovic-Todorovic V. The role of T-type calcium channels in peripheral and central pain processing. CNS Neurol Disord Drug Targets 2006;5:639–53.
- Snutch TP, David LS. T-type calcium channels: an emerging therapeutic target for the treatment of pain. Drug Develop Res 2006;67:404–15.
- Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev 2005;57:411–25.
- Perez-Reyes E, Cribbs LL, Daud A, et al. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature 1998;391:896–900.
- Cribbs LL, Lee JH, Yang J, et al. Cloning and characterization of alpha1H from human heart, a member of the T-type Ca2+ channel gene family. Circ Res 1998;83:103–9.
- Lee JH, Daud AN, Cribbs LL, et al. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J Neurosci 1999;19:1912–21.
- Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 2003;83:117–61.
- Iftinca MC, Zamponi GW. Regulation of neuronal T-type calcium channels. Trends Pharmacol Sci 2009;30:32–40.
- Bourinet E, Alloui A, Monteil A, et al. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J 2005;24:315–24.
- Messinger RB, Naik AK, Jagodic MM, et al. In vivo silencing of the Ca(V)3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain 2009;145:184–95.
- Talley EM, Cribbs LL, Lee JH, et al. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci 1999;19:1895–911.
- Na HS, Choi S, Kim J, et al. Attenuated neuropathic pain in Cav3.1 null mice. Mol Cells 2008;25:242–6.
- Choi S, Yu E, Hwang E, Llinás RR. Pathophysiological implication of CaV3.1 T-type Ca2+ channels in trigeminal neuropathic pain. Proc Natl Acad Sci USA 2016;113:2270–5.
- Jimenez C, Bourinet E, Leuranguer V, et al. Determinants of voltage-dependent inactivation affect Mibefradil block of calcium channels. Neuropharmacology 2000;39:1–10.
- Gomora JC, Daud AN, Weiergraber M, Perez-Reyes E. Block of cloned human T-type calcium channels by succinimide antiepileptic drugs. Mol Pharmacol 2001;60:1121–32.
- Todorovic SM, Prakriya M, Nakashima YM, et al. Enantioselective blockade of T-type Ca2+ current in adult rat sensory neurons by a steroid that lacks gamma-aminobutyric acid-modulatory activity. Mol Pharmacol 1998;54:918–27.
- Dogrul A, Gardell LR, Ossipov MH, et al. Reversal of experimental neuropathic pain by T-type calcium channel blockers. Pain 2003;105:159–68.
- Pathirathna S, Todorovic SM, Covey DF, Jevtovic-Todorovic V. 5alpha-reduced neuroactive steroids alleviate thermal and mechanical hyperalgesia in rats with neuropathic pain. Pain 2005;117:326–39.
- Latham JR, Pathirathna S, Jagodic MM, et al. Selective T-type calcium channel blockade alleviates hyperalgesia in ob/ob mice. Diabetes 2009;58:2656–65.
- McNulty MM, Hanck DA. State-dependent mibefradil block of Na + channels. Mol Pharmacol 2004;66:1652–61.
- Coste B, Crest M, Delmas P. Pharmacological dissection and distribution of NaN/Nav1.9, T-type Ca2+ currents, and mechanically activated cation currents in different populations of DRG neurons. J Gen Physiol 2007;129:57–77.
- SoRelle R. Withdrawal of Posicor from market. Circulation 1998;98:831–2.
- Shin MC, Kim CJ, Min BI, et al. A selective T-type Ca2+ channel blocker R(-) efonidipine. Naunyn Schmiedebergs Arch Pharmacol 2008;377:411–21.
- Shipe WD, Barrow JC, Yang ZQ, et al. Design, synthesis, and evaluation of a novel 4-aminomethyl-4-fluoropiperidine as a T-type Ca2+ channel antagonist. J Med Chem 2008;51:3692–5.
- Xiang Z, Thompson AD, Brogan JT, et al. The discovery and characterization of ML218: a novel, centrally active T-Type calcium channel inhibitor with robust effects in STN neurons and in a rodent model of Parkinson’s disease. ACS Chem Neurosci 2011;2:730–42.
- Zhang Q, Xia Z, Joshi S, et al. Optimization of ADME properties for sulfonamides leading to the discovery of a T-type calcium channel blocker, ABT-639. ACS Med Chem Lett 2015;6:641–4.
- Tringham E, Powell KL, Cain SM, et al. T-type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Sci Transl Med 2012;4:121ra19.
- Siegrist R, Pozzi D, Jacob G, et al. Structure-activity relationship, drug metabolism and pharmacokinetics properties optimization, and in vivo studies of new brain penetrant triple T-type calcium channel blockers. J Med Chem 2016;59:10661–75.
- Remen L, Bezencon O, Simons L, et al. Preparation, antiepileptic activity, and cardiovascular safety of dihydropyrazoles as brain-penetrant T-type calcium channel blockers. J Med Chem 2016;59:8398–411.
- Jarvis MF, Scott VE, McGaraughty S, et al. A peripherally acting, selective T-type calcium channel blocker, ABT-639, effectively reduces nociceptive and neuropathic pain in rats. Biochem Pharmacol 2014;89:536–44.
- Choe W, Messinger RB, Leach E, et al. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol 2011;80:900–10.
- Kim T, Choi J, Kim S, et al. The biochemical activation of T-type Ca2+ channels in HEK293 cells stably expressing alpha1G and Kir2.1 subunits. Biochem Biophys Res Commun 2004;324:401–8.
- Monteil A, Chemin J, Bourinet E, et al. Molecular and functional properties of the human alpha(1G) subunit that forms T-type calcium channels. J Biol Chem 2000;275:6090–100.
- Chaplan SR, Bach FW, Pogrel JW, et al. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53:55–63.
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 1980;20:441–62.
- Choi Y, Yoon YW, Na HS, et al. Behavioral signs of ongoing pain and cold allodynia in a rat model of neuropathic pain. Pain 1994;59:369–76.
- Janssen PA, Niemegeers CJ, Schellekens KH, et al. The pharmacology of penfluridol (R 16341) a new potent and orally long-acting neuroleptic drug. Eur J Pharmacol 1970;11:139–54.
- Woo HM, Lee YS, Roh EJ, et al. Synthesis and biological evaluation of 4-piperidinecarboxylate and 4-piperidinecyanide derivatives for T-type calcium channel blockers. Bioorg Med Chem Lett 2011;21:5910–5.
- Nam G, Choi KI, Kim HR, et al. Pyrazolylmethylamine compounds as calcium channel modulators and preparation method thereof. U.S. Patent 8,299,072 B2; Oct. 30, 2012.
- Cho YS, Min SJ, Pae AN, et al., Pyrazolyl piperazine compounds as calcium channel antagonists. Kr. Patent 10-1351338 B1; Jan. 14, 2014.
- Doddareddy MR, Jung HK, Lee JY, et al. First pharmacophoric hypothesis for T-type calcium channel blockers. Bioorg Med Chem 2004;12:1605–11.
- Kim JH, Keum G, Chung H, Nam G. Synthesis and T-type calcium channel-blocking effects of aryl(1,5-disubstituted-pyrazol-3-yl)methyl sulfonamides for neuropathic pain treatment. Eur J Med Chem 2016;123:665–72.
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature 2006;440:463–9.
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992;50:355–63.
- Aley KO, Levine JD. Rapid onset pain induced by intravenous streptozotocin in the rat. J Pain 2001;2:146–50.