
Abstract
A series of new 1,3-diaryltriazene sulfonamides was synthesised by reaction of diazonium salt of metanilamide (3-aminobenzene sulfonamide) with substituted aromatic amines. The obtained new compounds were assayed as inhibitors of four physiologically and pharmacologically relevant human (h) isoforms of carbonic anhydrases (CA, EC 4.2.1.1), specifically, hCA I, hCA II, and hCA VII (cytosolic isoforms), as well as the tumour-associated membrane-bound isoform hCA IX. All isoforms investigated here were inhibited by the newly synthesised 1,3-diaryltriazene sulfonamide derivatives from the micromolar to the nanomolar range. The cytosolic isoforms were inhibited with Kis in the range of 92.3–8371.1 nM (hCA I), 4.3–9194.0 nM (hCA II), and 15.6–9477.8 nM (hCA VII), respectively. For the membrane-bound tumour-associated isoform hCA IX, the KI-s ranged between 50.8 and 9268.5 nM. The structure–activity relationship (SAR) with these newly synthesised metanilamide derivatives are discussed in detail.
Introduction
Carbonic anhydrases (CAs, also known as carbonate dehydratase, EC 4.2.1.1) are metalloenzymes present in Archaea, prokaryotes and eukaryotes, that catalyse the efficient interconversion of CO2 to HCO3− and protons via a ping-pong mechanism under physiological conditionsCitation1–6. This physiologically very simple, but highly relevant reaction plays an important role for the regulation of many physiologic processes in all living organisms. Up to now, seven genetically distinct CA families (α-, β-, γ-, δ-, ζ-, η, and θ-CAs) were described in various taxa, for all of them with numerous isoforms being present in all the investigated organismsCitation1–6.
In humans, 15 different isoforms have been described, all belonging to the α-CA family, with some of them being cytosolic (hCA I-III, VII, and XIII), others membrane-bound (hCA IV, IX, XII, and XIV), two mitochondrial (hCA VA and VB), as well as one of them secreted in saliva and milk (hCA VI). Since these isoforms play an important role in acid–base regulation, gluconeogenesis and other biosynthetic reactions, electrolyte secretion, bone resorption/calcification, and tumorigenicity, their inhibition/activation may be exploited in several diseases, including, glaucoma, obesity, neuropathic pain, arthritis, Alzheimers’ disease, and more recently cancerCitation1–6.
Primary sulfonamides and their isoesters (sulfamides, sulfamates) are the most widely studied CA inhibitors since the late 50’s and some of them have been used as drugs for decades. More recently, one of the sulfonamide-based CA inhibitor (CAI), which is the ureido-substituted derivative SLC-0111 (), was possessing a highly effective hCA IX/XII inhibitory action, reached to Phase I/II clinical trials for the treatment of advanced, metastatic solid cancersCitation7–9.
Figure 1. Clinically used triazene substituted compounds (TMZ and DTIC) and efficient CAI SLC-0111 (phase I/II trials for the advanced metastatic solid tumours).
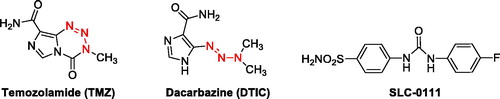
Triazenes are a diverse group of compounds which are amenable to many synthetic transformations and are also used for different applications, such as natural product synthesis, combinatorial chemistry, and biomedical applicationsCitation10. On the other hand, triazene compounds of clinical interest (such as Temozolomide and Dacarbazine), are a group of anticancer alkylating agents, with excellent pharmacokinetic properties and limited toxicityCitation10 ().
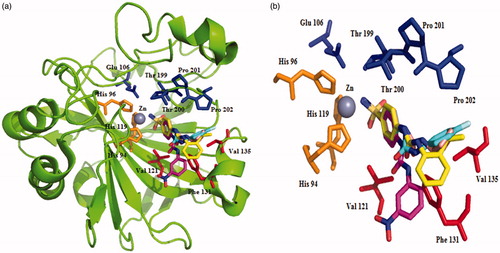
The X-ray crystal structure of SLC-0111 bound to hCA II as well as of four of its congeners, with various tail moieties was reportedCitation9. As shown in , the benzenesulfonamide fragment of molecules is rather superimposable for the four derivatives, whereas the ureido fragment allows a quite flexible orientation of the tail moieties in various parts of the active site, depending on nature and substitution pattern of the R moietyCitation9. This has as a consequence the fact that some of these compounds show a rather impressive isoform specificity. For example, SLC-0111 is an effective inhibitor of only hCA IX, XII, and XIV, being a weak inhibitor of off-target isoforms such as hCA I, II, or IV.Citation9
Figure 2. Ribbon diagram (a) and active site detail of the adducts with ureido-substituted benzenesulfonamide CAIs (b); SLC-0111 (cyan, pdb: 3N4B), 4–(3-(3-nitrophenyl)ureido) benzenesulfonamide (pink, pdb: 3N2P), 4–(3-(2-isopropylphenyl)ureido) benzenesulfonamide (yellow, pdb: 3N3J) and 4–(3-cyclopentylureido) benzenesulfonamide (light orange, pdb: 3MZC) (superimposed)Citation9. Figure made using PyMol (Delano Scientific).
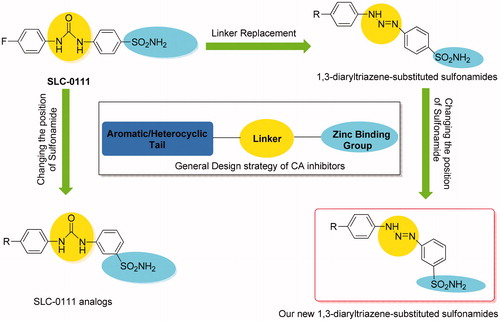
In continuation of our recent interest in CAIsCitation11, in this work, we report the synthesis and hCA I, II, VII, and IX inhibitory activity of new 1,3-diaryltriazene sulfonamides 4(a–h) obtained from the reaction of the diazonium salt of metanilamide with different substituted aromatic amines ().
Figure 3. General CA inhibitor design structure and design strategy of the reported 1,3-diaryltriazen-substituted sulfonamide derivatives starting from SLC-0111.
Materials and methods
General
All chemicals and anhydrous solvents were purchased from Sigma-Aldrich, Merck, Alfa Aesar and TCI and used without further purification. Melting points (mp) were determined with SMP30 melting point apparatus in open capillaries and are uncorrected. FT-IR spectra were recorded by using Perkin Elmer Spectrum 100 FT-IR spectrometer. Nuclear Magnetic Resonance (1H-NMR and 13C-NMR) spectra of compounds were recorded using a Bruker Advance III 300 MHz spectrometer in DMSO-d6 and TMS as an internal standard operating at 300 MHz for 1H-NMR and 75 MHz for 13C-NMR. Thin layer chromatography (TLC) was carried out on Merck silica gel 60 F254 plates.
General procedure for preparation 1,3-diaryltriazene sulfonamide derivatives 4(a-h)
A solution of metanilamide 1 (5 mmol) in 1.5 ml of conc. hydrochloric acid and 3 ml of water was cooled to 0–5 °C, sodium nitrite (7 mmol) in 3 ml of water was added dropwise to this solution during about 15–20 min under continuous stirring. The mixture was stirred about 20 min at 0–5 °C, and diazonium solution was added to aniline solution (prepared by 5 mmol anilines in 5 ml of MeOH) by adjusting the pH around 6–7 with simultaneous addition of saturated sodium acetate. Then, the reaction mixture was stirred 3 h at 0–5 °C and overnight at room temperature in dark. The obtained colorful mixture was filtered off, washed several times with cold water and the crystallized from ethanol. The final desired products 4(a–h) were dried under vacuum, kept under dark and fully characterised by FT-IR, 1H-NMR, 13C-NMR, and melting points.
3–(3-(4-fluorophenyl)triaz-1-en-1-yl) benzenesulfonamide (4a).
Yield: 85%; Color: light brown; mp: 140–142 °C; FT-IR (cm−1): 3333, 3251 (NH2), 1599, 1497 (asymmetric), 1314, 1145 (symmetric) (S=O), 1094; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 12.73 (s, 1H, –NH–), 7.87 (s, 1H, Ar-H), 7.75–7.48 (m, 5H, Ar-H), 7.46 (s, 2H, –SO2NH2), 7.28 (t, 2H, J = 2.2, Ar-H): 13C-NMR (DMSO-d6, 75 MHz, δ ppm): 158.3, 151.5, 145.7, 138.6, 130.6, 124.3, 123.6, 119.8, 116.3, 115.8.
4–(3-(3-sulfamoylphenyl)triaz-2-en-1-yl) benzoic acide (4b).
Yield: 70%; Color: yellow; mp: 161–163 °C; FT-IR (cm−1): 3373, 3245 (NH2), 1605, 1526, 1405 (asymmetric), 1335, 1161 (symmetric) (S=O), 1097; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 12,88 (br.s, 1H, -COOH), 12.75 (s, 1H, –NH–), 7.92 (s, 1H, Ar-H), 7.79–7.52 (m, 5H, Ar-H), 7.46 (s, 2H, –SO2NH2), 7.30 (t, 2H, J = 2.3, Ar-H): 13C-NMR (DMSO-d6, 75 MHz, δ ppm): 179.6, 159.5, 151.2, 146.2, 139.5, 130.8, 124.7, 123.3, 119.5, 116.2, 115.1.
3–(3-(4-cyanophenyl)triaz-1-en-1-yl) benzenesulfonamide (4c).
Yield: 75%; Color: light yellow; mp: 170–172 °C; FT-IR (cm−1): 3369, 3266 (NH2), 2218 (CN), 1606, 1521 (asymmetric), 1326, 1139 (symmetric) (S=O), 1094; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 13.01 (s, 1H, –NH–), 8.01 (s, 1H, Ar-H), 7.85–7.72 (m, 5H, Ar-H), 7.71–7.61 (m, 2H, Ar-H), 7.50 (s, 2H, –SO2NH2),: 13C-NMR (DMSO-d6, 75 MHz, δ ppm): 160.1, 151.8, 146.6, 139.8, 131.0, 125.2, 123.9, 119.8, 118.2, 116.6, 115.3.
3–(3-(4-butoxyphenyl)triaz-1-en-1-yl) benzenesulfonamide (4d).
Yield: 78%; Color: brown; mp: 140–143 °C; FT-IR (cm−1): 3362, 3267 (NH2), 1596, 1503 (asymmetric), 1333, 1147 (symmetric) (S=O), 1092; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 12.92 (s, 1H, –NH–), 7.96 (s, 1H, Ar-H), 7.83–7.70 (m, 5H, Ar-H), 7.69–7.62 (m, 2H, Ar-H), 7.49 (s, 2H, –SO2NH2), 3.92 (t, 2H, –OCH2CH2CH2CH3), 2.00–1.95 (m, 2H, –OCH2CH2CH2CH3), 1.68–1.60 (m, 2H, -OCH2CH2CH2CH3), 0.98 (t, 3H, –OCH2CH2CH2CH3): 13C-NMR (DMSO-d6, 75 MHz, δ ppm): 159.8, 151.4, 146.1, 139.5, 131.3, 125.7, 123.2, 119.5, 116.2, 115.5, 64.8, 32,5, 19.9, 15.7.
3–(3-(4-methoxyphenyl)triaz-1-en-1-yl) benzenesulfonamide (4e).
Yield: 82%; Color: dark red; mp: 126–128 °C; FT-IR (cm−1): 3337, 32578 (NH2), 1598, 1498 (asymmetric), 1303, 1147 (symmetric) (S=O), 1092; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 13.00 (s, 1H, –NH–), 7.99 (s, 1H, Ar-H), 7.82–7.75 (m, 5H, Ar-H), 7.73–7.66 (m, 2H, Ar-H), 7.51 (s, 2H, –SO2NH2),: 13C-NMR (DMSO-d6, 75 MHz, δ ppm): 159.7, 150.8, 146.7, 139.3, 131.2, 125.7, 123.6, 119.5, 116.8, 115.5, 55.3.
3–(3-(3,5-dimethylphenyl)triaz-1-en-1-yl)benzenesulfonamide (4f).
Yield: 78%; Color: orange; mp: 162–164 °C; FT-IR (cm−1): 3381, 3264 (NH2), 1602, 1488 (asymmetric), 1307, 1137 (symmetric) (S=O), 1090; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 8.13 (s, 1H, Ar-H), 7.95–7.66 (m, 2H, Ar-H), 7.72–7.66 (m, 2H, Ar-H), 7.51 (s, 2H, –SO2NH2), 6.45 (s, 2H, Ar-H), 2.50 (s, 6H, -CH3): 13C-NMR (DMSO-d6, 75 MHz, δ ppm): 160.3, 151.2, 146.9, 139.6, 131.6, 125.8, 123.5, 119.8, 116.2, 115.1, 21.8.
3–(3-(3,4-dimethoxyphenyl)triaz-1-en-1-yl)benzenesulfonamide (4g).
Yield: 75%; Color: dark brown; mp: 113–115 °C; FT-IR (cm−1): 3311, 3215 (NH2), 1621, 1549, 1489 (asymmetric), 1378, 1150 (symmetric) (S=O), 1089; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 8.35 (s, 1H, Ar-H), 8.17–8.12 (m, 1H, Ar-H), 7.76–7.59 (m, 3H, Ar-H), 7.52 (s, 2H, –SO2NH2), 7.40 (s, 1H, Ar-H), 6.47 (s, 1H, Ar-H), 3.88 (s, 3H, -OCH3), 3.80 (s, 3H, -OCH3),: 13C-NMR (DMSO-d6, 75 MHz, δ ppm): 160.8, 151.7, 146.5, 139.3, 138.2, 131.3, 130.2, 125.3, 123.2, 119.3, 116.8, 115.5, 55.8, 55.6.
3–(3-(3,4-dichlorophenyl)triaz-1-en-1-yl)benzenesulfonamide (4h).
Yield: 68%; Color: light yellow; mp: 122–124 °C; FT-IR (cm−1): 3415, 3332, 3322 (NH2), 1602, 1524, 1470 (asymmetric), 1323, 1150 (symmetric) (S=O), 1121; 1H-NMR (DMSO-d6, 300 MHz, δ ppm): 8.38 (s, 1H, Ar-H), 8.21–8.16 (m, 1H, Ar-H), 7.75–7.60 (m, 3H, Ar-H), 7.50 (s, 2H, –SO2NH2), 7.44 (s, 1H, Ar-H), 6.48 (s, 1H, Ar-H): 13C-NMR (DMSO-d6, 75 MHz, δ ppm): 161.1, 152.5, 146.9, 139.8, 138.5, 131.7, 130.5, 125.6, 123.1, 119.5, 116.3, 115.1.
CA inhibition assay
An SX.18 MV-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the inhibition of various CA isozymesCitation12. Phenol Red (at a concentration of 0.2 mM) has been used as an indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.4) as a buffer, 0.1 M Na2SO4 or NaClO4 (for maintaining constant the ionic strength; these anions are not inhibitory in the used concentration), following the CA-catalyzed CO2 hydration reaction for a period of 5–10 s. Saturated CO2 solutions in water at 25 °C were used as substrate. Stock solutions of inhibitors were prepared at a concentration of 10 mM (in DMSO-water 1:1, v/v) and dilutions up to 0.01 nM done with the assay buffer mentioned above. At least seven different inhibitor concentrations have been used for measuring the inhibition constant. Inhibitor and enzyme solutions were preincubated together for 10 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. Triplicate experiments were done for each inhibitor concentration, and the values reported throughout the paper is the mean of such results. The inhibition constants were obtained by nonlinear least-squares methods using the Cheng–Prusoff equation, as reported earlier, and represent the mean from at least three different determinationsCitation13–17. All CA isozymes used here were recombinant proteins obtained as reported earlier by our groupCitation18,Citation19.
Results and discussion
Chemistry
The rationale behind the design of these new 1,3-diaryltriazene sulfonamide derivatives presented in this work is based on our recent reportCitation11, in which we showed that novel 1,3-diraryltriazene-substituted sulfonamide derivatives possess interesting CA inhibitory properties. These compounds showed potent inhibition against the cytosolic hCA II, with great selectivity versus hCA I, hCA VII, and hCA IX inhibition. Thus, we decided to apply the same procedure by changing of position of the sulfonamide moiety from para to meta, in order to investigate whether the potency comes from triazene linker or the position of the sulfonamide zinc-binding group.
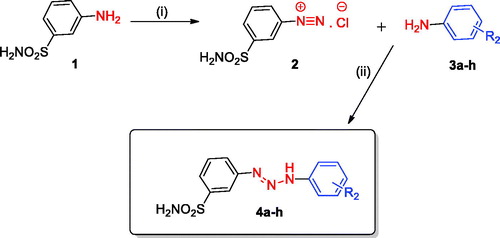
A series of structurally diverse 1,3-diaryltriazene sulfonamide derivatives were synthesised according to general synthetic route shown in Scheme 1Citation11,Citation18. Briefly, the diazonium salt derived of metanilamide was reacted with different substituted aromatic amines, leading to the formation of 1,3-diaryltriazene sulfonamides. The chemical structures of these novel 1,3-diaryltriazene sulfonamide derivatives reported here were confirmed by using several analytical and spectral data (see experimental part for details).
Scheme 1. General synthetic route for the synthesis of the 1,3-diaryltriazene sulfonamide derivatives 4(a–h). Reagent and conditions: (i) H2O, HCl, NaNO2, 0–5 °C, 30 min, (ii) Substituted aromatic anilines 3(a–h), MeOH, H2O, sodium acetate, 0–5 °C 3h, r.t. overnight.
CA inhibition studies
The newly synthesised 1,3-diaryltriazene sulfonamides 4(a–h) were evaluated as inhibitors of four physiologically relevant CA isoforms, the cytosolic hCA I, hCA II, and hCA VII, and the transmembrane tumour-associated hCA IX, by a stopped-flow CO2 hydrase assayCitation12. The clinically used sulfonamide acetazolamide (AAZ) was used as a positive control.
The following structure-activity relationship (SAR) may be drawn regarding the inhibition data of , for the series of 1,3-diaryltriazene substituted sulfonamides 4(a–h):
Table 1. In vitro hCA I, hCA II, hCA VII, and hCA IX inhibition data with 1,3diaryltriazene-substituted sulfonamides 4(a–h) investigated here, and standard sulfonamide inhibitor Acetazolamide (AAZ) by a stopped flow CO2 hydrase assayCitation12.
The ubiquitous cytosolic isoform hCA I, which is highly abundant among others in the gastrointestinal tract and red blood cells, was moderately inhibited by all compounds investigated here, with inhibition constants in the range of 92.3–8371.1 nM. Compound 4b (possessing a 4-COOH moiety) showed the best inhibition potency against hCA I, with a KI of 92.3 nM. Interestingly, the 3,4-disubstituted compounds 4g (3,4-diMeO) and 4h (3,4-diCl) displayed the lowest hCA I inhibition activity among this series, with Kis of 3853.9 and 8371.1 nM, respectively.
An interesting inhibition profile with the reported 1,3-diaryltriazene-substituted matanilamide derivatives was observed for the physiologically dominant isoform hCA II, for which Kis spanning between 4.3 and 9194.0 nM were obtained. The most effective inhibitor was 4f, which has the 3,5-dimethyl substitution pattern and a KI of 4.3 nM, being almost 3 times more effective compared to the standard inhibitor AAZ ().
Another cytosolic isoform, hCA VII, mostly present in the brain, was inhibited by most of the new compounds investigated here in the micromolar range, except the compound 4f which had a KI of 15.6 nM.
The inhibition potential of novel 1,3-diaryltriazene-substituted metanilamide derivatives 4(a–h) against hCA IX was not satisfactory since all the compounds reported here were rather inefficient hCA IX inhibitors, with KIs in the range of 50.8–9268.5 nM (compared to AZA which has an inhibition constant of 25 nM).
Conclusions
We investigated a series of 1,3-diaryltriazene-subsituted sulfonamide derivatives as CA inhibitors, continuing our most recent research on 1,3-diaryltriazene based compounds. The compounds were synthesised by reaction of diazonium salt of metanilamide with substituted aromatic amines. The new compounds discovered here were assessed as CAIs, against several pharmacologically relevant isoforms, namely hCA I, hCA II, hCA VII (cytosolic isoforms), as well as membrane-bound tumor-associated isoform hCA IX. Only compound 4f showed potent inhibition against hCA II and hCA VII with Kis of 4.3 and 15.6 nM, respectively. Since hCA II is an important drug target for several diseases such as, glaucoma, retinis pigmentosa, and edema, and hCA VII recently validated antineuropathic pain target, some of these compounds might be improved and used potent CAIs and potential drug candidates.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- (a) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88; (b) Capasso C., Supuran CT. An overview of the alpha-beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32; (c) Supuran CT, Carbon-versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95.
- (a) Supuran CT, Capasso C. The eta-class carbonic anhydrases as drug targets for antimalarial agents. Expert Opin Ther Targets 2015;1:551–63; (b) Del Prete S, De Luca V, De Simone G, et al. Cloning, expression and purification of the complete domain of the g-carbonic anhydrase from Plasmodium falciparum. J Enzyme Inhib Med Chem 2016;31:54–9; (c) Vullo D, Del Prete S, Fisher GM, et al. Sulfonamide inhibition studies of theg-class carbonic anhydrase from the malaria pathogen Plasmodium falciparum. Bioorg Med Chem 2015;23:526–31.
- (a) Supuran C.T. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60; (b) Supuran C.T. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32; (c) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81; (d) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov 2011;10:767–77; (e) Supuran CT, Vullo D, Manole G, Casini A, Scozzafava A. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68.
- (a) Akocak S, Ilies MA. Next-generation primary sulfonamide carbonic anhydrase inhibitors. In: Supuran C.T., Cappasso C., eds. Targeting Carbonic Anhydrases. London: Future Science; 2014:35–51; (b) Akocak S, Alam MR, Shabana AM, et al. PEGylated Bis-sulfonamide carbonic anhydrase inhibitors can efficiently control the growth of several carbonic anhydrase IX-expressing carcinomas. J Med Chem 2016;59:5077–88; (c) Shabana AM, Mondal UK, Alam R., et al. pH-Senstive multiligand gold nanoplatform targeting carbonic anhydrase IX enhances the delivery of Doxorubicin to hypoxic tumor spheroids and overcomes the hypoxia-induced chemoresistance. ACS Appl Mater Interfaces 2018;10:17792–808.
- Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev 2018; in press.
- (a) Gul HI, Yamali C, Yesilyurt F, et al. Microwave-assisted synthesis and bioevaluation of new sulfonamides. J Enzym Inhib Med Ch 2017;32:369–74; (b) Gulçin İ., Abbasova M., Taslimi P., et al. Synthesis and biological evaluation of aminomethyl and alkoxymethyl derivatives as carbonic anhydrase, acetylcholinesterase and butyrylcholinesterase inhibitors. J Enzyme Inhib Med Chem 2017;32:1174–82; (c) Guzel-Akdemir O., Akdemir A., Karali N., et al. Discovery of novel isatin-based sulfonamides with potent and selective inhibition of the tumor-associated carbonic anhydrase isoforms IX and XII. Org Biomol Chem 2015;13:6493–9; (d) Alterio V, Di Fiore A, D'Ambrosio K, Supuran CT, De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68; (e) Abbate F, Winum JY, Potter BV, Casini A, Montero JL, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with EMATE, a dual inhibitor of carbonic anhydrases and steroid sulfatase. Bioorg Med Chem Lett 2004;14:231–34.
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902.
- Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res 2011;71:3364–76.
- Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371.
- (a) Kimball DB, Haley MM., Triazenes: a versatile tool in organic synthesis. Angew Chem Int Ed 2002;41:3338–51; (b) Marchesi F, Turriziani M, Tortorelli G, et al. Triazene compounds: mechanism of action and related DNA repair systems. Pharmacol Res 2007;56:275–87.
- (a) Akocak S, Lolak N, Nocentini A, et al. Synthesis and biological evaluation of novel aromatic and heterocyclic bis-sulfonamide Schiff bases as carbonic anhydrase I, II, VII and IX inhibitors. Bioorg Med Chem 2017;25:3093–7; (b) Akocak S, Lolak N, Bua S, et al. Synthesis and biological evaluation of novel N,N′-diaryl cyanoguanidines acting as potent and selective carbonic anhydrase II inhibitors. Bioorg Chem 2018;77:245–51; (c) Lolak N, Akocak S., Bua S, et al. Design and synthesis of novel 1,3-diaryltriazene-substituted sulfonamides as potent and selective carbonic anhydrase II inhibitors. Bioorg Chem 2018;77:542–47.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Draghici B, Vullo D, Akocak S, et al. Ethylene bis-imidazoles are highly potent and selective activators for isozymes VA and VII of carbonic anhydrase, with a potential nootropic effect. Chem Commun 2014;50:5980–3; (b) Akocak S, Lolak N, Vullo D, et al. Synthesis and biological evaluation of histamine Schiff bases as carbonic anhydrase I, II, IV, VII, and IX activators. J Enzyme Inhib Med Chem 2017;32:1305–12; (c) Buzás GM, Supuran CT. The history and rationale of using carbonic anhydrase inhibitors in the treatment of peptic ulcers. In memoriam Ioan Puşcaş (1932–2015). J Enzyme Inhib Med Chem 2016;31:527–33; (d) Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol 2011;2:34; (e) Nishimori I, Onishi S, Takeuchi H, Supuran CT. The alpha and beta classes carbonic anhydrases from Helicobacter pylori as novel drug targets. Curr Pharm Des 2008;14:622–30.
- (a) Senturk M, Gulcin I, Beydemir S, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9; (b) Carradori S, Secci D, De Monte C, et al. A novel library ofsaccharin and acesulfame derivatives as potent and selective inhibitors of carbonic anhydrase IX and XII isoforms. Bioorg Med Chem 2016;24:1095–105; (c) Nocentini A, Bua S, Lomelino CL, et al. Discovery of new sulfonamide carbonic anhydrase IX inhibitors incorporating nitrogenous bases. ACS Med Chem Lett 2017;8:1314–19.
- (a) Ilies MA, Vullo D, Pastorek J, et al. Carbonic anhydrase inhibitors. Inhibition of tumor-associated isozyme IX by halogenosulfanimamide and halogenophenylaminobenzolamide derivatives. J Med Chem 2003;46:2187–96; (c) Ward C, Langdon SP, Mullen P, et al. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat Rev 2013;39:171–79; (d) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett. 2005;15:3102–08; (e) Casey JR, Morgan PE, Vullo D, et al. Carbonic anhydrase inhibitors. Design of selective, membrane-impermeant inhibitors targeting the human tumor-associated isozyme IX. J Med Chem 2004;47:2337–47.
- (a) Krall N, Pretto F, Decurtins W, et al. A small‐molecule drug conjugate for the treatment of carbonic anhydrase ix expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–35; (b) Rehman SU, Chohan ZH, Gulnaz F, et al. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzyme Inhib Med Chem 2005;20:333–40; (c) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: Structure‐activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73; (d) Dubois L, Peeters S, Lieuwes NG, et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 2011;99:424–31; (e) Chohan ZH, Munawar A, Supuran CT. Transition metal ion complexes of Schiff-bases. Synthesis, characterization and antibacterial properties. Met Based Drugs 2001;8:137–43; (f) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal β-class (Cab) and γ-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–08.
- (a) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–38; (b) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016; 31: 689–94; (c) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem. 2017;32:1002–11; (d) Alper Türkoğlu E, Şentürk M, Supuran CT, Ekinci D. Carbonic anhydrase inhibitory properties of some uracil derivatives. J Enzyme Inhib Med Chem 2017;32:74–7; (e) Soydan E, Güler A, Bıyık S, et al. Carbonic anhydrase from Apis mellifera: purification and inhibition by pesticides. J Enzyme Inhib Med Chem 2017;32:47–50.
- (a) Unsalan S., Cikla P., Kucukguzel SG., et al. Synthesis and characterization of triazenes derived from sulfonamides. Marmara Pharm J 2011;15:11–7; (b) Zovko TC., Brozovic A., Piantanida I., et al. Synthesis and biological evaluation of 4-nitro-substituted 1,3-diaryltriazenes as a novel class of potent antitumor agents. Eur J Med Chem 2011;46:2971–83; (c) Hill DT., Stanley KG., Williams JE., et al. 1,3-Diaryltriazenes: a new class of anorectic agents. J Med Chem 1983;26:865–9.
- (a) Diaz JR, Fernández Baldo M, Echeverría G, et al. A substituted sulfonamide and its Co (II), Cu (II), and Zn (II) complexes as potential antifungal agents. J Enzyme Inhib Med Chem 2016;31:51–62; (b) Menchise V, De Simone G, Alterio V, et al. Carbonic anhydrase inhibitors: stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J Med Chem 2005;48:5721–7; (c) Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors—part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents in rabbits. Eur J Med Chem 1998;33:247–54; (d) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8; (e) Şentürk M, Gülçin İ, Beydemir Ş, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9; (f) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47.