
Abstract
A series of new Olaparib derivatives was designed and synthesized, and their inhibitory activities against poly (ADP-ribose) polymerases-1 (PARP-1) enzyme and cancer cell line MDA-MB-436 in vitro were evaluated. The results showed that compound 5l exhibited the most potent inhibitory effects on PARP-1 enzyme (16.10 ± 1.25 nM) and MDA-MB-436 cancer cell (11.62 ± 2.15 μM), which was close to that of Olaparib. As a PARP-1 inhibitor had been reported to be viable to neuroprotection, in order to search for new multitarget-directed ligands (MTDLs) for the treatment of Alzheimer’s disease (AD), the inhibitory activities of the synthesized compounds against the enzymes AChE (from electric eel) and BChE (from equine serum) were also tested. Compound 5l displayed moderate BChE inhibitory activity (9.16 ± 0.91 μM) which was stronger than neostigmine (12.01 ± 0.45 μM) and exhibited selectivity for BChE over AChE to some degree. Molecular docking studies indicated that 5l could bind simultaneously to the catalytic active of PARP-1, but it could not interact well with huBChE. For pursuit of PARP-1 and BChE dual-targeted inhibitors against AD, small and flexible non-polar groups introduced to the compound seemed to be conducive to improving its inhibitory potency on huBChE, while keeping phthalazine-1-one moiety unchanged which was mainly responsible for PARP-1 inhibitory activity. Our research gave a clue to search for new agents based on AChE and PARP-1 dual-inhibited activities to treat Alzheimer’s disease.
Introduction
Poly (ADP-ribose) polymerases-1 (PARP-1) is a pivotal nuclear enzyme ubiquitously expressed in eukaryotic cells. It can detect DNA single-strand break and catalyse the addition of ADP-ribose units to acceptor proteins, thus facilitating DNA repair process and promoting cell survivalCitation1,Citation2. Inhibition of PARP-1 accelerates the damage of injured DNA, which especially leads to synthetic lethality in DNA-repairing-deficient cancer cells, for example BRCA1/2-deficient cells. Therefore, PARP-1 has become an attractive antitumor targetCitation3–5. PARP-1 inhibitors can be utilized not only as a single agent for the treatment of BRCA1- or BRCA2-deficient cancers, but in combination with DNA-damaging therapeutics (radiation or chemotherapy) to improve their potencies by blocking DNA-repairing process. Then, PARP-1 inhibitors have been extensively investigated as potential anticancer drugsCitation6,Citation7.
In addition to being used for cancer treatment, recent researches indicate that PARP-1 inhibitors have potentially therapeutic value in Alzheimer's disease (AD)Citation8–10. PARP-1 utilizes nicotinamide adenine dinucleotide (NAD+) as substrate to catalyse the transfer of ADP-ribose units to nuclear target proteins upon oxidative stress and DNA injury. Over-activation of PARP-1 will lead to cellular depletion of NAD+ and ATP, and then, energy deficiency slows glycolysis and mitochondrial respiration rates, resulting in controlled nerve cell death. This PARP-related specific neurodegenerative mechanism was termed as parthanatosCitation10. Meanwhile, over-activated PARP-1 will increase its interaction with transcription regulators such as nuclear receptors, sirtuins, and other metabolic transcription factors. Further, it will also lead to the impairment of mitochondrial function that seems to be an early and critically important event in the pathogenesis of ADCitation9,Citation11. Although the role of PARP-1 in human AD is not fully elucidated and many mechanisms are under investigations, accumulating evidences suggest that PARP-1 inhibitors are viable to neuroprotection and PARP-1 has emerged as a potential therapeutic target for ADCitation12,Citation13. As for AD, it still imposes a great threat to human health. Currently, most of the approved drugs for AD treatment are acetylcholinesterase (AChE) inhibitors, namely donepezil, tacrine, rivastigmine and galantamine. Nevertheless, these drugs modestly alleviate the symptoms but cannot cure brain damage or stop neuronal degenerationCitation14. Therefore, discovery for effective anti-AD drugs remains an enormous challenge to medicinal chemistry communities. In fact, in addition to AChE, there is another cholinesterase, butyrylcholinesterase (BChE), in the synaptic gap of neuronal cells to efficiently hydrolyse acetylthiocholine (ACh), and its inhibitors have been reported to be more beneficial to AD in later stages of AD than AChE inhibitorsCitation15–17. In the light of the multifactorially pathological mechanisms of AD, developing multitarget-directed ligands (MTDLs) may be a more promising approach for its treatment. Some researches and reviews of MTDLs for AD have already been reportedCitation18,Citation19, but almost none of which is related to both PARP-1 and cholinesterase inhibitorsCitation20. In this regard, the PARP-1 and cholinesterase dual-targeted inhibitor of AD are worthy to be investigated.
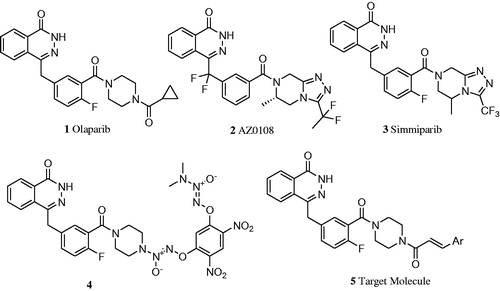
Olaparib is the first-in-class PARP-1 inhibitor marketed in 2014 as monotherapy for advanced BRCA-deficient ovarian cancer. However, its anticancer activity was not potent enoughCitation7,Citation21, and it showed dose-limiting toxicity that was more pronounced than that seen with the chemotherapeutic agents alone when it was utilized in combination with chemotherapeutic agents for cancer treatmentCitation22. To overcome these shortcomings, several modifications based on structural skeleton of Olaparib have been reported ()Citation21–24. The reported structure–activity relationship (SAR) revealed that 4-benzyl phthalazinone was the core scaffold responsible for moderate PARP-1-inhibiting potency, and a substituted piperazine at the meta position of benzyl moiety was conducive to enhancing activity and maintaining good oral bioavailabilityCitation7,Citation25. In view of the important therapeutic value of PARP-1 inhibitor for cancer and potentially for AD treatment, it was interesting and deserved to further explore structural optimization of Olaparib based on the existed SAR. With this aim in mind, we decided to keep the 4-benzyl phthalazinone group unchanged and replace the cyclopropane group of Olaparib by substituted aryl vinyl ones to constitute 3-aromatic α, β-unsaturated carbonyl moiety. On the one hand, the aryl vinyl groups are hydrophobic, just like cyclopropane group, which is helpful to improve oral bioavailabilityCitation7; on the other hand, 3-aromatic α, β-unsaturated carbonyl moiety is widely included into natural products, especially within chalcone framework that possesses a variety of bioactivities, including anticancer, inhibition of cholinesterases and neuroprotectionCitation26–28. Through the structural optimizations, we hoped to find a more potent PARP-1 inhibitor, at the same time, in order to explore new MTDLs for the treatment of AD, the inhibitory activities against AChE and BChE enzymes of these compounds were also evaluated. Here, we described the synthesis and bioactivities assays of 15 Olaparib derivatives; meanwhile, molecular dockings were conducted to investigate their interaction fashion with the corresponding proteins.
Figure 1. The structures of Olaparib analogs and target molecules.
Materials and methods
Chemistry
All reagents and solvents were obtained from commercially available sources and were used without further purification. Reaction progress was monitored using analytical thin layer chromatography (TLC) on precoated silica gel GF254 (Qingdao Haiyang Chemical Plant, Qing-Dao, China) plates and spots were detected under UV light (254 nm). Melting points were determined on a WRS-2B digital melting point apparatus and uncorrected. IR spectra were recorded using a Nicolet 380 Fourier-transform infrared (FTIR) spectrophotometer (Thermo, USA) from KBr pellets. 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE III-600 NMR spectrometer (Bruker Biospin Co., Switzerland) with tetramethylsilane (TMS) as the internal standard and CDCl3 or DMSO-d6 as solvent and known chemical shifts of residual proton signals of deuterated solvents (1H NMR: δ: 2.50 for DMSO-d6 and δ: 3.33 for H2O) or carbon signals of deuterated solvents (13C NMR: δ: 39.52 for DMSO-d6) as internal standard. MS (ESI) measurement was conducted on an Agilent 1100 LC-MS spectrometer (Agilent, Palo Alto, USA).
Dimethyl (3-oxo-1, 3-dihydro-isobenzofuran-1-yl)-phosphonate (7)
Compounds 7, 8 and 9 were prepared according to the literature procedureCitation24,Citation29. A mixture of 2-formylbenzoic acid 6 (2.0 g, 15.0 mmol) and dimethyl phosphate (4.0 g, 36.0 mmol) was heated to 100 °C in oil bath and stirred for 8 h. Subsequently, the mixture was cooled to room temperature and then poured into ice-cold water (20 mL), which was followed by extraction with dichloromethane (3 × 30 mL). The combined organic extracts were dried over MgSO4 and were evaporated in vacuum to produce a white solid which was crystallized in ethanol to yield 2.25 g compound 7. Yield: 62.3%, mp: 96.1–98.2 °C. 1H-NMR (CDCl3, 600 MHz), δ: 7.93 (d, J = 7.68 Hz, 1H, ArH), 7.76–7.70 (m, 2H, ArH), 7.58 (t, J = 7.56 Hz, 1H, ArH), 5.70 (d, J = 10.98 Hz, 1H, CH), 3.91 (d, J = 10.92 Hz, 3H, OCH3), 3.58 (d, J = 10.62 Hz, 3H, OCH3).
2-Fluoro-5-[(3-oxo-2-benzofuran-1-ylidene) methyl] benzonitrile (8)
To a mixture of 7 (4.3 g, 17.8 mmol) and 2-fluoro-5-formylbenzonitrile (2.6 g, 17.5 mmol) in anhydrous THF (40 mL) was added triethylamine (1.8 mL, 13 mmol) dropwise in 30 min, and the temperature was maintained below 15 °C. The reaction mixture was slowly warmed to room temperature. The progress of the reaction was monitored by TLC. After completion of the reactions, the reaction mixture was then concentrated in vacuo. The residue was slurried in water (50 mL) for 30 min, and the solid was collected by filtration, was washed with diethyl ether (2 × 10 mL) and was dried to afford 3.6 g of 8 in 78.5% yield. The material was carried forward without further purification.
2-Fluoro-5-[(4-oxo-3H-phthalazin-1-yl) methyl] benzoic Acid (9)
To a stirred suspension of 8 (2.1 g, 8.0 mmol) in water (20 mL) was added aqueous NaOH (10 mol/l, 3.5 mL) with subsequent heating to 90 °C for 1 h. The reaction mixture was cooled to 70 °C into which hydrazine hydrate (5.4 mL, 94.0 mmol) was added, and the mixture was stirred for 18 h at 70 °C before TLC indicated the reaction was completed. The mixture was cooled to ambient temperature and was acidified with HCl (4 mol/L) to pH 4, and then, the suspension was filtered and was washed with diethyl ether and dried to get 2.2 g of 9 as a light red powder. Yield: 93.7%, mp: 208.4–210.1 °C。1H-NMR (600 MHz, DMSO-d6), δ: 13.05 (s, 1H, COOH), 12.56 (s, 1H, NH), 8.26–8.20 (m, 1H, ArH), 7.94 (d, J = 8.0 Hz, 1H, ArH), 7.91–7.84 (m, 1H, ArH), 7.83–7.75 (m, 2H, ArH), 7.58–7.50 (m, 1H, ArH), 7.20 (m, 1H, ArH), 4.32 (s, 2H, CH2).
General procedure for preparation of intermediates 13a–o
Compounds 10a–o, 11a–o, 12a–o and 13a–o were prepared according to the literature proceduresCitation29,Citation30. To a stirred solution of various substituted benzaldehydes (or 2-furaldehyde) (24.0 mmol) in pyridine (15 mL), malonic acid (4.9 g, 48.0 mmol) and ammonium acetate (0.2 g, 23 mmol) were added. The mixture was stirred for 5 h at 85 °C and then cooled to room temperature, slowly diluted with ice water (100 mL). The solution was acidified with HCl to pH 2, and the precipitate formed. After filtered, the solid was washed with water until it had no pyridine smell. It was dried, which was followed by recrystallization from ethanol to give 10a–o in 50–80% yields. Compounds 10a–o (8.0 mmol) were dissolved in DMF (0.25 mL) to which oxalyl chloride was added dropwise at 0–5 °C. The mixture was stirred for 1.5 h at room temperature and then evaporated under reduced pressure. The residue 11a–o was added dichloromethane (DCM, 15 mL) to dissolve and sealed for use in next step. Tert-butoxycarbonylpiperazine (1.5 g, 8.0 mmol) and triethylamine (1.1 mL, 8.0 mmol) were dissolved in DCM (15 mL). After dropwise addition of 11a–n at 0–5 °C, the mixture was stirred for 12 h at room temperature. The mixture was washed with saturated NaHCO3 solution (2 × 15 mL), brine (30 mL), dried over Na2SO4 and evaporated under reduced pressure, and then, the residue was purified by silica gel column chromatography to obtain 12a–o in 40–70% yields. Finally, 12a–o (12.0 mmol) was dissolved in DCM (30 mL) to which trifluoroacetic acid (78 mmol) was added, and then, the mixture was stirred for 8 h at room temperature. Water (30 mL) was added to the mixture, which was stirred for another 0.5 h and then separated. The water layer was extracted with DCM (3 × 10 mL), the combined organic layers were dried over Na2SO4 and concentrated and purified by silica gel column chromatography to give 13a–o as yellow oils in 50–80% yieldsCitation1. HNMR data of 12a–o were shown below:
Tert-butyl 4-((E)-3-phenylacryloyl)piperazine-1-carboxylate (12a)
A white solid, yield: 63.3%, mp: 131.1–132.2 °C, 1H NMR (600 MHz, CDCl3) δ: 7.68 (d, J = 15.4 Hz, 1H, =CH), 7.51 (dd, J = 7.8, 1.4 Hz, 2H, ArH), 7.35 (dd, J = 8.3, 6.6 Hz, 2H, ArH), 6.84 (d, J = 15.4 Hz, 1H, =CH), 3.66–3.47 (m, 8H, NCH2), 1.47 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3-(3,4-dimethoxyphenyl)acryloyl]piperazine-1-carboxylate (12b)
A white solid, yield: 55.3%, mp: 124.9–125.1 °C, 1H NMR (600 MHz, CDCl3) δ: 7.87 (d, J = 15.6 Hz, 1H, =CH), 7.02 (s, 1H), 6.91 (d, J = 15.6 Hz, 1H, =CH), 6.83 (dt, J = 17.9, 5.9 Hz, 2H, ArH), 3.80 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.65–3.62 (m, 4H, NCH2), 3.50–3.36 (m, 4H, NCH2), 1.45 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3-(4-methoxyphenyl)acryloyl]piperazine-1-carboxylate (12c)
A yellow solid, yield: 56.3%, mp: 127.1–128.8 °C, 1H NMR (600 MHz, CDCl3) δ: 7.90 (d, J = 15.6 Hz, 1H, =CH), 7.47 (d, J = 7.6 Hz, 2H, ArH), 7.33–7.28 (m, 2H, ArH), 6.94 (d, J = 15.6 Hz, 1H, =CH), 3.88 (s, 3H, OCH3), 3.68–3.62 (m, 4H, NCH2), 3.53–3.16 (m, 4H, NCH2), 1.46 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3-(4-methylphenyl)acryloyl]piperazine-1-carboxylate (12d)
A light yellow solid, yield: 58.3%, mp: 149.6–151.3 °C, 1H NMR (600 MHz, DMSO-d6) δ: 7.88 (d, J = 15.6 Hz, 1H, =CH), 7.82–7.79 (m, 2H, ArH), 7.59–7.56 (m, 2H, ArH), 6.91 (d, J = 15.6 Hz, 1H, =CH), 3.77–3.48 (m, 6H, NCH2), 3.21–3.19 (m, 2H, NCH2), 2.31 (s, 3H, CH3), 1.45 (s, 9H, C (CH3)3);
Tert-butyl 4-[(E)-3-(2-methoxyphenyl)acryloyl]piperazine-1-carboxylate (12e)
A white solid, yield: 58.1%, mp: 114.7–115.8 °C, 1H NMR (600 MHz, CDCl3) δ: 7.91 (d, J = 15.6 Hz, 1H, =CH), 7.47 (d, J = 7.6 Hz, 1H, ArH), 7.33–7.28 (m, 1H, ArH), 6.98–6.88 (m, 3H, ArH and = CH), 3.87 (s, 3H, OCH3), 3.68–3.62 (m, 4H, NCH2), 3.53–3.16 (m, 4H, NCH2), 1.46 (s, 9H, C (CH3)3);
Tert-butyl 4-[(E)-3-(2,5-dimethoxyphenyl)acryloyl]piperazine-1-carboxylate (12f)
A light yellow solid, yield: 60.2%, mp: 147.0–147.6 °C, 1H NMR (600 MHz, CDCl3) δ: 7.85 (d, J = 15.6 Hz, 1H, =CH), 6.99 (d, J = 2.9 Hz, 1H, ArH), 6.92 (d, J = 15.6 Hz, 1H, =CH), 6.83 (dt, J = 17.9, 5.9 Hz, 2H, ArH), 3.80 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.65–3.61 (m, 4H, NCH2), 3.50–3.36 (m, 4H, NCH2), 1.45 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3-(4-fluorophenyl)acryloyl]piperazine-1-carboxylate (12g)
A white solid, yield: 62.1%, mp: 143.6–144.1 °C, 1H NMR (600 MHz, CDCl3) δ: 7.64 (d, J = 15.4 Hz, 1H, =CH), 7.54–7.45 (m, 2H, ArH), 7.09–7.00 (m, 2H, ArH), 6.76 (d, J = 15.4 Hz, 1H, =CH), 3.65–3.32(m, 8H, NCH2), 1.46 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3–(4-trifluoromethylphenyl)acryloyl]piperazine-1-carboxylate (12h)
A white solid, yield: 56.2%, mp: 173.1–173.6 °C, 1H NMR (600 MHz, CDCl3) δ: 7.68 (d, J = 15.4 Hz, 1H, =CH), 7.64–7.58 (m, 4H, ArH), 6.92 (d, J = 15.5 Hz, 1H, =CH), 3.66–3.48 (m, 8H, NCH2), 1.47 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3-(3-fluorophenyl)acryloyl]piperazine-1-carboxylate (12i)
A white solid, yield: 55.3%, mp: 121.8–122.2 °C, 1H NMR (600 MHz, CDCl3) δ: 7.62 (d, J = 15.4 Hz, 1H, =CH), 7.32 (d, J = 5.8 Hz, 1H, ArH), 7.26–7.20 (m, 1H, ArH), 7.03 (t, J = 8.5 Hz, 1H, ArH), 6.83 (d, J = 15.4 Hz, 1H, =CH), 3.65–3.47 (m, 8H, NCH2), 1.46 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3-(4-chlorophenyl)acryloyl]piperazine-1-carboxylate (12j)
A white solid, yield: 58.2%, mp: 187.1–187.5 °C, 1H NMR (600 MHz, DMSO-d6) δ: 7.73 (d, J = 8.4 Hz, 2H, ArH), 7.49–7.44 (m, 3H, ArH and = CH), 7.25 (d, J = 15.4 Hz, 1H), 3.58–3.31 (m, 8H, NCH2), 1.38 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3-(4-bromophenyl)acryloyl]piperazine-1-carboxylate (12k)
A white solid, yield: 65.3%, mp: 207.7–208.4 °C, 1H NMR (600 MHz, DMSO-d6) δ: 7.65 (d, J = 8.4 Hz, 2H, ArH), 7.57 (d, J = 8.4 Hz, 2H, ArH), 7.43 (d, J = 15.4 Hz, 1H, =CH), 7.26 (d, J = 15.4 Hz, 1H, =CH), 3.58–3.39 (m, 4H, NCH2), 3.32 (s, 4H, NCH2), 1.38 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3-(3-nitrophenyl)acryloyl]piperazine-1-carboxylate (12l)
A white solid, yield: 52.4%, mp: 145.1–145.8 °C, 1H NMR (600 MHz, DMSO-d6) δ: 7.80 (d, J = 6.6 Hz, 1H, ArH), 7.72 (d, J = 15.4 Hz, 1H, =CH), 7.67–7.62 (m, 2H, ArH), 7.59 (t, J = 7.2 Hz, 1H, ArH), 7.45 (d, J = 15.4 Hz, 1H, =CH), 3.60–3.39 (m, 4H, NCH2), 3.35–3.31 (m, 4H, NCH2), 1.39 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3-(4-nitrophenyl)acryloyl]piperazine-1-carboxylate (12m)
A white solid, yield:55.6%, mp: 212.1–213.3 °C, 1H NMR (600 MHz, DMSO-d6) δ: 7.65 (d, J = 8.4 Hz, 2H, ArH), 7.57 (d, J = 8.4 Hz, 2H, ArH), 7.43 (d, J = 15.4 Hz, 1H, =CH), 7.26 (d, J = 15.4 Hz, 1H, =CH), 3.58–3.31 (m, 4H, NCH2), 3.32 (s, 4H, NCH2), 1.38 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3-(3,4,5-trimethoxyphenyl)acryloyl]piperazine-1-carboxylate (12n)
A light yellow solid, yield:52.7%, mp: 137.3–138.2 °C, 1H NMR (600 MHz, CDCl3) δ: 7.58 (d, J = 15.3 Hz, 1H, =CH), 6.75–6.70 (m, 3H, ArH and = CH), 3.90–3.83 (m, 9H, OCH3), 3.69–3.64 (m, 4H, NCH2), 3.47 (s, 4H, NCH2), 1.46 (s, 9H, C (CH3)3).
Tert-butyl 4-[(E)-3-(furan-2-yl)acryloyl]piperazine-1-carboxylate (12o)
A brown yellow solid, yield: 42.3%, mp: 103.7–104.7 °C, 1H NMR (600 MHz, CDCl3) δ: 7.45 (d, J = 15.1 Hz, 1H, =CH), 7.42 (d, J = 1.4 Hz, 1H, furan-H), 6.75 (d, J = 15.1 Hz, 1H, =CH), 6.53 (d, J = 3.4 Hz, 1H, furan-H), 6.43 (dd, J = 3.4, 1.8 Hz, 1H, furan-H), 3.63 (t, J = 9.6 Hz, 4H, NCH2), 3.45 (t, J = 7.3 Hz, 4H, NCH2), 1.45 (s, 9H, C (CH3)3).
General procedure for preparation of target compounds 5a–o
To a solution of 9 (1.2 g, 4.1 mmol) in DCM (30 mL) were added N,N-Diisopropylethylamine (DIPEA, 1.1 mL, 6.2 mmol) and hexafluorophosphate benzotriazole tetramethyl uranium (HBTU, 2.4 g, 6.2 mmol), followed by an appropriate solution of 13a–o (4.1 mmol) in DCM. The reaction mixture was stirred at room temperature for 18 h and was then poured into water (40 mL), which was followed by extraction with DCM (3 × 20 mL). The combined organic extracts were dried over Na2SO4 and evaporated under reduced pressure to give crude product. The pure product was isolated by silica gel column chromatography as a white or yellow powder. Data of these compounds were shown below:
(E)-4-{[3-[4-(3-phenylacryloyl) piperazine-1-carbonyl]-4-fluorophenyl]methyl}-2H-phthalazin-1-one (5a)
A white solid, yield: 18.3%, mp: 270.8–272.4 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.57 (s, 1H), 8.23 (d, J = 7.8 Hz, 1H), 7.95 (d, J = 8.1 Hz, 1H), 7.87 (t, J = 8.2 Hz, 1H), 7.80 (s, 1H), 7.67–7.70 (m, 2H), 7.48 (d, J = 15.4 Hz, 1H), 7.47–7.35 (m, 4H), 7.29–7.14 (m, 3H), 4.31 (s, 2H), 3.77–3.48 (m, 6H), 3.20 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 165.06, 164.49, 159.82, 157.63, 156.01, 145.27, 142.29, 135.49, 135.28, 133.94, 132.20, 132.00, 130.07, 129.53, 129.41, 129.20, 128.48, 128.34, 126.51, 125.89, 124.06, 123.92, 118.46, 116.46, 47.36, 46.77, 45.61, 45.04, 36.88; IR (KBr, cm−1): 3435, 3011, 2911, 1649, 1606, 1217, 769; ESI-MS: 495.8 [M − H]−.
(E)-4-{[3-[4–(3-(3,4-dimethoxyphenyl)acryloyl) piperazine-1-carbonyl]-4-fluorophenyl]methyl}-2H-phthalazin-1-one (5b)
A white solid, yield: 20.3%, mp: 197.9–199.3 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.57 (s, 1H), 8.23 (d, J = 7.8 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.87 (t, J = 7.6 Hz, 1H), 7.80 (t, J = 7.3 Hz, 1H), 7.45–7.30 (m, 4H), 7.24–7.19 (m, 3H), 6.94 (d, J = 8.3 Hz, 1H), 4.31 (s, 2H), 3.79–3.76(m, 7H), 3.63–3.48 (m, 5H), 3.20(s, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 165.35, 164.48, 159.83, 157.63, 156.01, 150.79, 149.36, 145.28, 142.72, 135.27, 133.93, 132.25, 131.99, 129.53, 129.42, 128.34, 128.30, 126.51, 125.88, 123.94, 122.87, 116.43, 115.69, 111.95, 110.80, 56.13, 55.98, 47.41, 46.87, 45.62, 45.07, 36.88; IR (KBr, cm−1): 3443, 3010, 2909, 1638, 1593, 1261, 835; ESI-MS: 555.8 [M − H]−.
(E)-4-{[3-[4–(3-(4-methoxyphenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H-phthalazin-1-one (5c)
A white solid, yield: 19.7%, mp: 212.5–213.2 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.23 (d, J = 7.8 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.87 (td, J = 7.7, 1.3 Hz, 1H), 7.80–7.69 (m, 3H), 7.44–7.33 (m, 4H), 7.20–7.17 (m, 2H), 7.04 (d, J = 8.3 Hz, 1H), 6.95 (t, J = 7.4 Hz, 1H), 4.31 (s, 2H), 3.82 (s, 3H), 3.62–3.47 (m, 6H), 3.20 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 165.34, 164.49, 159.83, 157.89, 157.63, 156.01, 145.27, 136.91, 135.27, 133.93, 132.24, 132.19, 131.58, 129.53, 128.34, 126.51, 125.88, 123.80, 120.98, 118.20, 116.43, 112.05, 56.03, 47.34, 46.80, 45.58, 45.12, 36.88; IR (KBr, cm−1): 3455, 3014, 2911, 1660, 1633, 1269, 847; ESI-MS: 526.1 [M − H]−.
(E)-4-{[3-[4-(3-(4-methylphenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H-phthalazin-1-one (5d)
A white solid, yield: 15.8%, mp: 245.6–245.9 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.24 (d, J = 7.8 Hz, 1H), 7.94 (d, J = 8.04 Hz, 1H), 7.90 (t, J = 11.94 Hz, 1H), 7.87 (t, J = 7.38 Hz, 1H), 7.59–7.56 (m, 2H), 7.48–7.36 (m, 3H), 7.23–7.19 (m, 4H), 4.31 (s, 2H), 3.76–3.47 (m, 6H), 3.21–3.18 (m, 2H), 2.30 (s, 3H); 12C NMR (150 MHz, DMSO-d6) δ: 165.18, 164.48, 159.82, 157.63, 156.01, 145.27, 142.33, 139.86, 135.28, 133.93, 132.75, 132.20, 131.99, 129.80, 129.44, 128.47, 128.34, 126.51, 125.88, 124.06, 117.27, 116.43, 47.37, 46.77, 45.59, 45.15, 36.88, 21.41; IR (KBr, cm−1): 3433, 3111, 2906, 1649, 1606, 1277, 847; ESI-MS: 509.8 [M − H]−.
(E)-4-{[3-[4-(3-(2-methoxyphenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl}-2H-phthalazin-1-one (5e)
A white solid, yield: 15.2%, mp: 243.8–244.0 °C, 1H NMR (600MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.23 (d, J = 7.7 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.89–7.84 (m, 1H), 7.83–7.62 (m, 3H), 7.43–7.33 (m, 3H), 7.23–7.18 (m, 2H), 7.03 (d, J = 8.2 Hz, 1H), 6.95 (t, J = 7.5 Hz, 1H), 4.30 (s, 2H), 3.82 (s, 3H), 3.73–3.47 (m, 6H), 3.23–3.18 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 165.35, 164.49, 159.83, 157.89, 157.71, 156.00, 145.28, 136.88, 135.27, 133.93, 132.19, 131.98, 131.58, 129.53, 129.42, 128.34, 126.51, 125.87, 123.94, 123.80, 120.97, 118.20, 116.44, 112.05, 56.03, 47.32, 46.77, 45.59, 45.11, 36.88; IR (KBr, cm−1): 3434, 3012, 2908, 1661, 1634, 1268, 980; ESI-MS: 525.4 [M − H]−.
(E)-4-{[3-[4–(3-(2,5-dimethoxyphenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H-phthalazin-1-one (5f)
A white solid, yield: 23.9%, mp: 133.9–134.6 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.23 (d, J = 7.9 Hz, 1H), 7.94 (d, J = 8.1 Hz, 1H), 7.86 (t, J = 7.6 Hz, 1H), 7.82–7.71 (m, 2H), 7.43–7.27 (m, 3H), 7.24–7.10 (m, 2H), 6.97–6.91 (m, 2H), 4.30 (s, 2H), 3.76(s, 3H), 3.73(s, 3H) 3.62–3.47 (m, 6H), 3.23–3.19 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 165.28, 164.46, 159.81, 157.61, 153.63, 152.28, 145.26, 136.70, 135.25, 133.92, 132.23, 131.97, 129.51, 128.32, 126.50, 125.87, 124.41, 123.94, 118.41, 116.97, 116.41, 113.28, 112.97, 56.50, 54.03, 47.38, 46.74, 45.57, 45.09, 36.86; IR (KBr, cm−1): 3432, 3109, 2909, 1641, 1565, 1224, 844; ESI-MS: 556.6 [M − H]−.
(E)-4-{[3-[4–(3-(4-fluorophenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H -phthalazin-1-one (5g)
A white solid, yield: 15.1%, mp: 272.7–272.9 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.23 (d, J = 8.9 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.86 (t, J = 7.6 Hz, 1H), 7.84–7.67 (m, 3H), 7.47 (d, J = 15.3 Hz, 1H), 7.4–7.31 (m, 2H), 7.27–7.07 (m, 4H), 4.30 (s, 2H), 3.83–3.45 (m, 6H), 3.24–3.17 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 164.87, 164.49, 159.83, 157.64, 156.01, 145.27, 140.97, 134.81, 133.93, 132.14, 132.00, 130.47, 129.53, 129.43, 128.34, 126.51, 125.88, 124.04, 123.92, 123.28, 119.39, 116.29, 47.37, 46.77, 45.69, 45.12, 36.88; IR (KBr, cm−1): 3435, 3109, 2908, 1654, 1276, 839; ESI-MS: 513.2 [M − H]−.
(E)-4-{[3-[4–(3-(4-trifluoromethylphenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H -phthalazin-1-one (5h)
A light yellow solid, yield: 19.3%, mp: 213.6–214.0 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.23 (d, J = 7.8 Hz, 1H), 7.96–7.84 (m, 3H), 7.83–7.76 (m, 2H), 7.73 (d, J = 8.3 Hz, 2H), 7.53 (d, J = 15.4 Hz, 1H), 7.47–7.29 (m, 3H), 7.22 (t, J = 9.0 Hz, 1H), 4.31 (s, 2H), 3.82–3.46 (m, 6H), 3.22–3.17 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 164.66, 164.52, 159.83, 157.43, 155.87, 145.27, 140.44, 139.56, 133.93, 131.99, 129.85, 129.53, 129.44, 129.10, 128.34, 126.51, 126.03, 125.88, 121.50, 116.39, 47.34, 46.79, 45.64, 45.18, 36.88; IR (KBr, cm−1): 3453, 3069, 2919, 1658, 1614, 1280, 981; ESI-MS: 563.2 [M − H]−.
(E)-4-{[3-[4–(3-(3-fluorophenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H -phthalazin-1-one (5i)
A white solid, yield: 16.5%, mp: 262.7–263.5 °C, 1H NMR (600MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.23 (d, J = 7.8 Hz, 1H), 7.94 (d, J = 8.1 Hz, 1H), 7.89–7.84 (m, 1H), 7.82–7.74 (m, 1H), 7.66–7.58 (m, 1H), 7.53–7.31 (m, 5H), 7.23–7.17 (m, 3H), 4.31 (s, 2H), 3.77–3.47 (m, 6H), 3.22–3.17(m 2H); 13C NMR (150 MHz, DMSO-d6) δ: 164.79, 164.46, 163.82, 162.16, 159.82, 145.27, 140.91, 138.13, 135.27, 133.94, 132.26, 132.00, 131.17, 129.53, 129.43, 128.34, 126.51, 125.89, 120.12, 116.80, 116.66, 114.58, 114.40, 47.38, 46.81, 45.62, 45.13, 36.88; IR (KBr, cm−1): 3435, 3109, 2907, 1651, 1609, 1251, 974; ESI-MS: 513.2 [M − H]−.
(E)-4-{[3-[4–(3-(4-chlorophenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H -phthalazin-1-one (5j)
A white solid, yield: 17.2%, mp: 239.2–240.4 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.23 (d, J = 7.3 Hz, 1H), 7.94 (d, J = 8.1 Hz, 1H), 7.89–7.84 (m, 1H), 7.80–7.70(m, 3H), 7.49–7.40 (m, 4H), 7.38–7.26 (m, 2H), 7.21 (t, J = 9.0 Hz, 1H), 4.30 (s, 2H), 3.81–3.43 (m, 6H), 3.22–3.17 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 164.88, 164.50, 159.83, 157.63, 156.01, 145.27, 140.88, 135.26, 134.51, 134.47, 133.92, 132.27, 132.22, 131.98, 130.21, 129.53, 129.21, 128.34, 126.51, 125.87, 124.01, 119.33, 116.29, 47.31, 46.76, 45.63, 45.09, 36.89; IR (KBr, cm−1): 3435, 3108, 2908, 1657, 1492, 1273, 842; ESI-MS: 529.2 [M − H]−.
(E)-4-{[3-[4–(3-(4-bromophenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H -phthalazin-1-one (5k)
A white solid, yield: 18.2%, mp: 225.7–226.0 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.23 (d, J = 7.74 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.88–7.84 (m, 1H), 7.80 (t, J = 7.3 Hz, 1H), 7.67–7.62 (m, 2H), 7.57 (d, J = 8.46 Hz, 2H), 7.46–7.18 (m, 5H), 4.30 (s, 2H), 3.75–3.47 (m, 6H), 3.19–3.13 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 164.87, 164.50, 159.83, 157.64, 156.01, 145.27, 140.97, 135.27, 134.81, 133.93, 132.27, 132.22, 132.14, 131.99, 130.47, 129.53, 129.41, 128.34, 126.51, 125.87, 123.92, 123.28, 119.39, 116.29, 47.41, 46.53, 45.69, 45.12, 36.89; IR (KBr, cm−1): 3461, 3064, 2915, 1657, 1612, 1359, 1286, 1224; ESI-MS: 573.1 [M − H]−.
(E)-4-{[3-[4–(3-(3-nitrophenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H -phthalazin-1-one (5l)
A white solid, yield: 16.9%, mp: 266.8–268.2 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.58 (d, J = 30.2 Hz, 1H), 8.23 (d, J = 7.8 Hz, 1H), 8.19–8.08 (m, 2H), 7.94 (d, J = 8.1 Hz, 1H), 7.86 (td, J = 7.7, 1.3 Hz, 1H), 7.81–7.80 (m, 1H), 7.67 (t, J = 8.0 Hz, 1H), 7.59 (d, J = 15.4 Hz, 1H), 7.50–7.53 (m, 1H), 7.43–7.35 (m, 3H), 7.22 (t, J = 9.0 Hz, 1H), 4.31 (s, 2H), 3.80–3.49 (m, 6H), 3.21 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 164.61, 159.82, 157.72, 156.47, 148.80, 145.26, 139.92, 137.41, 135.29, 135.00, 133.92, 132.22, 131.98, 130.66, 129.52, 129.40, 128.34, 126.51, 125.87, 124.29, 122.55, 121.61, 47.74, 46.76, 45.67, 45.10, 36.89; IR (KBr, cm−1): 3436, 3109, 2907, 1645, 1596, 1277, 841; ESI-MS: 540.9 [M − H]−.
(E)-4-{[3-[4–(3-(4-nitrophenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H -phthalazin-1-one (5m)
A white solid, yield: 17.3%, mp: 224.0–224.5 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.59 (s, 1H), 8.24 (t, J = 8.3 Hz, 3H), 8.01–7.95 (m, 3H), 7.89–7.82 (m, 2H), 7.58 (d, J = 15.4 Hz, 1H), 7.45–7.37 (m, 3H), 7.23 (t, J = 8.94 Hz, 1H), 4.32 (s, 2H), 3.81–3.51 (m, 6H), 3.27–3.18 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 164.45, 159.83, 157.59, 156.02, 148.01,145.28, 142.10, 139.74, 135.29, 133.94, 132.26, 132.01, 129.52, 129.44, 128.33, 126.52, 125.89, 124.32, 123.99, 123.07, 116.47, 116.33, 47.33, 46.73, 45.68, 45.20, 36.88; IR (KBr, cm−1): 3436, 3072, 2926, 1766, 1648, 1274, 842; ESI-MS: 539.7 [M − H]−.
(E)-4-{[3-[4–(3-(3,4,5-trimethoxyphenyl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H -phthalazin-1-one (5n)
A white solid, yield: 26.8%, mp: 235.3–236.2 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.23 (d, J = 7.1 Hz, 1H), 7.94 (d, J = 8.1 Hz, 1H), 7.90–7.84 (m, 1H), 7.80 (d, J = 7.7 Hz, 1H), 7.47–7.34 (m, 3H), 7.22 (t, J = 9.0 Hz, 2H), 7.03–7.00 (m, 2H), 4.31 (s, 2H), 3.79 (s, 7H), 3.71–3.46 (m, 9H), 3.22–3.16 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 165.13, 164.46, 159.81, 157.62, 155.99, 153.46, 145.26, 142.82, 139.31, 135.27, 133.93, 132.25, 132.19, 131.98, 131.04, 129.51, 128.32, 126.50, 125.87, 124.03, 123.92, 117.37, 116.41, 106.13, 60.52, 56.50, 54.04, 47.32, 46.80, 45.57, 44.90, 36.86; IR (KBr, cm−1): 3435, 3109, 2908, 1642, 1270, 843; ESI-MS: 586.2 [M − H]−.
(E)-4-{[3-[4-(3-(furan-2-yl) acryloyl) piperazine-1-carbonyl]-4-fluorophenyl] methyl} -2H -phthalazin-1-one (5o)
A light yellow solid, yield: 17.8%, mp: 152.5–155.0 °C, 1H NMR (600 MHz, DMSO-d6) δ: 12.56 (s, 1H), 8.23 (d, J = 7.8 Hz, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.86 (t, J = 7.1 Hz, 1H), 7.80 (td, J = 7.5, 0.78 Hz, 1H), 7.60 (d, J = 1.3 Hz, 1H), 7.43–7.31 (m, 3H), 7.21 (t, J = 9.0 Hz, 1H), 6.92–6.83 (m, 2H), 6.57 (dd, J = 3.2, 1.8 Hz, 1H), 4.30 (s, 2H), 3.67–3.47 (m, 6H), 3.19–3.10 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 164.69, 164.48, 159.83, 157.63, 156.01, 151.57, 145.39, 145.28, 135.25, 133.94, 132.25, 132.20, 131.99, 129.57, 129.53, 129.44, 129.42, 128.33, 126.51, 125.89, 124.04, 116.44, 116.30, 114.65, 112.98, 47.02, 46.59, 45.55, 45.18, 36.88; IR (KBr, cm−1): 3431, 3102, 2908, 2865, 1645, 1275, 844; ESI-MS: 485.5 [M − H]+.
Biological activity
PARP inhibition assay
PARP enzyme inhibition was measured using an HT F Homogeneous 96-well PARP Inhibition Assay Kit (Trevigen, Cat# 4690–096-K, Gaithersburg, USA), according to the manufacturer’s protocol. The various synthesized compounds were dissolved in DMSO and then serially diluted to the required concentrations with distilled water, keeping the final concentration of DMSO lower than 1%. Olaparib was used as positive control. Fluorescence values under the condition of excitation wavelength (544 nm) and emission wavelength (590 nm) were measured using a multiwell spectrophotometer (Molecular Devices SpectraMax M5 microplate reader, Careforde, Chicago, USA). Then, the standard curve was drawn and the inhibition rate of each test compound was calculated. IC50 value of each compound was calculated using GraphPad Prism 6 software according to the above results.
MTT assay for cell growth inhibition
The anti-proliferative activities of synthesized compounds were assessed against breast cancer cell line MDA-MB-436 by the 3-(4, 5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The MDA-MB-436 cell was purchased from Chinese Academy of Sciences. Briefly, cells were seeded in 96-well plates at a density of 4 × 104 cells per mL in Hyclone RPMI medium supplemented with 10% foetal bovine serum and then treated with different concentrations of synthesized compounds in DMSO. Olaparib was used as the positive control. After treated with compounds for 72 h, cells were incubated with 20 μL of 5 mg/mL MTT for 4 h. The culture medium was removed, and 150 μL DMSO was added to dissolve the formazan crystals. Wells containing only media were used for background correction. The optical density was measured spectrophotometrically at 490 nm. EC50 values were calculated using GraphPad Prism 6 software. Data were expressed as mean ± SEM.
Anti-cholinesterase activity assays
The acetylcholinesterase (eeAChE, from the electric eel), butyrylcholinesterase (eqBChE, from equine serum), 5, 5’-dithiobis-2-nitrobenzoic acid (Ellman's reagent, DTNB), S-butyrylthiocholine iodide (BTCI), acetylthiocholine iodide (ATCI), and Donepezil hydrochloride and Neoeserine methyl sulfate were purchased from Sigma-Aldrich.
The capacity of the test compounds 5a–o to inhibit the AChE and BChE activities were assessed by the Ellman's method in 96-well plates with slight modificationCitation31. The Donepezil and Neoeserine were used as reference drugs. The test compounds were dissolved in a minimum volume of DMSO (1%) and were diluted using sodium phosphate buffer (0.1 M, pH 7.4). A reaction mixture containing 20 μL sodium phosphate buffer, 100 μL DTMB (1 mM) and 40 μL AChE (0.2 U/Ml)/BChE (0.3 U/mL) was incubated with 20 μL of various concentrations of test compounds at 37 °C for 15 min followed by the addition of the substrate (20 μL) acetylthiocholine iodide (1 mM) or S-butyrylthiocholine iodide (1 mM) and incubation for another 3 min. The activities were determined by using a Varioskan Flash Multimode Reader (Thermo, Miami, FL, USA) at 412 nm. The inhibition per cent was calculated by the following expression: (1-Ai/Ac) × 100, where Ai and Ac were the absorbance obtained for AChE/BChE in the presence and absence of inhibitors, respectively. The concentration of compound producing 50% of enzyme activity inhibition (IC50) was calculated by nonlinear regression analysis of the response–concentration (log) curve, using the Graph Pad Prism program package (Graph Pad Software; San Diego, CA). Results are expressed as the mean ± SEM of at least three different experiments performed in triplicate.
Molecular docking
Molecular docking studies were performed using the AutoDock Vina 1.1.2Citation32 along with the AutoDockTools 1.5.6 (ADT) and AutoGridFR 1.0. The structures of compounds 5c, 5i, 5l were drawn and then subjected to energy minimization with the molecular mechanics (MM2) using the ChemBioOffice 13.0 (CambridgeSoft, PerkinElmer Inc., MA, USA). The ligands were further processed and saved in the pdbqt format for docking by ADT program. The X-ray crystallographic structures of PARP-1 (PDB Code 5DS3)Citation33 and hBChE (PDB Code 5NN0)Citation34 were obtained from the online PDB database (RCSB) (http://www.rcsb.org). Both receptors were firstly prepared using the Acceryls Discovery Studio 2017 R2 (Accelrys Inc., San Diego, CA, USA) involving removal of water molecules, non-bonded inhibitors and cofactors. Subsequently, both proteins were edited using the ADT program to add the polar hydrogen atoms and Kollman charges as means to rigidify protein structures and also saved in the same pdbqt format as the ligands 5c, 5i, 5l. The torsions for ligands and receptors were set to be rotatable and rigid respectively. The AutoGridFR software was applied to determine the binding pocket and the grid coordinateCitation35. After that, docking was performed using the default settings of Vina and only the minimum predicted Gibbs binding energy pose was considered for each case. Analysis and visualization of the docking results were done using the PyMOL version 1.78 for 3D diagrams (The PyMOL Molecular Graphics System, Version 1.6. Schrödinger, LLC.) and the LigPlot + version 1.45Citation36 for 2D ones.
Results and discussion
Chemistry
The synthetic route for the desired compounds 5a–o is described in Scheme 1. 2-Formyl benzoic acid 6 was used as starting material for the preparation of the key intermediate 9, which firstly reacted with dimethyl phosphate to afford the dimethyl (1, 3-dihydro-3-oxoisobenzofuran-1-yl)-1-phosphonate 7, and then, the Wittig reaction of 7 with 2-fluoro-5-formylbenzonitrile was carried out to give compound 8. The benzoic acid derivative 9 was finally obtained after hydrolysis of 8. Condensations of propanedioic acid with various substituted benzaldehydes or furan-2-carbaldehyde followed by decarboxylation reactions furnished compounds 10a–o. Treatment of 10a–o with oxalyl chloride followed by 1-boc-piperazine gave amides 12a–o, which were subsequently deprotected to provide compounds 13a–o. Finally, intermediates 9 reacted with 13a–o to afford the target compounds 5a–o.
Scheme 1. Synthesis route for target compounds. Reactions and conditions: (a) dimethyl phosphite, 100 °C, 8 h, 62.3%; (b) 2-fluoro-5-formylbenzonitrile, Et3N, anhydrous THF, 20 °C, 16 h, 92.6%; (c) (i) H2O, 10mol/l NaOH, 90 °C, 1 h, (ii) NH2NH2·H2O, 70 °C, 18 h, 4 mol/l HCl, 93.7%; (d) propane diacid, NH4Ac, 85 °C, 5 h, 50.0–80.0%; (e) oxalyl chloride, DMF, 20 °C, 1.5 h; (f) Tert-butoxycarbonylpiperazine, Et3N, DCM, 20 °C, 12 h, 40.0–70.0%; (g) TFA, DCM, 20 °C, 8 h, 50.0–80.0%; (h) HBTU/DIPEA, DCM, 20 °C, 18 h, 15.7–26.8%;
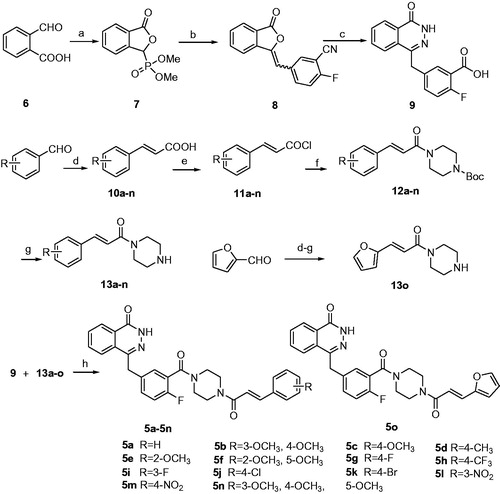
The structures of newly synthesized target compounds 5a–o were confirmed by various spectral techniques. The IR spectra of 5a–o exhibited absorption bands at around 3400 cm−1 which was assigned to NH group stretching vibration. In the 1H NMR spectra of 5a–o the N–H appeared at about 12.56 ppm. In the 1H NMR spectra of 12a–o, the vinylic protons appeared as doublet at 7.40–7.90. The large coupling constant (J = 15.4 Hz) of the vinylic protons confirmed the trans configuration.
Biology
The in vitro PARP-1 enzyme inhibitory activities of all the target compounds were firstly screened at the fixed concentration of 0.5 μM and then, the ones with inhibitory rates ≥80% were selected to further determine their IC50 value. The results are summarized in . It showed that all compounds possessed PARP-1 inhibitory activities, but none of them were more potent than Olaparib. 5l and 5m exhibited more potent inhibitory effects on PARP-1 than the other compounds, which might attribute to the nitro group on the phenyl ring. Introduction of steric groups into the phenyl ring of aromatic propylene moiety, e.g. the dimethoxy in 5b, 5f or the trimethoxy group in 5n, decreased the inhibitory potency; nevertheless, the less steric group in 5o with a furyl group also resulted in decreased activity. The cellar inhibitory potencies of all target compounds were tested against breast cancer cell line MDA-MB-436 by MTT assay and the results are shown in . It indicated that 5c, 5g, 5h, 5i, 5l and 5m had similar effects, among which 5l exhibited the most potent inhibitory activity. On the cellular level, incorporation groups containing fluorine (5g, 5h, 5i) helped to increase potency of target compounds perhaps improving bioavailability.
Table 1. PARP-1 inhibitory activity of compounds 5a–5o.
Table 2. MDA-MB-436 cell inhibitory activities of compounds 5a–5o.
In order to explore the potential of being MTDLs for AD, the in vitro inhibitory effects of all target compounds on Electrophorus electricus AChE (EeAChE) and equine serum butyrylcholinesterase (eqBChE) were investigated according to the methodCitation31. Two drugs, Donepezil and Neostigmine, were used as the standards. The results are summarized in . The IC50 values suggested that most of the synthesized compounds exhibited little-to-moderate inhibitory activities against cholinesterases. Furthermore, a clear trend appeared that with exception of compound 5c all the other compounds showed better inhibition of BChE than AChE. Although the inhibitory potency against AChE of compound 5m was weaker than Donepezil and Neostigmine, its inhibitory potency against BChE (5.93 μM) is more potent than the two drugs (7.64 and 12.01 μM, respectively). Comparison of 3, 4-dimethanoxy derivative 5b with 3, 4, 5-trimethanoxy one 5n, 2-methanoxy compound 5e with 2, 5-dimethanoxy one 5f, demonstrated the relatively compact group decreased its inhibitory activities against cholinesterase. Halogen-substituted compounds 5g–k having F, CF3, Cl, Br on the benzene ring appear less active than the unsubstituted compound 5a. Both compounds 5l and 5m with a strong electron-withdrawing nitro group showed the most potent activities among these derivatives.
Table 3. AChE and BChE inhibitory activities of compounds 5a–5o.
The two important cholinesterases, both AChE and BChE, can hydrolyse acetylthiocholine (ACh) in the synaptic gap of neuronal cells, which results in the loss of neurotransmitter and the decline of cognition during AD. The role of BChE for ACh hydrolysis is only of minor impact in healthy brain, but in advanced AD the level of AChE drops dramatically down to 90% and the levels of BChE in certain parts of the brain increase significantly to compensate the loss of neuronal AChECitation37. Besides that, it has been shown that BChE (over-)expression is associated with senile plaques and the transformation of non-fibrillar to fibrillary Aβ plaques. Hence, inhibition of BChE may be more therapeutically valuable than inhibition of AChE in later stages of AD and researches on selective BChE inhibitors are paid wide attention to in recent yearsCitation38,Citation39. In this study, the synthesized compounds to some degree showed inhibitory selectivity over BChE, although their potencies were not strong enough as nanomolar level, which at least gave a clue to search for new skeleton frame responsible for multifunctional PARP-1 and cholinesterase inhibition.
In biological tests, compound 5l showed more potent inhibitory activity than 5m against PARP-1, while 5m exhibited a little more potent than 5l against AChE and BChE; meanwhile, these synthesized compounds show inhibitory selectivity over BChE, and taking all together, 5l was chosen to carry on molecular docking with PARP-1 and BChE proteins to obtain an insight into the molecular level of the ligand binding. Additionally, in order to further investigate the interaction fashion of these new compounds with PARP-1, molecular docking of 5c and 5i with PARP-1was performed.
Molecular modelling studies
Docking simulations were performed for 5c, 5i and 5l into the PARP-1 (PDB ID: 5DS3) and 5l into the hBChE (PDB ID: 5NNO) active sites using the AutoDock Vina 1.1.2. The molecular docking protocols were validated through re-docking of the co-crystallized ligands in the vicinity of the PARP-1 and hBChE active site. It reproduced all the key interactions accomplished by the co-crystallized ligand with the key amino acids in the active site as shown in the crystal, indicating the suitability of the used setup for the docking study.
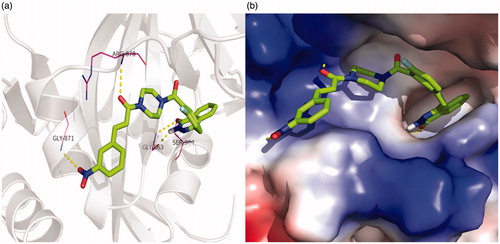
Regarding PARP-1, molecular docking of 5l in PARP-1 revealed an optimal binding mode characterized by affinity of the minimum Gibbs binding energy with –11.9 kcal/mol. As illustrated in the 2D diagrams plotted by LigPlot+ (), docking simulation of 5l showed that it fitted into the enzyme active site almost at the same position of co-crystallized ligand Olaparib. Three hydrogen bonds in 5l-PARP-1 complex were formed in the phthalazinone ring which was well recognized as a scaffold mainly responsible for PARP-1 inhibitory activity ( and ): the carbonyl group as H-bond acceptor with the Gly863 A-NH group (3.04 Å) and Ser904 A-OH group (2.92 Å), the –NH group as H-bond donor with Gly863 A-C=O group (2.88 Å). The fourth identical hydrogen bond was established between the C=O group from acryloyl moiety and Arg878 A-NH group (3.04 Å). The newly introduced nitro group formed a hydrogen bond with Gly871 A-NH group ( and ). However, another C=O group connected to the piperazine moiety did not form a hydrogen bond with the Tyr896 A-NH group as compared with Olaparib, which perhaps was one of the reasons why 5l exhibited a less potent effect on PARP-1 inhibition than Olaparib as shown in vitro experiments. Besides the interaction of hydrogen bond, compound 5l formed hydrophobic interactions with residues Tyr907, Lys903, Phe897, Tyr896, His862, Ile895, Gly894, Leu877 and Asn868. The minimum Gibbs binding energies of molecular docking of 5c and 5i into PARP-1 were –11.9 and –11.8 kcal/mol respectively. As shown in , the phthalazinone rings of both compounds 5c () and 5d () formed corresponding two and three hydrogen bonds with PARP-1which were similar with those of 5l and Olaparib: the carbonyl group as H-bond acceptor with the Ser904 A-OH group or Gly863 A-NH group, the –NH group as H-bond donor with Gly863 A – C=O group. However, within the other moieties of 5c and 5i just one position formed hydrogen bond with PARP-1 compared to two positions within those of 5l and Olaparib. The differences were maybe the reason why 5c and 5l exhibited weaker potency than 5l. On the basis of the docked conformation, the protein contact potential was generated for the compound 5l () depicting the whole surface of the protein. Red and blue colours showed negative and positive charges, respectively. In this depiction, it was shown that compound 5l was bound inside a cavity formed at the interface of the enzyme.
Figure 2. Ligplot images showing the interactions of the Olaparib (a), 5l (b), 5c (c) and 5i (d) with the enzyme PARP-1. The commonly interacting amino acid residues in both interactions were encircled in red circles.
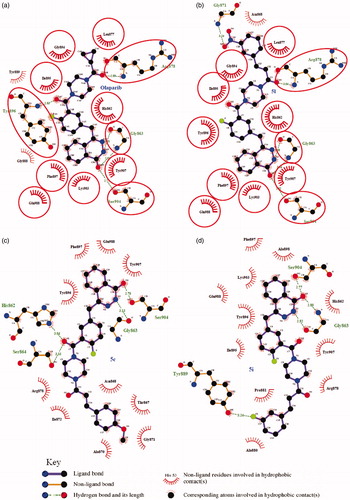
Figure 3. (a) 3D representation of different interactions of compound 5l with residues in the binding sites of PARP-1. The compound was rendered in green stick model and the residues were rendered in purple sticks. Hydrogen bonds were indicated with yellow dashed lines. (b) A vacuum electrostatics depiction of PARP-1 bound to derivative 5l, showing protein contact potential. Surface coloring was according to the electrostatic potential: red, white, and blue correspond to negative, neutral, and positive potential, respectively. The vacuum electrostatics/protein contact potential was generated by PyMOL. The 5l was depicted by sticks.
The crystal structure of the recombinant human BChE (PDB ID: 5NNO) complexed with ligand 92H has been reported recentlyCitation34. The compound 92H, N-((1–(2, 3-dihydro-1H-inden-2-yl) piperidin-3-yl)methyl)-N-(2-(dimethyl-amino)ethyl)-2-naphthamide, shows high selectivity and an extremely potent inhibitory effect on huBChE with picomolar affinity (IC50=1.03 ± 0.04 nM); therefore, 5NN0 was used for docking to analyse the key binding interactions of the synthesized compound 5l for huBChE inhibition.
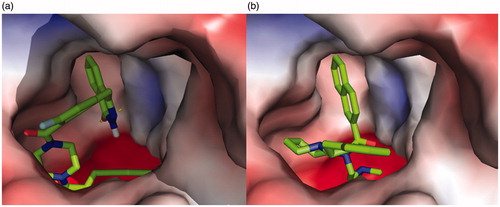
The molecular docking of 5l into huBChE revealed an optimal binding mode characterized by the affinity of the minimum Gibbs binding energy with –13.5 kcal/mol. As illustrated in the 2D diagrams plotted by LigPlot+ (), docking simulation of 5l showed that it interacted with the enzyme active site at local positions same as the reference 92H did. The naphthalene ring of 92H and the phthalazin-1-one moiety of 5l all formed hydrophobic interactions with residues Leu286, Gly116, Gly117 and Trp231. The –(CH2)2NMe2 side chain of 92H formed hydrophobic interactions with residues Trp82 and Glu197 as well as the fluorophenyl group of 5l did. In particular, the Trp231 and Trp82 were two important residues located, respectively, in the acyl-binding pocket and choline-binding pocket of huBChE with which strong interactions could result in an excellently inhibitory effect on huBChE. In spite of this, there were some differences between 5l and 92H about the interactions with residues of huBChE, which perhaps led to a comparatively weaker activity of 5l. Two hydrogen bonds, the carbonyl group as H-bond acceptor with the His438 A-NH group (2.84 Å) and the nitro group as H-bond acceptor with the Tyr128 –OH group (3.22 Å), were established between 5l and huBChE, which may be weaken the hydrophobic interaction of 5l with the key residues Trp82 and Trp231. On the other hand, the 2, 3-dihydro-1H-inden moiety of 92H formed hydrophobic interactions with residues Ile69 and Asp70 while 5l did not. As shown in the vacuum electrostatics depiction of huBChE bound to 5l () and 92H (), although both 5l and 92H were bound inside a cavity formed at the interface of the enzyme, it was obvious for 5l to be closely packed. Therefore, it was inferred that the compound with small and flexible groups except for the phthalazin-1-one moiety contributed to enhance its inhibitory activity against huBChE in the pursuit of the PARP-1 and AChE dual-targeted inhibitor.
Figure 4. Ligplot image showing the interactions of the 92H (a) and 5l (b) with the enzyme huBChE. Arcs with red lines represented amino acid hydrophobic contacts; green dashed lines represents hydrogen bonds. The commonly interacting amino acid residues in both interactions were encircled in red circles.
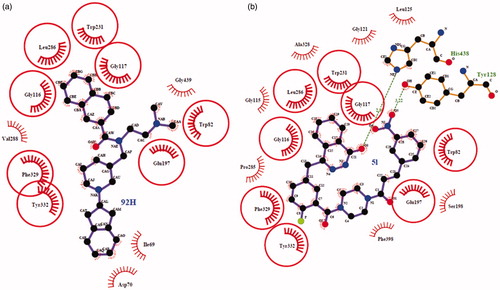
Figure 5. A vacuum electrostatics depiction of huBChE bound to derivative 5l (a) and reference 92H (b), showing protein contact potential. Surface colouring was according to the electrostatic potential: red, white, and blue correspond to negative, neutral, and positive potential, respectively. The vacuum electrostatics/protein contact potential was generated by PyMOL. The 5l and 92H are depicted by sticks.
Generally, the results of respective docking of 5c, 5i and 5l in complexly PARP-1 and 5l with huBChE indicated that 5l could interact well with the active sites of PARP-1 protein, but it did not do well with that of huBChE, which provided a basis and valuable information for further studies aimed at identifying inhibitors of both PARP-1 and huBChE.
Conclusion
Considering that inhibition of PARP-1 has therapeutical values not only for cancer but also for AD, a series of 15 novel Olaparib analogues was synthesized, and their inhibitory activities against the enzymes PARP-1, AChE (from electric eel), BChE (from equine serum) and the cancer cell line MDA-MB-436 were tested. Among these synthesized compounds, 5l exhibited the most potent inhibitory effects on PARP-1 (16.10 ± 1.25 nM) enzyme and MDA-MB-436 (11.62 ± 2.15 μM) cancer cell, which were just a little weaker than Olaparib. Meanwhile, 5l displayed moderate BChE inhibitory activity (9.16 ± 0.91 μM) which was stronger than Neostigmine (12.01 ± 0.45 μM) and selectivity for BChE over AChE to some degree. Molecular docking researches implied that the formation of hydrogen bond was necessary for PARP-1 inhibitory potency, but it was not important for BChE inhibitory activity. While keeping phthalazin-1-one unchanged, small and flexible non-polar groups seemed to be conducive to improving inhibitory potency on BChE. Our research gave a clue to search for new agents based on AChE and PARP-1 dual-inhibited activities to treat Alzheimer's disease.
supplemental_file.doc
Download MS Word (1.4 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
Reference
- Rouleau M, Pate A, Hendzel MJ. PARP inhibition: PARP1 and beyond. Nat Rev Cancer 2010;10:293–301.
- Guha M. PARP inhibitors stumble in breast cancer. Nat Biotechnol 2011;29:373–4.
- O’Sullivan Coyne G, Chen A, Kummar S. Delivering on the promise: poly ADP ribose polymerase inhibition as targeted anticancer therapy. Curr Opin Oncol 2015;27:475–81.
- Livraghi L, Garber JE. PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med 2015;13:188
- Pommier Y, OConnor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 2016;8:362ps17.
- Jones P, Wilcoxen K, Rowley M, Toniatti C. Niraparib: a poly (ADP-ribose) polymerase (PARP) inhibitor for the treatment of tumors with defective homologous recombination. J Med Chem 2015;58:3302–14.
- Wang Y. -Q, Wang P-Y, Wang Y-T, et al. An update on poly (ADP-ribose)polymerase-1 (PARP-1) inhibitors: opportunities and challenges in cancer therapy. J Med Chem 2016;59:9575–98.
- Mehrabadi AR, Korolainen MA, Odero G, et al. Poly (ADP-ribose) polymerase-1 regulates microglia mediated decrease of endothelial tight junction integrity. Neurochem Int 2017;108:266–71.
- Salech FPDP, SanMartín CD, Rogers NK, et al. PARP-1 and p53 regulate the increased susceptibility to oxidative death of lymphocytes from MCI and AD patients. Front Aging Neurosci 2017;9:310.
- David KK, Andrabi SA, Dawson TM, et al. Parthanatos, a messenger of death. Front Biosci (Landmark Ed) 2009;14:1116–28.
- Alluri SR, Riss PJ. Poly (ADP-ribose) polymerase in neurodegeneration: radiosynthesis and radioligand binding in ARC-SWE tg mice. ACS Chem Neurosci 2018;9:1259–63.
- Raina R, Sen D. Can crosstalk between DOR and PARP reduce oxidative stress mediated neurodegeneration? Neurochem Int 2018;112:206–18.
- Ruan Q, Ruan J, Zhang W, et al. Targeting NAD + degradation: the therapeutic potential of flavonoids for Alzheimer's diseaseand cognitive frailty. Pharmacol Res 2018;128:345–58.
- Zemek F, Drtinova L, Nepovimova E, et al. Outcomes of Alzheimer's disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin Drug Saf 2014;13:759–74.
- Li Q, Yang H, Chen Y, et al. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer's disease. Eur J Med Chem 2017;132:294–309.
- Sawatzky E, Wehle S, Kling B, et al. Discovery of highly selective and nanomolar carbamate-based butyrylcholinesterase inhibitors by rational investigation into their inhibition mode. J Med Chem 2016;59:2067–82.
- Zengin M, Genc H, Taslimi P, et al. Novel thymol bearing oxypropanolamine derivatives as potent some metabolic enzyme inhibitors-their antidiabetic, anticholinergic and antibacterial potentials. Bioorg Chem 2018;81:119–26.
- Das S, Basu S. Multi-targeting strategies for Alzheimer's disease therapeutics: pros and cons. Curr Top Med Chem 2017;17:3017–61.
- Nepovimova E, Korabecny J, Dolezal R, et al. Tacrine-trolox hybrids: a novel class of centrally active, nonhepatotoxic multi-target-directed ligands exerting anticholinesterase and antioxidant activities with low in vivo toxicity. J Med Chem 2015;58:8985–9003.
- Hu XM, Dong W, Cui ZW, et al. In silico identification of AChE and PARP-1 dual-targeted inhibitors of Alzheimer's disease. J Mol Model 2018;24:151
- Yuan B, Ye N, Song SS, et al. Poly (ADP-ribose)polymerase (PARP) inhibition and anticancer activity of simmiparib, a new inhibitor undergoing clinical trials. Cancer Lett 2017;386:47–56.
- Maciag AE, Holland RJ, Kim Y, et al. Nitric oxide (NO) releasing poly ADP-ribose polymerase 1 (PARP-1) inhibitors targeted to glutathione S-transferase P1-overexpressing cancer cells. J Med Chem 2014;57:2292–302.
- Johannes JW, Almeida L, Daly K, et al. Discovery of AZ0108, an orally bioavailable phthalazinone PARP inhibitor that blocks centrosome clustering. Bioorg Med Chem Lett 2015;25:5743–7.
- Wang LX, Zhou XB, Xiao ML, et al. Synthesis and biological evaluation of substituted 4-(thiophen-2-ylmethyl)-2H-phthalazin-1-ones as potent PARP-1 inhibitors. Bioorg Med Chem Lett 2014;24:3739–43.
- Ferraris DV. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J Med Chem 2010;53:4561–84.
- Zhuang C, Zhang W, Sheng C, et al. Chalcone: a privileged structure in medicinal chemistry. Chem Rev 2017;117:7762–810.
- Liu H, Fan H, Gao X, et al. Design, synthesis and preliminary structure-activity relationship investigation of nitrogen-containing chalcone derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors: a further study based on Flavokawain B Mannich base derivatives. J Enzyme Inhib Med Chem 2016;31:580–9.
- Liu H, Liu L, Gao X, et al. Novel ferulic amide derivatives with tertiary amine side chain as acetylcholinesterase and butyrylcholinesterase inhibitors: the influence of carbon spacer length, alkylamine and aromatic group. Eur J Med Chem 2017;126:810–22.
- Menear KA, Adcock C, Boulter R, et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl] -2H-phthalazin- 1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem 2008;51:6581–91.
- Teixeira J, Silva T, Benfeito S, et al. Exploring nature profits: development of novel and potent lipophilic antioxidants based on galloyl-cinnamic hybrids. Eur J Med Chem 2013;62:289–96.
- Sang Z, Qiang X, Li Y, et al. Design, synthesis and evaluation of scutellarein-O-alkylamines as multifunctional agents for the treatment of Alzheimer's disease. Eur J Med Chem 2015;94:348–66.
- Apache license. Available from: http://vina.scripps.edu/, version 1.1.2, platform independent, Apache license.
- Dawicki-McKenna JM, Langelier MF, DeNizio JE, et al. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol Cell 2015;60:755–68.
- Košak U, Brus B, Knez D, et al. The Magic of Crystal Structure-Based Inhibitor Optimization: Development of a Butyrylcholinesterase Inhibitor with Picomolar Affinity and in Vivo Activity. J Med Chem 2018;61:119–39.
- OpenSource software license. Available from: http://adfr.scripps.edu/AutoDockFR/agfr.html, version 1.0, platform independent, LGPL OpenSource software license.
- Laskowski RA, Swindells M. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 2011;51:2778–86.
- Darvesh S. Butyrylcholinesterase as a diagnostic and therapeutic target for Alzheimer's disease. Curr Alzheimer Res 2016;13:1173–7.
- Dolles D, Hoffmann M, Gunesch S, et al. Structure-activity relationships and computational investigations into the development of potent and balanced dual-acting butyrylcholinesterase inhibitors and human canna-binoid receptor 2 ligands with pro-cognitive in vivo profiles. J Med Chem 2018;61:1646–63.
- Mansura H, Parham T, Ajdar M, et al. Synthesis, characterization, crystal structure, electrochemical studies and biological evaluation of metal complexes with thiosemicarbazone of glyoxylic acid. Polyhedron 2018;155:25–33.