
Abstract
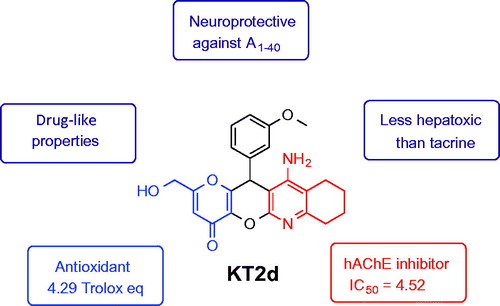
In view of the multifactorial nature of Alzheimer’s disease (AD), multitarget small molecules (MTSM) represent the most potent and attractive therapeutic strategy to design new drugs for Alzheimer’s disease therapy. The new MTSM KojoTacrines (KTs) were designed and synthesized by juxtaposition of selected pharmacophoric motifs from kojic acid and tacrine. Among them, 11-amino-2-(hydroxymethyl)-12-(3-methoxyphenyl)-7,9,10,12-tetrahydropyrano [2',3':5,6] pyrano[2,3-b]quinolin-4(8H)-one (KT2d) was identified as less-hepatotoxic than tacrine, at higher concentration, a moderate, but selective human acetylcholinesterase inhibitor (IC50 = 4.52 ± 0.24 µM), as well as an antioxidant agent (TE = 4.79) showing significant neuroprotection against Aβ1–40 at 3 µM and 10 µM concentrations. Consequently, KT2d is a potential new hit-ligand for AD therapy for further biological exploration.
Graphical Abstract
Introduction
The World Health Organization claims that 46 million people worldwide suffer from Alzheimer’s disease (AD). It is likely that this number may significantly rise as the life expectancy increasesCitation1,Citation2. AD is a multifactorial neurodegenerative disorder, inducing a progressive memory loss, decline in language skills, and other cognitive impairmentsCitation3,Citation4. Although the aetiology of the disease is still not fully understoodCitation5, low levels of acetylcholine (ACh), the aggregation of amyloid-beta peptide (Aβ), hyperphosphorylation of tau protein, oxidative stress, and accumulation of biometals (Cu, Fe, Zn) are thought to play key roles in the pathophysiology, progress and development of ADCitation6,Citation7. AD patients are currently treated either with the N-methyl-D-aspartate receptor antagonist, memantineCitation8, or donepezil, rivastigmine, and galantamineCitation9, three acetylcholinesterase inhibitors (AChEIs), but with limited therapeutic success. Consequently, there is an urgent need for new and more efficient drugs for AD therapy.
Because of the complex nature of AD, multitarget small molecules (MTSM), able to interact simultaneously with the different enzymatic systems or receptors involved in the pathology, have emerged as one of the most promising therapeutic strategies in order to design and identify new drugs for ADCitation10–15. Following this paradigm, we have prepared a number of new MTSM by multicomponent reactions (MCR)Citation16–19 as AChEIs, showing neuroprotective effect and strong antioxidant activity.
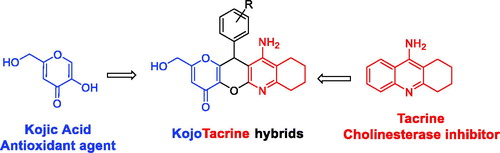
In this article, we report the synthesis and biological evaluation of novel MTSM, called KojoTacrines (KTs), resulting from the combination of selected motifs present in kojic acid (KA) and tacrine (). Tacrine was the first marketed and approved AChEI for AD therapy although discontinued shortly after its approval due to its hepatotoxicityCitation20. Despite this, tacrine has been largely used as a template to design new non-hepatotoxic analogues for ADCitation13,Citation15. Our current choice for tacrine to build the new anti-Alzheimer agents is based on the fact that (1) tacrine derivatives are easily available by simple synthetic protocols; (2) tacrine is a potent ChE inhibitor, able thus to increase the level of neurotransmitter ACh in the brain, and consequently, it has been assumed that (new) tacrine derivatives should behave similarly with particular effects depending on their structure; (3) we have demonstrated that it is possible to design and prepare non-hepatotoxic tacrine derivatives showing ChEs inhibitionCitation21; and (4) last but not least, the number of published papers dealing with tacrine derivatives as anti-Alzheimer should be also considered as a strong point in order to support our choice. So, the purpose of this preliminary communication is to identify a new tacrine-based scaffold and its derivatives. KA, a naturally occurring fungal metabolite, is a recognized antioxidant agentCitation22,Citation23. Thus, considering KA ability to scavenge reactive oxygen species, 12 new kojo tacrines (KTs) were designed, synthesized, and evaluated as potential cholinesterase inhibitors (ChEIs) enhanced with antioxidant properties. As a result, 11-amino-2-(hydroxymethyl)-12–(3-methoxyphenyl)-7,9,10,12-tetrahydropyrano[2', 3':5,6]pyrano[2,3-b]quinolin-4(8H)-one (KT2d) was identified as a hit-agent, less hepatotoxic than tacrine, completely selective against AChE, showing potent antioxidant properties and displaying a very significant neuroprotection against amyloid beta (Aβ).
. Design of KojoTacrine hybrids.
Methods
Chemistry
The synthetic method of only compound KT2d is described here, for the rest of compounds readers are invited to refer to Supporting Information section.
Amino-2-(hydroxymethyl)-12–(3-methoxyphenyl)-7,9,10,12-tetra hydropyrano[2',3':5,6] pyrano[2,3-b]quinolin-4(8H)-one (2d)
A solution of previously described compound 1dCitation24 (0.5 g, 1.5 mmol) in 1,4-dioxane (30 ml) with cyclohexanone (0.23 g, 2.25 mmol) and AlCl3 (0.31 g, 2.25 mmol) was heated at 110 °C for 2 h and monitored by TLC (CH2Cl2/MeOH, 8/2, v/v). When the reaction was complete, the reaction mixture was diluted with a solution of CH2Cl2/water (1/1) and treated with an aqueous solution of sodium hydroxide (10%) until pH 11–12. The mixture was stirred for 20 min and then extracted with CH2Cl2, dried over anhydrous sodium sulfate, filtered, and the solvent was evaporated to generated the desired product 2d (0.36 g, 59%): mp >260 °C; IR (KBr) ν 3470 (OH), 3330 and 3250 (NH2), 1725 (C = O) cm−1; 1H NMR (DMSO-d6, 400 MHz) δ 1.72 (m, 4H), 2.18–2.22 (m, 1H), 2.33–2.37 (m, 1H), 2.55–2.66 (m, 2H), 3.72 (s, 3H, OCH3), 4.20–4.28 (m, 2H), 5.32 (s, 1H), 5.69 (br s, 2H, NH2), 6.32 (s, 1H), 6.77–6.40 (m, 2Harom), 6.97 (s, 1Harom), 7.21–7.25 (m, 1Harom); Citation13C NMR (DMSO-d6, 100 MHz) δ 22.4 (CH2), 22.7 (CH2), 23.4 (CH2), 32.5 (CH2), 40.6 (CH), 55.5 (OCH3), 59.8 (CH2), 96.7 (C), 111.7 (CH), 112.7 (CH), 112.8 (C), 114.9 (CH), 120.2 (CH), 130.4 (CH), 137.8 (C), 142.6 (C), 150.0 (C), 152.1 (C), 153.7 (C), 154.3 (C), 159.7 (C), 168.1 (C), 171.04 (C). Anal. Calcd for C23H22N2O5: C, 67.97; H, 5.46; N, 6.89. Found: C, 68.21; H, 5.38; N, 7.01.
Biological evaluation
The methods for the in vitro cytotoxicity in HepG2 cells, the inhibition of EeAChE, eqBuChE, and hChE, the neuroprotective assay, as well as the ORAC assay have been briefly described
In vitro cytotoxicity of KojoTacrines in HepG2 cellsCitation17,Citation18,Citation25
The culture of HepG2 cells were incubated at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. The culture medium was refreshed after 24 h of incubation and 100 µl of six different concentrations (1–1000 µM) of test compounds or DMSO (0.1%) were then added. The cell viability was monitored by colourimetric assay with 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) Citation26.
Inhibition of EeAChE and eqBuChE of compounds KTs 2a–d, 2f–h, and 2lCitation17,Citation27
Ellman’s protocol was followed to perform the anticholinesterasic activityCitation28. Briefly, the reaction was carried out in a final volume of 3 ml of a phosphate-buffered solution (0.1 M) at pH =8.0 containing (2.625 µl, 0.35 mM) of 5,5’-dithiobis-2-nitrobenzoic acid (DTNB), 40 µl 1% w/v Bovine Albumin Serum phosphate-buffered solution (BSA), (29 µl, 0.035 U/ml) of EeAChE or (60 µl, 0.05 U/ml) of eqBuChE, and 3 µl of tested compound, which were incubated for 10 min. Then, the acetylthiocholine iodide or butyrylthiocholine was added, and the reaction was incubated for additional 15 min. The activity in absence of compound was used as control. The absorbances were measured in a spectrometric plate reader at 412 nm and the IC50 were determined using GraphPad Prism 5.
Inhibition of human cholinesterasesCitation17,Citation27
Inhibitory activities of the tested compounds were expressed as IC50, i.e., concentration of compound required for 50% reduction in cholinesterase activity using Ellman’s methodCitation28. The assay was carried out in medium (100 µl) containing 10 µl of cholinesterase obtained from human erythrocyte hemolyzate, out of blood samples, 40 µl of 0.1 M PBS (pH 7.4), 20 µl of 0.01 M DTNB, 10 µl of different concentrations of tested compound (10−3–10−9 M). This blend was preincubated for 5 min and 20 µl of 0.01 M substrate (ATCh or BTCh) was added to initiate the reaction. The activity was determined by measuring the increase in absorbance at 412 nm at 37 °C in 2 min intervals using Multi-mode microplate reader Synergy 2 (Vermont, USA). The IC50 values are expressed as a mean ± SEM.
Neuroprotection assayCitation29
SH-SY5Y cell culture and treatment with compounds 2b–d and 2f. The cultures of human SH-SY5Y neuroblastoma cells kept under a CO2/air (5%/95%) humidified atmosphere at 37 °C for 24 h were treated with 100 µl of the test compounds or DMSO (0.1%) in DMEM/F12 (1:1) medium supplemented with 1% fetal bovine serum, 1X non-essential amino acids, 100 units/ml penicillin and 10 mg/ml streptomycin. Treatment medium was renewed every day for up to 48 h of treatment. MTT assay was then performed.
Effect of 2b–d and 2f on O/R, Αβ1–40 peptide-induced cell death in SH-SY5Y cells
SH-SY5Y cells were seeded in 96-well culture plates at a density of 8 × 104 cells per well in DMEM/F12 (1:1) medium supplemented with 10% fetal bovine serum, 1X non-essential amino acids, 100 units/mL penicillin and 10 mg/mL streptomycin (Dutscher, France). After 24 h of incubation, the cultures were treated with 100 µl of the test compounds or DMSO (0.1%) in the same medium. Following 24 h, the cells were co-incubated with oligomycin A (10 µM)/rotenone (30 µM) (Sigma, France) or Aβ1–40 (30 µM) with or without the tested compounds 2b–d and 2f at nontoxic concentrations against cells for an additional period of 24 h. MTT assay was then performed to determine the % of cell viability.
Oxygen radical absorbance capacity assayCitation30,Citation31
The antioxidant activity of KTs was determined using the ORAC-FL method described by Dávalos et al.Citation30 using fluorescein as a fluorescent probe with minor modification. Briefly, fluorescein and antioxidant were incubated in a black 96-well microplate (Nunc) for 15 min at 37 °C. The 2,2’-azobis(amidinopropane) dihydrochloride (AAPH) was then added quickly using the built-in injector of Varioskan Flash plate reader (Thermo Scientific). The fluorescence was measured at 485 nm (excitation wavelength) and 535 nm (emission wavelength) every min for 60 min.
Molecular modelling
Molecular modelling of compound KT2d
For docking experiments, Autodock VinaCitation32 software was employed. Compounds (R)-KT2d and (S)-KT2d were prepared with Discovery Studio, version 2.1, software package, using standard bond lengths and bond angles. With the CHARMm force fieldCitation33 and partial atomic charges, the molecular geometries of the compounds were energy-minimized using the adopted-based Newton–Rapson algorithm until the rms gradient was below 0.01 kcal (mol-Å)−1. Three-dimensional crystal structure of hAChE complexed with fasciculin-II (PDB: 1B41) was retrieved from the Protein Data Bank (PDB). Before docking the ligands into the protein, this was prepared by removing all water molecules, heteroatoms, any co-crystallized solvent, and the ligand. Protein model tool in Discovery Studio, version 2.1, software package was used to assign proper bonds, bond orders, hybridization, and charges. CHARMm force field was applied using the receptor-ligand interactions tool in Discovery Studio, version 2.1, software package. AutoDockTools (ADT; version 1.5.4) was used to add hydrogens and partial charges for proteins and ligands using Gasteiger charges. Selected side chains into the target macromolecule are allowed to change their conformations at the same time as the ligand that is being docked. Using the AutoTors module, the macromolecule side chains chosen to be flexible are Trp286, Tyr124, Tyr337, Tyr72, Asp74, Thr75, Trp86 and Tyr341. The docking box was displayed using ADT and it is big enough to include whole protein target (“blind docking”). A grid box of 60 × 60 × 72 with grid point spacing of 1 Ǻ was positioned at the middle of the protein (x = 116.546; y = 110.33; z = −134.181). Default parameters were used except num_modes, which was set to 40. The AutoDock Vina docking procedure used was previously validatedCitation25. The scoring function of AutoDock Vina was chosen, and the docking poses for each ligand were analyzed by examining their relative total energy score. The more energetically favorable conformation was selected as the best pose. The structures of the macromolecule and ligands, as well as the docking results, were processed using Discovery Studio software.
Results and discussion
Chemistry
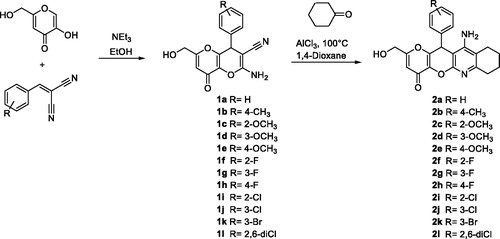
The synthesis of racemic KTs 11-amino-2-(hydroxymethyl)-12-aryl-7,9,10,12-tetrahydropyrano[2',3':5,6]pyrano[2,3-b]quinolin-4(8H)-ones (2a–l) was carried out in two steps by using easily available precursors, as shown in Scheme 1. Thus, KA was reacted with selected arylidenemalonitriles, in the presence of catalytic amounts of triethylamine, in ethanol, at 80 °C, for 5 min, to give the known 2-amino-6-(hydroxymethyl)-8-oxo-4-phenyl-4,8-dihydropyrano[3,2-b]pyran-3-carbonitriles 1a–l in very good yieldsCitation34. Next, Friedländer-type reactionCitation35 of compounds 1a–l with cyclohexanone, under the usual experimental conditionsCitation35, afforded the expected racemic KTs 2a–l (Scheme 1).
Scheme 1. Synthesis of racemic KojoTacrines 2a-l
All new KT ligands showed analytical and spectroscopic data in good agreement with their structures (see Experimental Part).
Biological evaluation
In vitro cytotoxicity of KTs 2a–l in HepG2 cells
Hepatotoxicity is the most critical biological concern when dealing with new tacrine derivatives. In this sense, all new KTs were submitted to the in vitro cell viability test (MTT assay) using human hepatocellular carcinoma cell line (HepG2)Citation26, as the most accepted probe to evaluate hepatotoxic effects. For this experiment, six different concentrations from 1 to 1000 µM were used. As shown in , it is important to note that no significant toxicity at concentrations up to 1000 µM was observed for KT2b; therefore, this compound can be considered as non-hepatotoxic. As shown, tacrine and KTs 2e, 2i–l start to exert a marked cytotoxicity at 300 µM, in contrast to KTs 2a–d and 2f–h, which showed low toxicity at this concentration; however, at 1000 µM, these KTs induce cell death but not as much as tacrine, remaining from 5- to 7-fold less toxic than tacrine.
Table 1. Effects of KojoTacrines 2a-l on human hepatocyte HepG2 cell viability.Table Footnotea
Based on the hepatotoxicity results, the next step was to investigate the capacity KTs 2a–d, 2f–h, and 2l, showing values >50% in the HepG2 cell viability test, for their ChE inhibition capacity and ORAC power.
Evaluation of AChE and BuChE inhibition of KTs 2a–d, 2f–h, and 2l
The inhibition of Electrophorus electricus AChE (EeAChE) and serum horse BuChE (eqBuChE) was carried out following Ellman’s assayCitation28, using the synthesized KTs and tacrine, as a positive control for comparative purposes.
As shown in , all KTs investigated are less potent ChEIs than tacrine. However, they are moderate, but selective AChEIs, in the micromolar range, showing IC50 (µM) values from 0.64 ± 0.06 (2d) to 3.44 ± 0.04 (2h). The most potent EeAChEIs was ligands 2d with IC50 values equal to 0.64, being only approximately 20-fold less active than tacrine (IC50 = 0.031 µM). For the inhibition of eqBuChE, the KTs showed micromolar inhibition ranging from 4.54 to 12.15 µM. The most potent was KT 2 g (IC50 = 4.54 µM). Regarding the structure-activity relationships, no evident and clear trends could be observed for the inhibition of EeAChE and eqBuChE.
Table 2. Inhibition of EeAChE, hACHE, eqBuChE, (IC50, μM)Table Footnotea, and ORAC-FL values for KTs 2a-d, 2f-h, 2l, tacrine, and ferulic acid (FA). 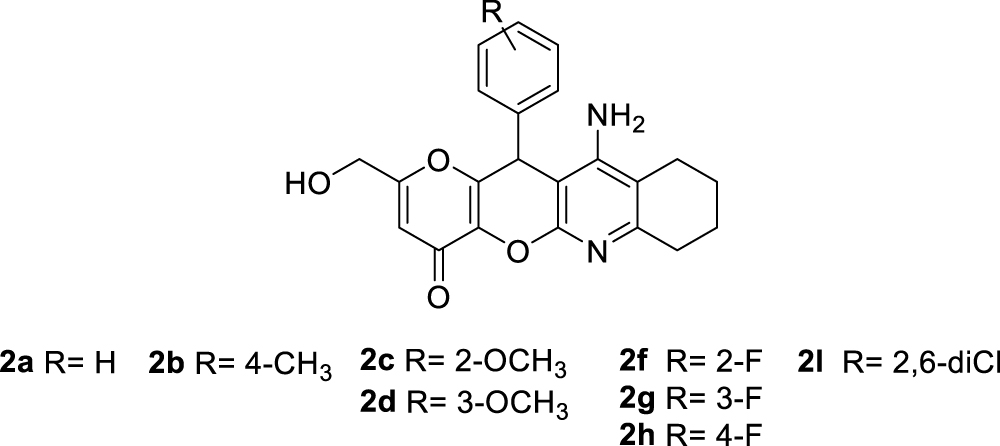
Next, the most balanced KTs 2b–d and 2f, based on the hepatotoxicity data and inhibitory activities against EeAChE and eqBuChE, were evaluated for their ability to inhibit human AChE (hAChE) and human BuChE (hBuChE). Regarding hBuChE the four products showed a percentage of inhibition under 50% at 10 µM and, consequently, their IC50 values were not determined. However, for hAChE, compound KT2d showed significant inhibition (IC50 = 4.52 µM), whereas the non-hepatotoxic KT2b showed a lower inhibitory activity equal to 13.7 µM, as it is shown in . At this point of our project, KT 2d had been identified as a promising compound on diverse biological tests, in spite that this ligand shows lower AChE inhibition than tacrine, because its hepatotoxic effects are lower, still showing a quite good, in the low micromolar range, and selective, AChE inhibition. A very finely balanced situation has been found. Next, as planned at the beginning of the project, the analysis of the antioxidant capacity of KT 2a–d, 2f–h, and 2l was carried out.
Evaluation of antioxidant power of KTs 2a–d, 2f–h, and 2l
The antioxidant ability of KTs 2a–d, 2f–h, and 2l was then evaluated using the Oxygen Radical Capacity (ORAC-FL) methodCitation30,Citation36 with tacrine and ferulic acid (FA)Citation29,Citation37 as negative and positive controls, respectively. Results are expressed in the Trolox equivalents (TE) unit in relation to radical scavenging properties of Trolox. As shown in , all evaluated KTs exhibited good radical scavenging properties, the ORAC values ranging from 2.58 TE (2a) to 6.14 TE (2l), comparing favourably with KA (2.51 TE) and ferulic acid (FA) (3.74 TE). The most potent antioxidant KTs were those bearing a methoxy group (2c) in position C2, and two chloro atoms in positions C2 and C6 (2l) showing 6.05 and 6.14 TE, respectively. Interestingly, the most balanced KT2d, regarding its hepatotoxicity, neuroprotection, cytotoxicity, and ChE inhibition, showed a very high 4.79 TE, 1.3-fold more potent than FA. Based on these interesting results, next, we decided to investigate the neuroprotective properties of some selected non-hepatotoxic KTs.
Neuroprotective capacity of KojoTacrines 2b–d and 2f
The ability to prevent the human neuroblastoma cell line SH-SY5Y cell death induced by toxic insults such as Αβ1–40, implicated in ROS production and apoptosis-related signalling pathwaysCitation38, and oligomycin/rotenone (O/R)Citation39, was then assayed to determine the neuroprotective capacity of the four less hepatotoxic KTs 2b–d and 2f at nontoxic concentration against SH-SY5Y 3 and 10 µM, using melatonin and KA as reference molecules. For the evaluation of the neuroprotective effect, the cytotoxicity of O/R (10 µM/30 µM) on SH-SY5Y cell viability was determined as well as the toxicity of the selected KTs 2b–d and 2f at. As shown in , only KT2d showed a significant and similar neuroprotection against Aβ1–40 insult at 3 and 10 µM and, therefore, slightly less active than KA. It must also be recognized that the non-hepatotoxic KT2b showed very poor neuroprotective activity on any of the toxic insults investigated.
Table 3. Neuroprotective activity of KTs 2b-d and 2f on Oligomycin/Rotenone (O at 10 µM)/Rotenone (R at 30 µM), and Aβ1–40 (30 µM) peptide-induced cell death in SH-SY5Y cells.
Finally, based on the results described above, KT2d was selected as our hit-compound. Thus, its docking analysis was carried out, using the program Autodock VinaCitation32 and the crystal structure (PDB: 1B41) of hAChE available as a complex with fasciculin II, in order to justify its binding properties on hAChE.
Molecular modelling of compound KT2d
Since compound KT2d has a chiral centre at the pyran ring, the reported biological data refer to the racemic mixture, and the observed whole result is a combination of the effects of both enantiomers. Nevertheless, to obtain more information about the binding properties, docking studies were performed on both (R)- and (S)-enantiomers of compound KT2d. The docking procedure was applied to the whole protein target, without imposing the binding site (“blind docking”). Flexible docking experiments have been performed for (R)- and (S)-enantiomers of KT2d. To address receptor flexibility in hAChE, the rearrangement of the side chains of eight residues, Trp286, Tyr124, Tyr337, Tyr72, Asp74, Thr75, Trp86, and Tyr341 was allowed. The movement of these residues may increase the size of the gorge and thus facilitate the access of bulky ligands to the catalytic siteCitation25,Citation40.
Both (R)- and (S)-enantiomers of compound KT2d were studied in docking simulations. The binding energy of (S)-enantiomer (−10.4 Kcal/mol) was lower than that of the (R)-enantiomer (−9.6 Kcal/mol). As depicted in and , both enantiomers are positioned in the enzyme active site in such way to interact with the aminoacids at the rim of the cavity and act as a barrier preventing the entry of the substrate into the active site. The fused-rings, part of compound KT2d, which has an approximately planar structure, make π–π-stacking with the aromatic aminoacids at the rim of the peripherial active site (PAS), including Trp286 and Tyr341. The phenyl ring showed π-alkyl hydrophobic interaction with Leu76 (Figures S1 and S2, see Supporting Information).
Figure 2. Binding mode of inhibitor (R)-KT2d at the active site of hAChE. Compound (R)-KT2d is pictured as ball and sticks in pink and the side chains of the mobile residues are represented in the pale pink. The sub-sites of the active site were coloured: oxyanion hole (OH) in fuchsia, anionic sub-site (AS) in orange, except Trp86, acyl binding pocket (ABP) in yellow, catalytic triad (CT) in green and PAS in light violet. Hydrogen bond interactions are represented as dashed black lines.
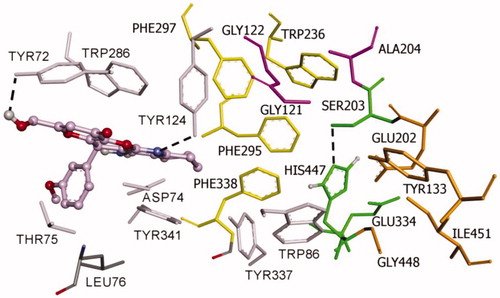
Figure 3. Docking pose of inhibitor (S)-KT2d at the active site of hAChE. Compound (S)-KT2d is illustrated as ball and sticks in blue and the side chains of the mobile residues are represented in pale blue. The subsites of the active site were coloured: Oxyanion hole (OH) in fuchsia, anionic sub-site (AS) in orange, except Trp86, acyl binding pocket (ABP) in yellow, catalytic triad (CT) in green, and peripheral anionic subsite (PAS) in light violet. Hydrogen bond interactions are represented as dashed black lines.
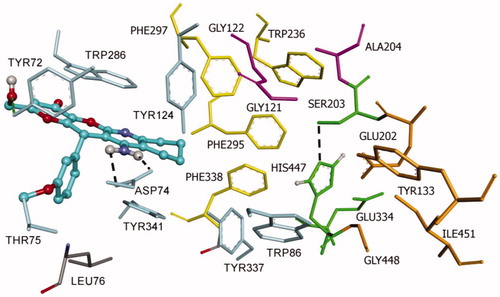
Furthermore, the binding is also supported by the formation of hydrogen bonds. For compound (R)-KT2d, a hydrogen bond between the Tyr124-OH and the nitrogen atom of the pyranotacrine moiety is established (). For compound (S)-KT2d, the amino group established hydrogen bond interactions with Asp74 carboxylate group (key residue of PAS) and the oxygen atom of the methoxy group establishes a hydrogen bond interaction with the backbone-NH-group of Thr75 (). Hydrogen bonding interactions with residue Tyr72 were observed for both enantiomers. As can be seen in Figure 5, another π-anion interaction has been established for (S)-KT2d enantiomer between the phenyl moiety and the Asp74.
On the basis of the set of interactions, which were identified between the (R)-enantiomer [ and Figure S1 (Supporting Information)], the (S)-enantiomer [ and Figure S2 (Supporting Information)] and the protein residues within the active site gorge of both enzymes, we conclude that the OH group and the phenyl ring are likely to be important features for these derivatives to exhibit hAChE inhibitory activities.
ADME of compounds KT2a–l
Theoretical calculations of the ADME (Absorption, Distribution, Metabolism, and Excretion) properties of both enantiomers of tacrine derivatives KT2a–l were accomplished using the QikProp module of Schrodinger suite (QikProp, version 3.8, Schrodinger, LLC, New York, NY, 2013) running in normal mode, for assessing the druggability. Drug kinetics and exposure of tissues to drug influences the pharmacological activity and the performance of a drug, which is ultimately determined by its ADME properties. Nearly 45 physically significant descriptors and pharmacologically relevant properties were predicted and investigated (, see Supporting Information).
In particular, for compound KT2d, the more significant observed data were the following: total Solvent Accessible Surface Area, in square angstroms, using a probe with a 1.4 Å radius (SASA = 653.798 and 670.621 for (R)- and (S)-enantiomer, respectively; limits 300.0–1000.0); estimated number of hydrogen bonds that would be accepted by the solute (donorHB =2.5 for both enantiomers; limits: 2.0–20.0); predicted octanol/water partition coefficient (QPlogPo/w = 2.716 and 2.662 for (R)- and (S)-enantiomer, respectively; limits −2.0–6.5); predicted aqueous solubility. S, in mol/dm3, is the concentration of the solute’s saturated solution that is in equilibrium with crystalline solid (QPlogS = −4.852 and −5.125 for (R)- and (S)-enantiomer, respectively; limits −6.5–0.5); Van der Waals surface area of polar nitrogen and oxygen atoms (PSA =107.282 and 107.713 for (R)- and (S)-enantiomer, respectively; limits 7.0–200.0); predicted brain/blood partition coefficient (QPlog BB = −1.083 and −1.289 for (R)- and (S)-enantiomer, respectively; limits −3.0–1.2); number of violations of Lipinski's Rule Of Five (molecular weight <500, QPlogPo/w < 5, number of hydrogen bond donor ≤5, number of hydrogen bond acceptors HB ≤10; ROF =0 for both enantiomers); number of violations of Jorgensen's rule of three (QPlogS > −5.7, QPCaco >22 nm/s, number of primary metabolites <7; ROT =1 for both enantiomers).
Conclusions
We are persuaded that the MTSM approach is the most appropriate therapeutic strategy to design new drugs for AD therapy. In this context, Lipinski’s rules must be kept in mind during drug discovery to ensure molecular properties concerning pharmacokinetics will enable candidate drugs to have a greater opportunity of achieving the market. Based on this concept, new single dual-targeted chemical entities have been designed and synthesized by juxtaposition of molecular frameworks from KA and tacrine. The new KTs are small molecules showing ChE inhibitory activity, antioxidant, and neuroprotective properties. Particularly, KT2d [IC50 (hAChE) = 4.52 ± 0.24 µM; TE = 4.79], is six-fold less-hepatotoxic than tacrine at 1000 µM, nontoxic for SH-SY5Y cells at 10 µM, exhibiting significant neuroprotection against Aβ1–40 at 3 µM and 10 µM; thus, KT2d is a potential new hit agent that deserves further investigation for AD therapy. Work is now in progress in our laboratory to separate the enantiomers of the racemic mixture of compound KT2d in order to analyze their corresponding in vitro and in vivo biological properties. Results will be reported elsewhere.
Supporting_Information_JEIMC_.pdf
Download PDF (5.1 MB)Disclosure statement
The authors declare no competing financial interest.
Additional information
Funding
Related Research Data
References
- Jameel E, Meena P, Maqbool M. Rational design, synthesis and biological screening of triazine-triazolopyrimidine hybrids as multitarget anti-Alzheimer agents. Eur J Med Chem 2017;136:36–51.
- Borroni E, Bohrmann B, Grueninger F, et al. Sembragiline: a novel, selective monoamine oxidase type B inhibitor for the treatment of Alzheimer's disease. J Pharmacol Exp Ther 2017;362:413–23.
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science 2006;314:777–81.
- Finder VH. Alzheimer's disease: a general introduction and pathomechanism. J Alzheimers Dis 2010;22:5–19.
- Savelieff MG, Lee S, Liu Y, Lim MH. Untangling amyloid-β, tau, and metals in Alzheimer's disease. ACS Chem Biol 2013;8:856–65.
- Bush AI. Drug development based on the metals hypothesis of Alzheimer's disease. J Alzheimers Dis 2008;15:223–40.
- Opazo C, Huang X, Cherny RA, et al. Metalloenzyme-like activity of Alzheimer's disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2). J Biol Chem 2002;277:40302–8.
- Wilkinson D, Wirth Y, Goebel C. Memantine in patients with moderate to severe Alzheimer’s disease: meta-analyses using realistic definitions of response. Dement Geriatr Cogn Disord 2014;37:71–85.
- Bond M, Rogers G, Peters J, et al. The effectiveness and cost-effectiveness of donepezil, galantamine, rivastigmine and memantine for the treatment of Alzheimer’s disease (review of Technology Appraisal No. 111): a systematic review and economic model. Health Technol Assess 2012;16:1–470. [10.3310/hta16210]
- Perez D, Martinez A, Gil C, Campillo N. From bitopic inhibitors to multitarget drugs for the future treatment of Alzheimer’s disease. Curr Med Chem 2015;22:3789–806.
- Agis-Torres A, Sollhuber M, Fernandez M, Sanchez-Montero JM. Multi-target-directed ligands and other therapeutic strategies in the search of a real solution for Alzheimer’s disease. Curr Neuropharmacol 2014;12:2–36.
- Cavalli A, Bolognesi ML, Minarini A, et al. Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem 2008;51:347–72.
- León R, Garcia AG, Marco-Contelles J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer's disease. Med Res Rev 2013;33:139–89.
- Rosini M, Simoni E, Caporaso R, Minarini A. Multitarget strategies in Alzheimer's disease: benefits and challenges on the road to therapeutics. Future Med Chem 2016;8:697–711.
- Ismaili L, Refouvelet B, Benchekroun M, et al. Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer's disease. Prog Neurobiol 2017;151:4–34.
- Benchekroun M, Ismaili L, Pudlo M, et al. Donepezil-ferulic acid hybrids as anti-Alzheimer drugs. Future Med Chem 2015;7:15–21.
- Benchekroun M, Romero A, Egea J, et al. The antioxidant additive approach for Alzheimer’s disease therapy: new ferulic (lipoic) acid plus melatonin modified tacrines as cholinesterases inhibitors, direct antioxidants, and nuclear factor (erythroid-derived 2)-like 2 activators. J Med Chem 2016;59:9967–73.
- Dgachi Y, Sokolov O, Luzet V, et al. Tetrahydropyranodiquinolin-8-amines as new, non hepatotoxic, antioxidant, and acetylcholinesterase inhibitors for Alzheimer’s disease therapy. Eur J Med Chem 2017;126:576–89.
- Dgachi Y, Ismaili L, Knez D, et al. Synthesis and biological assessment of racemic benzochromenopyrimidinimines as antioxidant, cholinesterase, and Aβ1 − 42 aggregation inhibitors for Alzheimer’s disease therapy. ChemMedChem 2016;11:1318–27.
- Watkins PB, Zimmerman HJ, Knapp MJ, et al. HEpatotoxic effects of tacrine administration in patients with Alzheimer's disease. JAMA 1994;271:992–8.
- Boulebd H, Ismaili L, Martin H, et al. New (benz)imidazolopyridino tacrines as nonhepatotoxic, cholinesterase inhibitors for Alzheimer disease. Future Med Chem 2017;9:723–9.
- Gomes AJ, Lunardi CN, Gonzalez S, Tedesco AC. The antioxidant action of polypodium leucotomos extract and kojic acid: reactions with reactive oxygen species. Braz J Med Biol Res 2001;34:1487–94.
- Emami S, Hosseinimehr SJ, Taghdisi SM, Akhlaghpoor S. Kojic acid and its manganese and zinc complexes as potential radioprotective agents. Bioorg Med Chem Lett 2007;17:45–8.
- Kataev EA, Reddy MR, Reddy GN, et al. Supramolecular catalysis by β-cyclodextrin for the synthesis of kojic acid derivatives in water. New J Chem 2016;40:1693–7.
- Bartolini M, Pistolozzi M, Andrisano V, et al. Chemical and pharmacological studies on enantiomerically pure p-methoxytacripyrines, promising multi-target-directed ligands for the treatment of Alzheimer’s disease. ChemMedChem 2011;6:1990–7.
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986;89:271–7.
- Decker M. Homobivalent quinazolinimines as novel nanomolar inhibitors of cholinesterases with dirigible selectivity toward butyrylcholinesterase. J Med Chem 2006;49:5411–3.
- Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95.
- Benchekroun M, Bartolini M, Egea J, et al. Novel tacrine-grafted Ugi adducts as multipotent anti-Alzheimer drugs: a synthetic renewal in tacrine-ferulic acid hybrids. ChemMedChem 2015;10:523–39.
- Dávalos A, Gómez-Cordovés C, Bartolomé B. Extending applicability of the oxygen radical absorbance capacity (ORAC-fluorescein) assay. J Agric Food Chem 2004;52:48–54.
- Dgachi Y, Bautista-Aguilera OM, Benchekroun M, et al. Synthesis and biological evaluation of benzochromenopyrimidinones as cholinesterase inhibitors and potent antioxidant, non-hepatotoxic agents for Alzheimer’s disease. Molecules 2016;21:634.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61.
- Brooks BR, Bruccoleri RE, Olafson BD, et al. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 1983;4:187–217.
- Piao M-Z, Imafuku K. Convenient synthesis of amino-substituted pyranopyranones. Tetrahedron Lett 1997;38:5301–2.
- Marco-Contelles J, Pérez-Mayoral E, Samadi A, et al. Recent advances in the Friedländer reaction. Chem Rev 2009;109:2652–71.
- Ou B, Hampsch-Woodill M, Prior RL. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J Agric Food Chem 2001;49:4619–26.
- Fang L, Kraus B, Lehmann J, et al. Design and synthesis of tacrine-ferulic acid hybrids as multi-potent anti-Alzheimer drug candidates. Bioorg Med Chem Lett 2008;18:2905–9.
- Shi C, Zhao L, Zhu B, et al. Protective effects of Ginkgo biloba extract (EGb761) and its constituents quercetin and ginkgolide B against beta-amyloid peptide-induced toxicity in SH-SY5Y cells. Chem Biol Interact 2009;181:115–23.
- González-Muñoz GC, Arce MP, López B, et al. N-Acylaminophenothiazines: neuroprotective agents displaying multifunctional activities for a potential treatment of Alzheimer's disease. Eur J Med Chem 2011;46:2224–35.
- Martins C, Carreiras MC, León R, et al. Synthesis and biological assessment of diversely substituted furo[2,3-b]quinolin-4-amine and pyrrolo[2,3-b]quinolin-4-amine derivatives, as novel tacrine analogues. Eur J Med Chem 2011;46:6119–30.