
Abstract
Acetylcholinesterase (AChE) and Butyrylcholinesterase (BuChE) inhibitors are interesting compounds for different therapeutic applications, among which Alzheimer’s disease. Here, we investigated the inhibition of these cholinesterases with uracil derivatives. The mechanism of inhibition of these enzymes was observed to be due to obstruction of the active site entrance by the inhibitors scaffold. Molecular docking and molecular dynamics (MD) simulations demonstrated the possible key interactions between the studied ligands and amino acid residues at different regions of the active sites of AChE and BuChE. Being diverse of the classical AChE and BuChE inhibitors, the investigated uracil derivatives may be used as lead molecules for designing new therapeutically effective enzyme inhibitors.
1. Introduction
Alzheimer’s disease (AD) is defined as a neurodegenerative condition characterised by abnormal behaviour, intellectual reduction, being a major public health problem, especially due to the increasing elderly population in developed countriesCitation1,Citation2. In spite of the fact that AD pathogenesis has not been clarified as yet, one of the most important theories was the ‘‘cholinergic hypothesis”Citation3. A defect in the levels of acetylcholine (ACh) and butyrylcholine (BCh) acting as neuromediators was observed in the brains of patients with AD. The inhibition of AChE and BuChE enzymes that hydrolyse ACh and BCh neurotransmitters has become thus a treatment option of ADCitation3. For this reason, many research groups have conducted investigations of the inhibitory activity for these enzymes involved in AD pathogenesis. AChE catalyses the hydrolysis of ACh, which has an important role in cognition and memory. The observation of ACh depletion in AD patients due to the loss of cholinergic neurons constitutes a strategy for their treatment. Drugs such as tacrine, donepezil, galantamine, and rivastigmin are AChE enzyme inhibitors, mainly increasing the amount of ACh by blocking ACh hydrolysisCitation4. While this strategy works in about half of the patients for several years, curative therapy continues to be an unachieved goalCitation4,Citation5. These drugs interact with the active site of the AChE: tacrine, without altering the structure of the enzyme (being a reversible inhibitor), whereas rivastigmine changing it:Citation6,Citation7 the carbamoyl group of rivastigmine was found covalently bound to AChE, with the rest of the drug in the catalytic site and with its phenol functional group exposed to the solventCitation7–11.
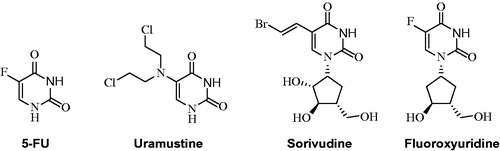
5-Fluorouracil (5-FU) is an uracil analogue used as an antineoplastic drug (antimetabolite). 5-FU interferes with DNA synthesis by blocking DNA polymerase and thymidylate synthetase enzymes. 5-FU and its metabolites have several different mechanisms of action. In vivo, 5-FU is converted to the active metabolite 5-fluoroxyuridine monophosphate (5-FUMP); replacing U, 5-FUMP incorporates into RNA and inhibits RNA processing, thereby inhibiting cell growth. Fluoroxyuridine is used to treat malignant neoplasms of the liver and gastrointestinal tract and hepatic metastases. Sorivudine is a uridine derivative with potent antiviral activity against herpes simplex and varicella zoster viruses. Sorivudine acts by inhibiting DNA polymerase by converting it into triphosphate form in cells. Uramustine, a uracil derivative, is an alkylating antineoplastic agent used in lymphatic malignancies that causes mainly gastrointestinal and bone marrow damageCitation12,Citation13. In this study, the in vitro inhibition properties and in silico calculations of these uracil derivatives 2–9 in their interaction with AChE and BuChE were investigated.
2. Materials and methods
2.1. Chemistry
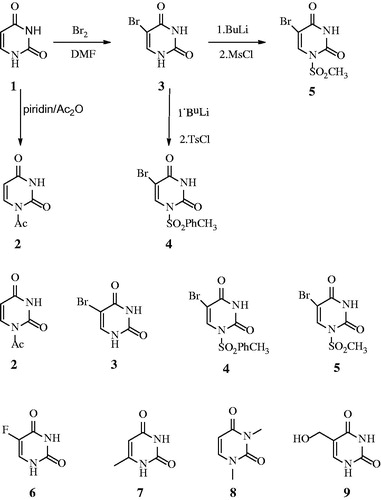
1-Acetyl-1H-pyrimidine-2,4-dione (2), 5-bromo-1H-pyrimidine-2,4-dione (3), 5-Bromo-1-(toluene-4-sulfonyl)-1H-pyrimidine-2,4-dione (4), 5-Bromo-1-methanesulfonyl-1H-pyrimidine-2,4-dione (5). Uracil derivatives 2–5 were synthesised according to ref Citation11Citation14. 5-Flourouracil (6), 6-methyluracil (7), 1,3-Dimethyluracil (8), 5-Hydroxymethyluracil (9), and other chemicals were obtained commercially from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
2.2. Biological activities
The AChE and BuChE enzymes inhibitory activities with the target uracil derivative 2–9 were determined by using the Ellman methodCitation15. Neostigmine was used as the reference drug in this study. The IC50 values obtained for compounds 2–9 are summarised in .
Table 1. IC50 values obtained from AChE and BuChE (µM). Docking scores of corresponding calculations are also shown in the table.
1 mg of each inhibitor was dissolved in 1 ml DMSO and then diluted to various concentrations with deionised water. To determine the cholinesterase inhibition activity, six serial dilutions of the inhibitors were measured. The reaction system was composed of 5–60 µL inhibitor sample, 200 µL buffer (1 M, pH 8.0: Tris-HCl buffer for the AChE assay and phosphate buffer for the BuChE assay), 50 µL DTNB (0.5 mM), 50 µL acetylthiocholine iodide/S-butyrylthiocholine chloride (10 mM), and 10 µL enzyme (0.28 units mL for the AChE assay and 0.32 units/mL for the BuChE assay). The reaction was initiated upon addition of the enzyme. The reaction system was prepared at room temperature in a quartz cuvette. The blank reading was composed of all chemicals except the inhibitorCitation16,Citation17.
The absorbance of the reaction mixture was measured at 412 nm within 5 min from the start of the reaction on a Thermo Scientific Evolution 200 Series (UV-VIS) spectrophotometer (Thermo Fischer Scientific, Waltham, MA, USA). The absorbance for each reaction mixture was measured three times within 5 min of adding the enzyme and the results are reported as mean ± standard deviation. The inhibition properties are reported as IC50 values which were determined graphically from inhibition curves of log inhibitor concentration vs. percent of inhibition. IC50 values represent the concentration of inhibitor required for 50% inhibition of the enzymeCitation16–20.
2.3. In silico studies
2.3.1. Ligand and protein preparation
Maestro Molecular Modeling Package2Citation1 was used for protein and ligand preparations. First, AChE (PDB ID: 4EY7)Citation22 and BuChE (PDB ID: 5DYW)Citation23 crystal structures were retrieved from the protein data bank, then AChE and BuChE amino acid sequences were downloaded in UniProtCitation24 to crosscheck and fix the unresolved residues in the crystal structures. Crosslink Proteins tool in Maestro was utilised to fill the missing amino acid residues in implicit solvent environment. “A” chain of each crystal structures was used for further steps. The missing elements in the proteins (e.g. hydrogen atoms and missing atoms) were added by Protein Preparation Wizard moduleCitation25. Water molecules near 5 Å of the ligands were kept and other water molecules were removed. The pKa prediction and protonation state of ligandsCitation21,Citation26 was predicted at pH 7. PROPKA was used to assign the protonation states of the protein residues at pH 7. Subsequently, restrained minimisation (with 0.30 Å RMSD heavy atom convergence) was realised for the systems with OPLS3 force field. Ligands were drawn with 3D Builder tool and subsequently Ioniser module in conjunction with LigPrep tool of Maestro molecular modelling suite was used for compound preparation and energy optimisation with OPLS3 force field.
2.3.2. Ligand docking
(i) GoldScore scoring function implemented in GOLD (Genetic Optimization for Ligand Docking, v.5.3) docking programmeCitation27 was used in order to obtain the predicted binding poses for protein-ligand complexes and binding energies of the studied ligands towards AChE and BuChE proteins. Protein binding sites of AChE and BuChE targets were defined according to their co-crystallised ligands allowing to cover the whole ligand binding cavity regions during the docking simulations. 50 poses were generated for each ligand where protein residues were treated as rigid bodies and ligands were treated flexible. Water molecules were set in toggle and spin states at the surrounding ligand sites. Search efficiency was set to 100% while 10,000 and 125,000 minimum and maximum operation values were selected, respectively. Early termination was turned off and diverse solution generation selection was invoked.
(ii) In addition, Glide/SP and Glide/XP docking algorithms in Maestro were also used for flexible ligand docking simulationsCitation28–31. Protein grid generation calculation steps (prior to docking) and both standard (SP) and extra precision (XP) docking settings were used with default values. Docking simulation boxes were defined from the centroids of their crystal ligand binding sites and maximum 50 poses were requested for each ligand.
2.3.3. MD simulations
The top-docking scored poses of molecule 4 complexed with AChE and BuChE were used in the MD simulations. The buffer size of the system box was set to 10 × 10 × 10 Å3, and the box shape was specified as orthorhombic. Explicit water molecules (SPC) were used in the preparation of the system, and also 0.15 M NaCl ion concentrations were added to it for the neutralisation of the system. In MD simulations, NPT ensemble at 310 K with Nosé-Hoover temperature coupling and at constant pressure of 1.01 bar via Martyna-Tobias-Klein pressure coupling was providedCitation26. All the systems were prepared and put through the MD simulations by using Desmond programme employing the OPLS2005 force field and RESPA integratorCitation28. There were no constraints on the generated systems and the initial velocity values are used as default. The prepared system was subjected to 100 ns of MD simulations run.
3. Results and discussion
In an earlier report from our group, the inhibitory ability of compounds 2–9 on human carbonic anhydrase (hCA) was investigatedCitation14. Some of these uracil derivatives demonstrated good to moderate inhibition profiles against hCA I and hCA IICitation14,Citation32. Inhibitors of carbonic anhydrase (CA) have been carried out in many therapeutic applications, especially antiglaucoma activity. It was thus decided to screen them against AChE and BuChE. AChE and BChE inhibitors are used in the treatment of many neurodegenerative diseases, especially Alzheimer's diseaseCitation8–11. Compounds 2–9 (), possessing different functional groups on the pyrimidine scaffold, were evaluated for their inhibitory activity of AChE and BuChE by means of the Ellman’s colorimetric assayCitation15. Neostigmine, commercially available cholinesterase inhibitor was used as the reference compound.
Figure 1. 2D structures of compounds 2–9.
The concentration of the uracil derivatives (inhibitors 2–9) required to inhibit 50% of AChE and BuChE activity was calculated from various inhibitor concentrations and reported in . A comparison of the IC50 values of 2–9 indicated that their inhibition was mixed in nature, IC50 values of the inhibitors ranged from 0.088 to 0.388 µM for AChE and from 0.137 to 0.544 µM for BuChE. The results demonstrated that the compounds showed IC50 values weaker compared to the reference compounds neostigmine (IC50 AChE = 0.136 µM and IC50 BuChE = 0.084 µM) against both AChE and BuChE. The strongest inhibition was observed with 4 (IC50 = 0.088 µM) against AChE but was 1.54-fold active compared to neostigmine. Compound 4 (IC50 = 0.137 µM) exhibited the strongest inhibition of BuChE; however, 1.63-fold less active compared to neostigmine. Thus, a computational study was performed in order to rationalizse the observed inhibitory activities. Compunds 2–9 docking scores ranged from −5.06 to −7.90 kcal/mol for AChE and −5.93 to −7.97 kcal/mol for BuChE (Glide/XP results).
Both GOLD and Glide docking results fit to experimental findings (). Scores of top docking poses of compound 4 show higher scores compared to other molecules. In GOLD, higher GoldScore Fitness scoring values represent tighter binding interactions. Results also show ligand efficiency scores (LIE) of studied molecules. In order to escape the affinity-biased selection and optimisation towards larger ligands, Hopkins et al.Citation33recommended to assess binding affinity in relation to number of heavy atoms in a molecule and introduced the term ligand efficiency (the average affinity contribution per atom is considered) instead of considering the affinity of the whole compound. This provides a way to compare the affinity of molecules corrected for their size. In our case, we used Glide/XP scores for the calculation of ligand efficiency scores (ligand efficiency: GlideScore/number of heavy atoms). Results show that compound 3 has top-scored LIE values both in AChE and BuChE.
represents the 2 D and 3 D ligand interaction diagrams of top-docking poses of compound 4 as well as a well-known AChE inhibitor donepezil at the binding pocket of the target. Both molecules interact common active site residues at the AChE; such as Phe295, Trp86, and Trp286. However, as compared to top-poses of Glide SP and Glide XP, top docking pose of GoldScore has an alternative orientations at the binding pocket. The Br-uracil fragment locates between the Ser125 and Glu202 residues where this orientation allows compound 4 to make hydrogen bonding interaction within Glu202 and π-π stacking interaction with Trp86 (). For BuChE, both three docking algorithms predict identical binding orientation of compound 4 () where Trp82 and Tyr 332 form π-π stacking interaction with the aromatic rings of the ligand while Glu197 and His438 residues make hydrogen bonding interactions with the Br-uracil fragment of the molecule.
Figure 2. (Top) 3D representation of compound 4 at the binding pocket of AChE (Glide/XP) (left) and GoldScore (right); 2D ligand interaction diagram of the top docking pose of donepezil (Glide/XP); (bottom) 2D ligand interactions diagrams of top docking poses of compound 4 at the binding pocket of AChE using Glide/SP, Glide/XP and GoldScore from left to right, respectively. Green and purple lines represent π-π stacking and hydrogen bonding interactions, respectively.
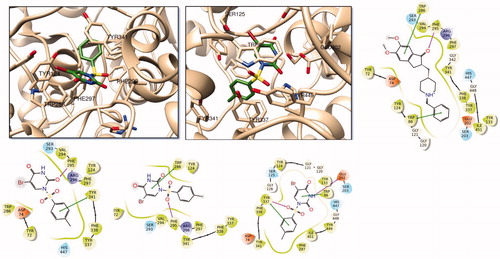
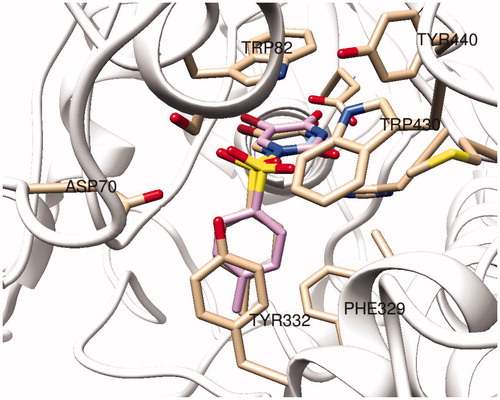
Figure 3. Superposition of top-docking poses of compound 4 at BuChE binding site, generated by Glide/SP (wheat), Glide/XP (blue), and GoldScore (pink).
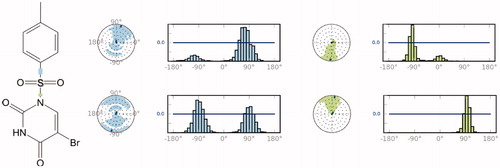
In order to investigate the structural and dynamical profiles of molecule 4 at the binding pockets of AChE and BuChE, MD simulations were performed for the top-docked poses attained from Glide/XP and GoldScore for AChE and Glide/XP for BuChE, using Desmond. and show a timeline representation of the interactions and contacts (H-bonds, hydrophobic, ionic, water bridges) of compound 4 at the binding pockets of AChE and BuChE. The top panel shows the total number of specific contacts the AChE makes with the molecule 4 over the course of the trajectory. The bottom panel represents which residues interact with the ligand 4 in each trajectory frame. Some residues make more than one specific contact with the ligand, which is represented by a darker shade of orange, according to the scale to the right of the plot. Mostly observed contacts at AChE are from Trp286, Phe295, Arg296, Phe338, and Tyr341. Corresponding interactions were Trp82, Glu197, Tyr332, His438, and Tyr441 at the BuChE. Interactions that occur more than 30.0% throughout the simulation are also shown and . The ligand torsions plot summarises the conformational evolution of every rotatable bond in the ligand 4 throughout the simulation (). The top panel shows the 2D schematic of a ligand with colour-coded rotatable bonds. Each rotatable bond torsion is accompanied by a dial plot and bar plots of the same colour. The radial plots describe the conformational change of the torsion throughout the MD simulations. The beginning of the simulation is in the centre of the radial plot and the time evolution is plotted radially outwards. The bar plots summarise the data on the dial plots, by showing the probability density of the torsion. Results show that rotatable bonds are quite stable throughout the simulations. The histogram plot and torsional analysis of ligand give detailed information into the conformational change of the ligand 4 at the binding sites of of AChE and BuChE.
Figure 4. Timeline representation of the interactions and contacts throughout the MD simulations of 4 at the binding pocket of AChE. Protein-ligand interactions are monitored throughout the MD simulations. These interactions are categorsed into four types: Hydrogen Bonds, Hydrophobic, Ionic, and Water Bridge. Interactions that occur more than 30.0% of the simulation time in the selected trajectory are shown in 2D interaction diagram.
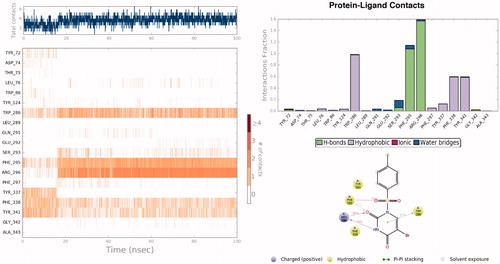
Figure 5. Timeline representation of the interactions and contacts throughout the MD simulations of 4 at the binding pocket of BuChE. Protein-ligand interactions are monitored throughout the MD simulations. These interactions are categorised into four types: Hydrogen Bonds, Hydrophobic, Ionic, and Water Bridge. Interactions that occur more than 30.0% of the simulation time in the selected trajectory are shown in 2D interaction diagram.
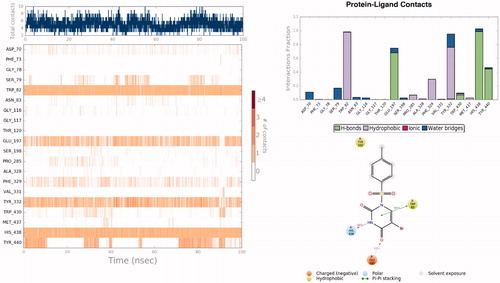
Figure 6. A schematic of detailed ligand atom (molecule 4) interactions with the AChE (top) and BuChE (bottom) residues. Interactions that occur more than 30.0% of the simulation time throughout the MD simulations.
AChE and BuChE are enzymes which play an important role in memory and cognition. They catalyse the hydrolysis of acetylcholine causing a loss of communication between nerve cells. This leads to a loss of brain function and causes AD. Treatment of AD relies on the restoration of the level of acetylcholineCitation8–11. Pharmaceutical research has thus been focusing on cholinesterase inhibitors as treatments for cognitive disorders. Commercially available medicines for AD suffer from drawbacks such as gastrointestinal upset and bioavailability problems and therefore new cholinesterase inhibitors are continuously being investigated. We thus screened uracil derivatives 2–9 for their inhibitory activity.
Uracil derivative 8 (IC50 = 0.388 µM) showed the least potent inhibitory activity against AChE. Decreasing the number of methyl groups on the aromatic ring showed an improvement of the IC50 values obtained, 7 (0.191 µM) with methyl group demonstrated a 2.03-fold decrease of inhibition activity while 9 (0.205 µM) with hydroxymethyl group showed a 1.89-fold decrease of inhibition activity compared to compound 8. However, compound 4 (0.088 µM) possessing 1-(toluene-4-sulfonyl) group showed better inhibitory activity compared to other seven molecules. The difference between the other tested uraciles and 4 is that this molecule is a more voluminous derivative. The stronger inhibition capability of uracil derivative 4 may suggest that the compound’s geometry is more suitable for enzyme interaction when (toluene-4-sulfonyl) group is N1. These results may indicate that the substituent position is more important for inhibition activity compared to toluene-4-sulfonyl groups present in the molecule. A more in-depth study will be done to investigate this theory. To determine the importance of the toluene-4-sulfonyl group on inhibitory activity, other seven uracils were compared to compound 8. Adding a mesylate group (4) to toluene-4-sulfonyl group (5) the inhibitory activity decreased 1.26-fold, suggesting that the toluene-4-sulfonyl moiety is an important functional group for enzyme activity. Compound 4 exhibited a 1.71-fold stronger inhibitory profile compared to uracil 3.
In the case of BuChE, uracil derivative 4 showed the most promising activity with an IC50 value of 0.137 µM. This is in agreement with the results observed for N1 position toluene-4-sulfonyl group substituted uracil. 3 (IC50 = 0.292 µM) is a slightly weaker inhibitor (2.13-fold) compared to uracil derivative 4 but possessed better inhibitory potential compared to the other compounds tested (see entry 6–9). 4 was a slightly better inhibitor (1.42-fold) compared to 5 (0.195 µM), this once again supports the N1 toluene-4-sulfonyl substituted 5 Br-uracil theory as discussed before. The weakest inhibitor amongst the set of compounds was 8 with an IC50 value of 0.544 µM. Tested uracil derivatives (2–9) showed similar results for both AChE and BuChE.
4. Conclusions
As discussed, the screening led to interesting results and can help with the development of more effective drugs to slow down or stop AD. We will expand the study to explore the structure activity relationship of uracil derivatives 2–9, in addition, a comparison of these uracil derivatives with other aromatic compounds will be investigated. These compounds could also be used as precursors or building blocks in the preparation of much more effective drug molecules.
Acknowledgements
This study was financed by Agri Ibrahim Cecen University Scientific Research Council, (project no: FEF.15.007) for (MS and MG).
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Bachman DL, Wolf PA, Linn RT, et al. Prevalence of dementia and probable senile dementia of the Alzheimer type in the Framingham Study. Neurology 1992;42:115–19.
- (a) Ulrich JD, Holtzman DM. TREM2 function in Alzheimer's disease and neurodegeneration. ACS Chem Neurosci 2016;7:420–7. (b) Taslimi P, Sujayev A, Mamedova S, et al. Synthesis and bioactivity of several new hetaryl sulfonamides. J Enzyme Inhib Med Chem 2017; 32:137–45. (c) Sonmez F, Zengin Kurt B, Gazioglu I, et al. Design, synthesis and docking study of novel coumarin ligands as potential selective acetylcholinesterase inhibitors. J Enzyme Inhib Med Chem 2017; 32:285–97. (d) Janjušević L, Karaman M, Šibul F, et al. The lignicolous fungus Trametes versicolor (L.) Lloyd (1920): a promising natural source of antiradical and AChE inhibitory agents. J Enzyme Inhib Med Chem 2017; 32:355–62.
- (a) Digiacomo M, Chen Z, Wang S, et al. Synthesis and pharmacological evaluation of multifunctional tacrine derivatives against several disease pathways of AD. Bioorg Med Chem Lett 2015;25:807–10. (b) Kim JH, Thao NP, Han YK, et al. The insight of in vitro and in silico studies on cholinesterase inhibitors from the roots of Cimicifuga dahurica (Turcz.) Maxim. J Enzyme Inhib Med Chem 2018;33:1174–80. (c) Tripathi RKP, M Sasi V, Gupta SK, et al. Design, synthesis, and pharmacological evaluation of 2-amino-5-nitrothiazole derived semicarbazones as dual inhibitors of monoamine oxidase and cholinesterase: effect of the size of aryl binding site. J Enzyme Inhib Med Chem 2018;33:37–57. (d) Gulçin İ, Abbasova M, Taslimi P, et al. Synthesis and biological evaluation of aminomethyl and alkoxymethyl derivatives as carbonic anhydrase, acetylcholinesterase and butyrylcholinesterase inhibitors. J Enzyme Inhib Med Chem 2017;32:1174–82. (e) Mocan A, Zengin G, Simirgiotis M, et al. Functional constituents of wild and cultivated Goji (L. barbarum L.) leaves: phytochemical characterization, biological profile, and computational studies. J Enzyme Inhib Med Chem 2017;32:153–68.
- (a) Telpoukhovskaia MA, Patrick BO, Rodríguez-Rodríguez C, Orvig C. In silico to in vitro screening of hydroxypyridinones as acetylcholinesterase inhibitors. Bioorg Med Chem Lett 2016;26:1624–8. (b) Qiu GL, He SS, Chen SC, et al. Design, synthesis and biological evaluation of tricyclic pyrazolo[1,5-c][1,3]benzoxazin-5(5H)-one scaffolds as selective BuChE inhibitors. J Enzyme Inhib Med Chem 2018;33:1506–15. (c) Lan JS, Hou JW, Liu Y, et al. Design, synthesis and evaluation of novel cinnamic acid derivatives bearing N-benzyl pyridinium moiety as multifunctional cholinesterase inhibitors for Alzheimer's disease. J Enzyme Inhib Med Chem 2017; 32:776–88. (d) Locatelli M, Zengin G, Uysal A, et al. Multicomponent pattern and biological activities of seven Asphodeline taxa: potential sources of natural-functional ingredients for bioactive formulations. J Enzyme Inhib Med Chem 2017;32:60–7. (e) Soyer Z, Uysal S, Parlar S, et al. Synthesis and molecular docking studies of some 4-phthalimidobenzenesulfonamide derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors. J Enzyme Inhib Med Chem 2017;32:13–19.
- (a) Schneider LS. Training of Alzheimer’s disease with cholinesterase inhibitors. Clin Geriatr Med 2001;17:337–58. (b) Bosak A, Knežević A, Gazić Smilović I, et al. Resorcinol-, catechol- and saligenin-based bronchodilating β2-agonists as inhibitors of human cholinesterase activity. J Enzyme Inhib Med Chem 2017;32:789–97. (c) Xie Q, Zheng Z, Shao B, et al. Pharmacophore-based design and discovery of (-)-meptazinol carbamates as dual modulators of cholinesterase and amyloidogenesis. J Enzyme Inhib Med Chem 2017;32:659–71.
- (a) Mangialasche F, Solomon A, Winblad B, et al. Alzheimer's disease: clinical trials and drug development. Lancet Neurol 2010;9:702–16. (b) Wu WY, Dai YC, Li NG, et al. Novel multitarget-directed tacrine derivatives as potential candidates for the treatment of Alzheimer's disease. J Enzyme Inhib Med Chem 2017;32:572–87.
- Giacobini E. Cholinesterase inhibitors stabilize Alzheimer's disease. Ann N Y Acad Sci 2000;920:321–7.
- (a) Dawson RM. Reversibility of the inhibition of acetylcholinesterase by tacrine. Neurosci Lett 1990;118:85–7. (b) Zhu J, Yang H, Chen Y, et al. Synthesis, pharmacology and molecular docking on multifunctional tacrine-ferulic acid hybrids as cholinesterase inhibitors against Alzheimer's disease. J Enzyme Inhib Med Chem 2018; 33:496–506. (c) Chen Y, Zhu J, Mo J, et al. Synthesis and bioevaluation of new tacrine-cinnamic acid hybrids as cholinesterase inhibitors against Alzheimer's disease. J Enzyme Inhib Med Chem 2018;33:290–302.
- (a) Bar-On P, Millard CB, Harel M, et al. Kinetic and structural studies on the interaction of cholinesterases with the anti-Alzheimer drug rivastigmine. Biochemistry 2002;41:3555–64. (b) Gao X, Tang J, Liu H, et al. Structure-activity relationship investigation of tertiary amine derivatives of cinnamic acid as acetylcholinesterase and butyrylcholinesterase inhibitors: compared with that of phenylpropionic acid, sorbic acid and hexanoic acid. J Enzyme Inhib Med Chem 2018;33:519–24. (c) Czarnecka K, Girek M, Maciejewska K, et al. New cyclopentaquinoline hybrids with multifunctional capacities for the treatment of Alzheimer's disease. J Enzyme Inhib Med Chem 2018;33:158–70. (d) Gao XH, Liu LB, Liu HR, et al. Structure-activity relationship investigation of benzamide and picolinamide derivatives containing dimethylamine side chain as acetylcholinesterase inhibitors. J Enzyme Inhib Med Chem 2018; 33:110–14.
- (a) Gao XH, Zhou C, Liu HR, et al. Tertiary amine derivatives of chlorochalcone as acetylcholinesterase (AChE) and buthylcholinesterase (BuChE) inhibitors: the influence of chlorine, alkyl amine side chain and α,β-unsaturated ketone group. J Enzyme Inhib Med Chem 2017;32:146–52. (b) Markowicz-Piasecka M, Huttunen KM, Sikora J. Metformin and its sulphonamide derivative simultaneously potentiate anti-cholinesterase activity of donepezil and inhibit beta-amyloid aggregation. J Enzyme Inhib Med Chem 2018;33:1309–22. (c) Piemontese L, Tomás D, Hiremathad A, et al. Donepezil structure-based hybrids as potential multifunctional anti-Alzheimer's drug candidates. J Enzyme Inhib Med Chem 2018;33:1212–24. (d) Roca C, Requena C, Sebastián-Pérez V, et al. Identification of new allosteric sites and modulators of AChE through computational and experimental tools. J Enzyme Inhib Med Chem 2018;33:1034–47. (e) Caliandro R, Pesaresi A, Cariati L, et al. Kinetic and structural studies on the interactions of Torpedo californica acetylcholinesterase with two donepezil-like rigid analogues. J Enzyme Inhib Med Chem 2018;33:794–803.
- (a) Feng B, Li X, Xia J, et al. Discovery of novel isoflavone derivatives as AChE/BuChE dual-targeted inhibitors: synthesis, biological evaluation and molecular modelling. J Enzyme Inhib Med Chem 2017;32:968–77. (b) Jiménez-González A, Quispe C, Bórquez J, et al. UHPLC-ESI-ORBITRAP-MS analysis of the native Mapuche medicinal plant palo negro (Leptocarpha rivularis DC. - Asteraceae) and evaluation of its antioxidant and cholinesterase inhibitory properties. J Enzyme Inhib Med Chem 2018;33:936–44. (c) Doytchinova I, Atanasova M, Valkova I, et al. Novel hits for acetylcholinesterase inhibition derived by docking-based screening on ZINC database. J Enzyme Inhib Med Chem 2018;33:768–76. (d) Mezeiova E, Spilovska K, Nepovimova E, et al. Profiling donepezil template into multipotent hybrids with antioxidant properties. J Enzyme Inhib Med Chem 2018;33:583–606. (e) Burčul F, Generalić Mekinić I, Radan M, et al. Isothiocyanates: cholinesterase inhibiting, antioxidant, and anti-inflammatory activity. J Enzyme Inhib Med Chem 2018;33:577–82.
- Palasz A, Ciez D. In search of uracil derivatives as bioactive agents. Uracils and fused uracils: synthesis, biological activity and applications. Eur J Med Chem 2015;97:582–611.
- Durdagi S, Senturk M, Guney M, et al. Design of novel uracil derivatives as inhibitors of carbonic anhydrase I & II, acetylcholinesterase, butyrylcholinesterase, and glutathione reductase using in silico, synthesis and in vitro studies. FEBS J 2016;283:106.
- Guney M, Cavdar H, Senturk M, et al. Synthesis and carbonic anhydrase inhibitory properties of novel uracil derivatives. Bioorg Med Chem Lett 2015;25:3261–3.
- Ellman GL, Courtney KD, Andres V, et al. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95.
- Zilbeyaz K, Stellenboom N, Guney M, et al. Effects of aryl methanesulfonate derivatives on acetylcholinesterase and butyrylcholinesterase. J Biochem Mol Toxicol 2018. https://doi.org/10.1002/jbt.22210.
- Cavusoglu K, Celik H, Senturk M, et al. Investigation of inhibitory effects of some sulfonamides on acethylcholinesterase enzyme. Acta Physiol 2016;218:57.
- (a) Işık S, Vullo D, Durdagi S, et al. Interaction of carbonic anhydrase isozymes I, II, and IX with some pyridine and phenol hydrazinecarbothioamide derivatives. Bioorg Med Chem Lett 2015;25:5636–41.9b) Borras J, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406; 9c) Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95. (d)Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12. (e) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018; 28:713–21. (f) Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008-2018). Expert Opin Ther Pat 2018; 28:729–40. (g) Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin Ther Pat 2018; 28:745–54.
- Urcar H, Senturk E, Senturk M, et al. Investigation of effects of some catecholamines on the activity of carbonic anhydrase enzyme purified from bovine kidney tissue. Acta Physiol 2016;218:57.
- Senturk E, Senturk M. Determination of cholinesterase inhibition potential of rose extract. Anadolu J Agr Sci 2018;33:237–40.
- Sastry GM, Adzhigirey Day T, Annabhimoju R, et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 2013;27:221–34.
- Cheung J, Rudolph MJ, Burshteyn F, et al. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J Med Chem 2012;55:10282–6.
- Kosak U, Brus B, Knez D, et al. Development of an in-vivo active reversible butyrylcholinesterase inhibitor. Sci Rep 2016;6:39495.
- The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res 2015;43:D204–12.
- Schrödinger Release 2018-3: Maestro, Schrödinger, LLC, New York, NY, 2018.
- Shelley JC, Cholleti A, Frye LL, et al. Epik: a software program for pK( a ) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des 2007;21:681–91.
- Jones G, Willett P, Glen RC, et al. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–48.
- Verdonk ML, Cole JC, Hartshorn MJ, et al. Improved protein-ligand docking using GOLD. Proteins 2003;52:609–23.
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47:1739–49.
- Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys 1994;101:4177.
- (a) Fidan I, Salmas RE, Arslan M, et al. Carbonic anhydrase inhibitors: design, synthesis, kinetic, docking and molecular dynamics analysis of novel glycine and phenylalanine sulphonamide derivatives. Bioorg Med Chem 2015;23:7353–8. (b) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (c) Alterio V, Di Fiore A, D'Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. (d) Abbate F, Winum JY, Potter BV, et al. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with EMATE, a dual inhibitor of carbonic anhydrases and steroid sulfatase. Bioorg Med Chem Lett 2004;14:231–4. (e) Capasso C, Supuran CT. An overview of the alpha-, beta-and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–332. (f)Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. (g)Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. (h)Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. (i) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. (j) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68.
- (a) Alper Türkoğlu E, Şentürk M, Supuran CT, et al. Carbonic anhydrase inhibitory properties of some uracil derivatives. J Enzym Inhib Med Chem 2017;32:74–7. (b) Şentürk M, Gülçin İ, Beydemir Ş, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9. (c) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012; 27:138–47. (d) Krall N, Pretto F, Decurtins W, et al. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5. (e) Rehman SU, Chohan ZH, Gulnaz F, Supuran CT. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzyme Inhib Med Chem 2005; 20:333–40. (f) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: structure‐activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73. (g) Dubois L, Peeters S, Lieuwes NG, et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 2011;99:424–31. (h) Chohan ZH, Munawar A, Supuran CT. Transition metal ion complexes of Schiff-bases. Synthesis, characterization and antibacterial properties. Met Based Drugs 2001;8:137–43. (i) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal β-class (Cab) and γ-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–8.
- Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today 2004;9:430–31.