
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Using histamine as lead molecule, a library of (hetero)aryl substituted thiazol-2,4-yl derivatives incorporating pyridine as proton shuttling moiety were obtained and investigated as activators of human carbonic anhydrase (CA, EC 4.2.1.1) isoforms I, II, VII and XIV. Some derivatives displayed good activating and selectivity profiles. This study provides an interesting opportunity to study the thiazole scaffold for the design of CA activators (CAAs), possibly acting on the central nervous system and targeting pathologies involving memory and learning impairments.
1. Introduction
Similar to many enzymes used as therapeutic targets, carbonic anhydrases (CAs, EC 4.2.1.1) are of particular interest considering the large spectra of physiological and pathologic processes in which they are involved. Because of their high catalytic activity, the 13 different active carbonic anhydrase isozymes can be modulated or with inhibitors, or with activators. CA inhibition has found potential in a range of therapeutic areas, and several inhibitors are exploited pharmacologically in clinic as diuretics, anticonvulsant drugs, antiglaucoma, anti-neuropathic pain agents, anti-arthritis agents, and ultimately also for the management of hypoxic tumorsCitation1,Citation2.
CA activation is known more than 80 years, but pharmacological applications started to be explored in the last 15 years on diseases in which CA activity is diminished, such as aging, memory disorder, cognition impairment or Alzheimer's disease. In animal models it has been shown that CA activators are able to enhance cognition, spatial memory and learningCitation3,Citation4. In the last years, CAAs drug design studies afforded a large number of potent but nonselective CA activators. Natural and non-natural amino-acids as well as aromatic/heterocyclic amines were among the first activators described in literature. First X-ray crystal structure of histamine as hCA II activator was reported in 1997Citation5.Several other X-ray structures with amines or amino-acids activators were published among which l- and d-His, l- and d-Phe, d-Trp and l-adrenalineCitation4.
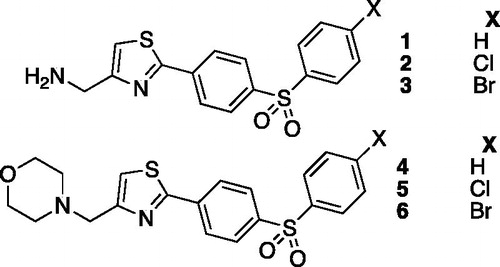
One of the main drawback with the activator of amine and amino-acid types is their lack of selectivity for various CA isoforms. Based on this fact, derivatized histamineCitation6,Citation7, substituted histidineCitation6,Citation7 and bis-imidazoles compoundsCitation8 were reported as CA activators. Heterocyclic scaffolds were also studied such as piperazineCitation9, diketopiperazineCitation10, indazole, pyrazole, oxazoleCitation11, triazoleCitation12 and thiadiazoleCitation13, leading to active carbonic anhydrase activators with enhanced selectivity. The thiazole scaffold is a privileged one in drug discovery, but very few CA activators incorporating this heterocyclic scaffold were reported so far. In 2001, thiazole activators compounds 1, 2 and 3 depicted in were reported as micromolar CA activators against hCA I and hCA II, whereas compounds 4, 5 and 6 were found to be inactiveCitation14.
Figure 1. Thiazole activators reported in Ref. (Citation14).
In order to expand the scope of using thiazole scaffold in the design of CA activators, we present here the synthesis and CA activation studies of a library of derivatives having (hetero)aryl substituted thiazol-2,4-yl scaffold and the pyridine group as proton shuttling moiety, which have been investigated on a panel of selected CA isozymes involved in brain function.
2. Materials and methods
2.1. Chemistry
General
All common reagents and solvents were obtained from commercial sources (Sigma-Aldrich Alfa Aesar or Acros organics) and used without further purification. Compounds were purified on a glass column using Merck Silica Gel 60 (230–400 mesh). Their purity and Mass spectra were determined on Surveyor MSQ Thermoelectron spectrometer (+cAPCI corona sid =30.00, det =1400.00 Full ms [100.00–1000.00]). Melting points were determined with a büchi 510 capillary apparatus and are uncorrected. 1H NMR spectra were recorded on a Bruker AC400P spectrometer using DMSO-d6 or CDCl3 as solvents. Chemical shifts are reported in δ units (parts per million) relative to (Me)4Si as internal standard, and coupling constants (J) are expressed in Hertz. Infrared spectra were obtained on a Perkin–Elmer FT-IR S1000 on KBr paths.
General procedure for the synthesis of thiazol-4-yl-methanamine arylsubstituted (13a-f) and thiazol-4-yl-ethanamine arylsubstituted (10a-c) and pyridinyl thiazol-4-yl acetamide (16a-b)
To a solution of acid (1.1 mmol) in dichloromethane (20 ml) was added triethylamine (1.2 mmol) at 0 °C. Hydroxybenzotriazole (1.2 mmol) and 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (1.2 mmol) were added to a solution. The reaction mixture was stirred at 0 °C 30 min. Corresponding primary amine (1 mmol) was added to a solution. The reaction was stirred at 0 °C for 1 h and at room temperature for 1 h. The reaction mixture was hydrolyzed (H2O) and extracted with dichloromethane. The organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure. The crude was purified by column chromatography (SiO2), or recrystallized from appropriate solvent.
N-{2-[4–(3-Chloro-phenyl)-thiazol-2-yl]-ethyl}-nicotinamide (10a)
White solid; Yield: 60%; mp: 91–93 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 3.31 (m, 2H), 3.70 (m, 2H), 7.38 (m, 1H), 7.48 (m, 2H), 7.89 (td, J = 7.57 Hz, J = 1.45 Hz, 1H), 7.99 (t, J = 1.61 Hz, 1H), 8.15 (m, 2H), 8.70 (dd, J = 4.95 Hz, J = 1.74 Hz, 1H), 8.89 (t, J = 5.53 Hz, 1H), 8.87 (dd, J = 2.18 Hz, J = 0.72 Hz, 1H); Citation13 C NMR (101 MHz, DMSO-d6) δ ppm 32.41, 38.59, 115.41, 123.38, 124.43, 125.49, 127.57, 129.77, 130.58, 133.51, 134.78, 136.05, 148.20, 151.80, 152.12, 164.85, 167.71; IR (neat cm−1) 1634 (CO), 3306 (NH); m/z 344.49 [M+H]+.
N-{2-[4–(3-Chloro-phenyl)-thiazol-2-yl]-ethyl}-isonicotinamide (10b)
White solid; Yield: 68%; mp: 156–158 °C; 1H NMR (400 MHz, CDCl3) δ 3.35 (t, J = 5.76 Hz, 2H), 3.96 (m, 2H), 7.36 (m, 2H), 7.44 (m, 1H), 7.71 (m, 3H), 7.92 (m, 2H), 8.77 (brs, 2H); Citation13 C NMR (101 MHz, DMSO-d6) δ ppm 31.94, 38.15, 117,49, 123.22, 128.89, 133.35, 134.80, 136.09, 146.66, 147.27, 149.81, 150.66, 152.09, 163.19, 168.54; IR (neat cm−1) 1637 (CO), 3271 (NH); LC/MS m/z 344.50 [M+H]+.
N-{2-[4–(3-Chloro-phenyl)-thiazol-2-yl]-ethyl}-2-pyridin-4-yl-acetamide (10c)
White solid; Yield: 67%; mp: 123–125 °C; 1H NMR (400 MHz, CDCl3) δ 3.23 (t, J = 6.02 Hz, 2H), 3.55 (s, 2H), 3.75 (m, 2H), 6.44 (brs, 1H), 7.18 (m, 2H), 7.23 (m, 3H), 7.65 (m, 1H), 8.47 (m, 1H), 8.47 (brs, 2H); Citation13 C NMR (101 MHz, DMSO-d6) δ ppm 26.95, 32.87 42.14, 116.12, 122.21, 127.87, 131.32, 131.78, 135.12, 144.56, 146.28, 148.52, 149.21, 151.11, 162.26, 167.61; IR (neat cm−1) 1639 (CO), 3300 (NH); LC/MS m/z 358.59 [M+H]+.
N-[(4–(2-Chloro-phenyl)-thiazol-2-yl)methyl]-isonicotinamide (13a)
White solid; Yield: 70%; mp: 121–123 °C. 1H NMR (400 MHz, DMSO-d6) δ ppm 4.83 (d, J = 5.67 Hz, 2H), 7.41 (m, 2H), 7.55 (dd, J = 7.46 Hz, J = 1.89 Hz, 1H), 7.82 (dd, J = 4.51 Hz, J = 1.45 Hz, 2H), 7.87 (dd, J = 7.42 Hz, J = 2.03 Hz, 1H), 8.01 (brs, 1H), 8.77 (dd, J = 4.51 Hz, 1.45 Hz, 2H), 9.76 (t, J = 5.82 Hz, 1H); Citation13 C NMR (101 MHz, DMSO-d6) δ ppm 41.01, 119.16, 121.15, 127.28, 129.46, 130.27, 130.82, 132.31, 132.66, 140.47, 150.18, 150.35, 165.03, 167.95; IR (neat cm−1) 1639 (CO), 3274 (NH); LC/MS m/z 330.79 [M+H]+.
N-[(4–(4-Chloro-phenyl)-thiazol-2-yl)methyl]-nicotinamide (13b)
White solid; Yield: 64%; mp: 173–175 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 4.81 (d, 6.11 Hz, 2H), 7.48 (m, 1H), 7.51 (m, 1H), 7.55 (ddd, J = 8.00 Hz, J = 4.95 Hz, J = 0.72 Hz, 1H), 7.97 (m, 2H), 8.09 (brs, 1H), 8.24 (m, 1H), 8.74 (dd, J = 4.80 Hz, 1.74 Hz, 1H), 9.06 (dd, J = 2.33 Hz, J = 0.72 Hz, 1H), 9.66 (t, J = 5.82 Hz, 1H); Citation13 C NMR (101 MHz, DMSO-d6) δ ppm 41.03, 115.05, 123.55, 127.55, 128.74, 129.09, 132.39, 132.82, 135.82, 135.00, 148.34, 152.34, 152.22, 152.45, 165.11, 169.52; IR (neat cm−1) 1631 (CO), 3242 (NH); LC/MS m/z 330.71 [M+H]+.
N-[(4–(3-Chloro-phenyl)-thiazol-2-yl)methyl]-nicotinamide (13c)
White solid; Yield: 77%; mp: 164–166 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 4.83 (d, J = 5.82 Hz, 2H), 7.39 (m, 1H), 7.47 (t, J = 7.86 Hz, 1H), 7.55 (ddd, J = 8.00 Hz, J = 4.80 Hz, J = 0.87 Hz, 1H), 7.92 (td, J = 7.71 Hz, 1.31 Hz, 1H), 8.01 (t, J = 1.74 Hz, 1H), 8.19 (brs, 1H), 8.26 (m, 1H), 8.74 (dd, J = 5.10 Hz, 1.45 Hz, 1H), 9.08 (dd, J = 2.33 Hz, J = 0.72 Hz, 1H), 9.67 (t, J = 5.97 Hz, 1H); Citation13 C NMR (101 MHz, DMSO-d6) δ ppm 41.04, 115.82, 123.53, 124.39, 125.47, 127.66, 129.09, 130.64, 133.56, 135.00, 135.95, 148.36, 152.07, 152.21, 165.15, 169.57; IR (neat cm−1) 1634 (CO), 3290 (NH); LC/MS m/z 330.20 [M+H]+.
N-[(4–(3-Chloro-phenyl)-thiazol-2-yl)methyl]-isonicotinamide (13d)
White solid; yield: 69%; mp: 132–134 °C; 1H NMR (400 MHz, DMSO-d6) δ 4.83 (d, J = 5.97 Hz, 2H), 7.41 (m, 2H), 7.50 (dd, J = 7.42 Hz, J = 1.74 Hz, 1H), 7.82 (dd, J = 4.51 Hz, J = 1.45 Hz, 2H), 7.92 (dd, J = 7.42 Hz, J = 2.02 Hz, 1H), 8.01 (s, 1H), 8.77 (dd, J = 4.51 Hz, J = 1.45 Hz, 2H), 9.77 (t, J = 5.82 Hz, 1H); Citation13 C NMR (101 MHz, DMSO-d6) δ ppm 41.59, 119.74, 121.72, 127.86, 136.04, 130.84, 131.39, 131.88, 133.24, 141.09, 150.76, 150.92, 165.61, 168.53; IR (neat cm−1) 1633 (CO), 3248 (NH); LC/MS m/z 330.19 [M+H]+.
N-[(4–(2-Methoxy-phenyl)-thiazol-2-yl)methyl]-isonicotinamide (13e)
White solid; Yield: 81%; mp: 138–140 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 3.91 (s, 3H), 4.82 (d, J = 5.82 Hz, 2H), 7.04 (m, 1H), 7.13 (d, J = 7.71 Hz, 1H), 7.33 (m, 1H), 7.82 (dd, J = 4.51 Hz, J = 1.47 Hz, 2H), 8.00 (brs, 1H), 8.14 (dd, J = 7.86 Hz, J = 1.74 Hz, 2H), 8.77 (dd, J = 4.36 Hz, J = 1.74 Hz, 1H), 9.74 (t, J = 6.11 Hz, 1H); Citation13 C NMR (101 MHz, DMSO-d6) δ ppm 55.41, 111.63, 117.63, 120.42, 121.14, 122.24, 128.96, 130.19, 132.21, 129.14, 140.52, 149.57, 150.35, 156.38, 164.38, 164.97, 166.93; IR (neat cm−1) 1643 (CO), 3303 (NH); LC/MS m/z 326.13 [M+H]+.
N-[(4–(2-Methoxy-phenyl)-thiazol-2-yl)methyl]-3-pyridin-3-yl-propionamide (13f)
White solid; Yield: 59%; mp: 200–202 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 2.67 (t, J = 6.98 Hz, 2H), 3.09 (t, J = 7.13 Hz, 2H), 3.91 (s, 3H), 4.54 (d, J = 6.11 Hz, 2H), 7.01 (m, 1H), 7.12 (m, 1H), 7.30 (m, 1H), 7.93 (brs, 1H), 7.95 (m, 1H), 8.09 (dd, J = 7.71 Hz, J = 1.74 Hz, 1H), 8.53 (dt, 8.15 Hz, 1.6 Hz, 1H), 8.77 (m, 1H), 8.87 (m, 1H), 8.99 (t, J = 6.97 Hz, J = 5.82 Hz, 1H); Citation13 C NMR (101 MHz, DMSO-d6) δ ppm 27.41, 34.96, 55.44, 111.64, 117.45, 120.42, 122.21, 126.78, 128.96, 129.14, 139.03, 140.87, 141.23, 146.25, 149.44, 156.36, 167.63, 170.96); IR (neat cm−1) 1657 (CO), 3329 (NH); LC/MS m/z 354.23 [M+H]+.
N-(3-Methoxy-benzyl)-2–(2-pyridin-3-yl-thiazol-4-yl)-acetamide (16a)
White solid; Yield: 68%; mp: 110–112 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 3.67 (s, 3H), 3.75 (s, 2H), 4.29 (d, J = 5.97 Hz, 2H), 6.82 (m, 3H), 7.21 (t, J = 7.86 Hz, 1H), 7.55 (ddd, J = 8.00 Hz, J = 4.8 Hz, J = 0.72 Hz, 1H), 7.58 (brs, 1H), 8.27 (m, 1H), 8.58 (t, J = 5.97 Hz, 1H), 8.66 (dd, J = 4.8 Hz, J = 1.45 Hz, 1H), 9.11 (dd, J = 2.30 Hz, J = 0.72 Hz, 1H); Citation13 C NMR (101 MHz, DMSO-d6) δ ppm 38.18, 42.01, 54.84, 111.91, 112.69, 117.68, 119.16, 124.12, 128.89, 129.18, 133.36, 140.36, 140.92, 146.69, 150.70, 152.13, 159.21, 168.61; IR (neat cm−1) 1642 (CO), 3292 (NH); LC/MS m/z 340.23 [M+H]+.
N-[2-Pyridin-3-yl)ethyl)-2–(2-(pyridin-3-yl)thiazol-4-yl]acetamide (16b)
White solid; Yield: 75%; mp: 117–119 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm 2.24 (s, 2H), 2.50 (t, J = 6.83 Hz, 2H), 3.09 (m, 2H), 7.01 (m, 1H), 7.30 (m, 3H), 7.91 (m, 1H), 7.98 (m, 1H), 8.15 (m, 2H), 8.39 (m, 1H), 8.83 (d, J = 1.6 Hz, 1H); Citation13 C NMR (101 MHz, DMSO-d6) δ ppm 31.94, 38.15, 40.26, 117.49, 123.22, 124.11, 128.89, 133.35, 134.80, 136.09, 146.66, 147.28, 149.81, 150.66, 152.09, 163.19, 168.54; IR (neat cm−1) 1635 (CO), 3282 (NH); LC/MS m/z 325.0 [M+H]+.
2.2. Carbonic anhydrase assays
A stopped-flow methodCitation15 has been used for assaying the CA catalyzed CO2 hydration activity with Phenol red as indicator, working at the absorbance maximum of 557 nm, following the initial rates of the CA-catalyzed CO2 hydration reaction for 10–100 s. For each activator, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of activator (0.1 mM) were prepared in distilled-deionized water and dilutions up to 0.1 nM were done thereafter with the assay buffer. The activation constant (KA), defined similarly with the inhibition constant KI, was obtained by considering the classical Michaelis–Menten equation (EquationEquation 1(1)
(1) ), which has been fitted by nonlinear least squares by using PRISM 3:
(1)
(1)
where [A]f is the free concentration of activator.
Working at substrate concentrations considerably lower than KM ([S]≪KM), and considering that [A]f can be represented in the form of the total concentration of the enzyme ([E]t) and activator ([A]t), the obtained competitive steady-state equation for determining the activation constant is given by EquationEquation 2(2)
(2) :
(2)
(2)
where v0 represents the initial velocity of the enzyme-catalyzed reaction in the absence of an activator. All CA isozymes used in the experiments were purified recombinant proteins obtained as reported earlier by our groupCitation6,Citation16–23.
3. Results and discussion
3.1. Chemistry
The small library of (Hetero)aryl substituted thiazol-2,4-yl derivatives was synthesized as follows, obviously considering histamine, a well investigated CA activatorCitation5, as lead molecule. The drug design rationale was to use the substituted thiazole-aminoethyl/aminomethyl scaffold known to possess affinity for the CA active site, by introducing a diverse proton-shuttling moiety (PSM) of the pyridine type, in order to generate new CA activators. Pyridine-carboxylic acids and pyridine-acetic acids were used to introduce this less investigated PSM in the molecules of the new CA activators reported here, as shown in Schemes 1–3.
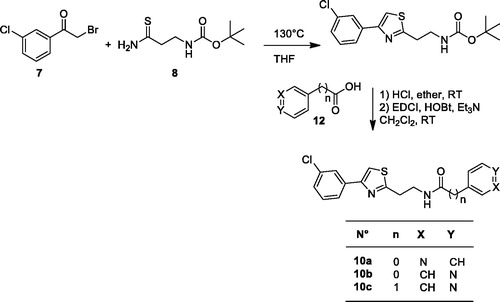
Scheme 1. Synthesis of thiazoles 10a–c.
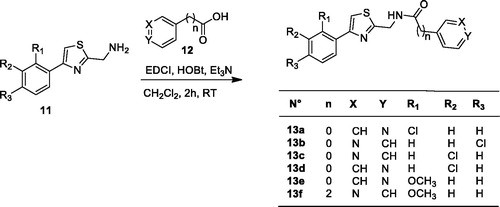
Scheme 2. Synthesis of thiazoles 13a–f.
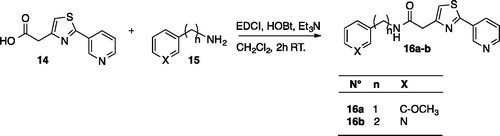
Scheme 3. Synthesis of thiazoles 16a–b.
To access compounds 10a–c, we used a strategy depicted in Scheme 1, using a three steps procedure: (i) condensation between tert-butyl N-(3-amino-3-thioxopropyl)carbamate 8 and 3-chlorophenacylbromide 7 commercially available in THF. (ii) The obtained carbamate was converted to the corresponding amine dihydrochloride by treatment with HCl (gas) at room temperature, (iii) coupling of the primary amine with the corresponding carboxylic acid to lead to target compounds 10a–c (Scheme 1).
Coupling between carboxylic acid 12 and the 4-arylthiazol-2-yl methamine in dichloromethane using EDCI as a coupling reagent, with HOBt and triethylamine as a base, led to derivatives 13a–f as illustrated in Scheme 2.
Compounds 16a–b were prepared using the same simple strategy by coupling the 2-(pyridin-3-yl)-thiazol-4-yl acetic acid 14 with amine 15.
All final derivatives were obtained in a good yield (59–81%) after purification by column chromatography (SiO2), or after recrystallization from appropriate solvent.
3.2. CA activation
Activation data against four physiologically relevant hCA isoforms, hCA I, II, VII and XIV, are shown in using histamine as standard activator.
Table 1. CA activation of isoforms hCA I, II, and VII (cytosolic) and XIV (membrane-associated) with compounds 10a–c, 13a–f, and 16a–b by a stopped-flow CO2 hydrase assay.
All the derivatives tested were active in the nanomolar range against the different isoforms tested.
The structure-activity relationship (SAR) is not easy to rationalize for each isoform. However, the following should be noted:
Isoform hCA I was the one which was most activated by the new compounds reported here, which showed KAs in the range of 6.0–63.4 µM (). The best activators, among which 13e, 13f and 16b incorporated either the isonicotinoyl or the nicotinoyl moieties as PSM, and the methoxy group on the phenyl, which when absent, led to activators with much less effective properties. Compound 16b on the other hand incorporates two PSMs of the 3-pyridyl type, whereas the structurally related derivative with only one such functionality, was 6.5 times less effective as hCA I activator. All compounds investigated here were less effective than histamine (standard derivative)Citation5 as activators.
Isoform hCA II, which is normally less sensitive to amine activatorsCitation4,Citation5, was moderately activated by all compounds investigated here, which showed KAs in the range of 68.1–115.6 µM (). Thus, they were more effective activators compared to histamine, but the activation was not highly significant in these high micromolar concentration ranges. Furthermore, the SAR is not very conclusive, since most of the derivatives showed a rather similar behavior, irrespective of the substitution pattern and the PSM present in their molecule.
The brain-associated cytosolic isoform CA VII showed a rather similar behavior to what mention above for CA II, except for 16a, which was an effective activator with a KA of 7.5 µM (5 times a better activator than histamine, ). This is the only compound incorporating one 3-pyridyl PSM and the 3-methoxy-phenylmethyl tail, among the investigated activators. The structurally related compound with a second 3-pyridyl moiety 16b, was around 9 times less effective as CA VII activator compared to 16a, proving that small structural changes are essential for the biological activity. In addition, 16a is also a CA VII-selective activator, since its affinity for the other investigated isoforms is in the range of 28.7–68.1 µM.
The transmembrane isoform hCA XIV, also present in the brain as well as in other tissues, was moderately activated by the investigated derivatives, which showed KAs in the range of 18.3–78.8 µM. Again the SAR is rather difficult to delineate since most compounds showed similar and modest activating properties.
4. Conclusion
In this paper, we report the synthesis of 2-(hetero)aryl thiazole and 4-(hetero)aryl thiazole compounds acting as activators of the hCAs (CAAs), obtained considering histamine as lead. Both the imidazole and the aminoethyl parts of the lead were extensively changed, replacing the first with a thiazole scaffold and extensively derivatizing the aliphatic amine part of the molecules, by introducing pyridyl type of PSMs (3- and 4-pyridyl). This new family of CAAs derivatives could be of interest in the drug design of agent acting on the central nervous system affecting pathologies mainly characterized from memory and learning impairments, considering the fact that some of them were low micromolar activators of CA I and VII, and one compound also showed CA VII-selective activator profile.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. b)Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- a) Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev 2018;38:1799–836. b) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- a) Temperini C, Scozzafava A, Supuran CT. Carbonic anhydrase activation and the drug design. Curr Pharm Des 2008;14:708–15. b)Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25.
- Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73.
- Briganti F, Mangani S, Orioli P, et al. Carbonic anhydrase activators: X-ray crystallographic and spectroscopic investigations for the interaction of isozymes I and II with histamine. Biochemistry 1997;36:10384–92.
- Akocak S, Lolak N, Vullo D, et al. Synthesis and biological evaluation of histamine Schiff bases as carbonic anhydrase I, II, IV, VII, and IX activators. J Enzyme Inhib Med Chem 2017;32:1305–12.
- Saada MC, Vullo D, Montero JL, et al. Mono- and di-halogenated histamine, histidine and carnosine derivatives are potent carbonic anhydrase I, II, VII, XII and XIV activators. Bioorg Med Chem 2014;22:4752–8.
- a) Draghici B, Vullo D, Akocak S, et al. Ethylene bis-imidazoles are highly potent and selective activators for isozymes VA and VII of carbonic anhydrase, with a potential nootropic effect. Chem Commun (Camb) 2014;50:5980–3. b) Angeli A, Chiaramonte N, Manetti D, et al. Investigation of piperazines as human carbonic anhydrase I, II, IV and VII activators. J Enzyme Inhib Med Chem 2018;33:303–8.
- a) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21. b) Canto de Souza L, Provensi G, Vullo D, et al. Carbonic anhydrase activation enhances object recognition memory in mice through phosphorylation of the extracellular signal-regulated kinase in the cortex and the hippocampus. Neuropharmacology 2017;118:148–56.
- Mollica A, Macedonio G, Stefanucci A, et al. Five- and six-membered nitrogen-containing compounds as selective carbonic anhydrase activators. Molecules 2017;22:2178.
- Maccallini C, Di Matteo M, Vullo D, et al. Indazole, pyrazole, and oxazole derivatives targeting nitric oxide synthases and carbonic anhydrases. Chem Med Chem 2016;11:1695–9.
- Le Duc Y, Licsandru E, Vullo D, et al. Carbonic anhydrases activation with 3-amino-1H-1,2,4-triazole-1-carboxamides: Discovery of subnanomolar isoform II activators. Bioorg Med Chem 2017;25:1681–6.
- Chhabria MT, Patel S, Modi P, Brahmkshatriya PS. Thiazole: a review on chemistry, synthesis and therapeutic importance of its derivatives. Curr Top Med Chem 2016;16:2841–62.
- Scozzafava A, Saramet I, Banciu MD, et al. Carbonic anhydrase activity modulators: synthesis of inhibitors and activators incorporating 2-substituted-thiazol-4-yl-methyl scaffolds. J Enzyme Inhib 2001;16:351–8.
- Khalifah RG. The carbon dioxide hydration activity of car-bonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Supuran CT, Barboiu M, Luca C, et al. Carbonic anhydrase activators. Part 14. Synthesis of mono- and bis-pyridinium salt derivatives of 2-amino-5-(2-aminoethyl)- and 2-amino-5-(3-aminopropyl)-1,3,4-thiadiazole, and their interaction with isozyme II. Eur J Med Chem 1996;31:597–606.
- Ilies MA, Banciu MD, Ilies M, et al. Carbonic anhydrase activators. Part 17. Synthesis and activation study of a series of 1- (1,2,4-triazole-(1H)-3-yl)-2,4,6-trisubstituted-pyridinium salts against isozymes I, II, and IV. Eur J Med Chem 1997;32:911–8.
- a) Licsandru E, Tanc M, Kocsis I, et al. A class of carbonic anhydrase I – Selective activators. J Enzyme Inhib Med Chem 2017;32:37–46. b) Angeli A, Vaiano F, Mari F, et al. Psychoactive substances belonging to the amphetamine class potently activate brain carbonic anhydrase isoforms VA, VB, VII, and XII. J Enzyme Inhib Med Chem 2017;32:1253–9.
- a) Stefanucci A, Angeli A, Dimmito MP, et al. Activation of β- and γ-carbonic anhydrases from pathogenic bacteria with tripeptides. J Enzyme Inhib Med Chem 2018;33:945–50. b) Angeli A, Kuuslahti M, Parkkila S, Supuran CT. Activation studies with amines and amino acids of the α-carbonic anhydrase from the pathogenic protozoan Trypanosoma cruzi. Bioorg Med Chem 2018;26:4187–90.
- a) Angeli A, Buonanno M, Donald WA, et al. The zinc – but not cadmium – containing ζ-carbonic from the diatom Thalassiosira weissflogii is potently activated by amines and amino acids. Bioorg Chem 2018;80:261–5. b) Angeli A, Del Prete S, Alasmary FAS, et al. The first activation studies of the η-carbonic anhydrase from the malaria parasite Plasmodium falciparum with amines and amino acids. Bioorg Chem 2018;80:94–8. c) Angeli A, Donald WA, Parkkila S, Supuran CT. Activation studies with amines and amino acids of the β-carbonic anhydrase from the pathogenic protozoan Leishmania donovani chagasi. Bioorg Chem 2018;78:406–10. d) Angeli A, Alasmary FAS, Del Prete S, et al. The first activation study of a δ-carbonic anhydrase: TweCAδ from the diatom Thalassiosira weissflogii is effectively activated by amines and amino acids. J Enzyme Inhib Med Chem 2018;33:680–5.
- a) Vullo D, De Luca V, Scozzafava A, et al. The first activation study of a bacterial carbonic anhydrase (CA). The thermostable α-CA from Sulfurihydrogenibium yellowstonense YO3AOP1 is highly activated by amino acids and amines. Bioorg Med Chem Lett 2012;22:6324–7. b)Innocenti A, Zimmerman SA, Scozzafava A, et al. Carbonic anhydrase activators: activation of the archaeal beta-class (Cab) and gamma-class (Cam) carbonic anhydrases with amino acids and amines. Bioorg Med Chem Lett 2008;18:6194–8. c) Vullo D, Del Prete S, Osman SM, et al. Comparison of the amine/amino acid activation profiles of the β- and γ-carbonic anhydrases from the pathogenic bacterium Burkholderia pseudomallei. J Enzyme Inhib Med Chem 2018;33:25–30. d)Vullo D, Del Prete S, Osman SM, et al. Burkholderia pseudomallei γ-carbonic anhydrase is strongly activated by amino acids and amines. Bioorg Med Chem Lett 2017;27:77–80. e) Borras J, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406.
- a) Krall N, Pretto F, Decurtins W, et al. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5. b) Rehman SU, Chohan ZH, Gulnaz F, Supuran CT. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzyme Inhib Med Chem 2005;20:333–40. c) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: Structure‐activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73. d) Dubois L, Peeters S, Lieuwes NG, et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 2011;99:424–31. e) Chohan ZH, Munawar A, Supuran CT. Transition metal ion complexes of Schiff-bases. Synthesis, characterization and antibacterial properties. Met Based Drugs 2001;8:137–43. f) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal β-class (Cab) and γ-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–8.
- a) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8. b) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3. c) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. d) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulfamates and sulfamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11.