
Abstract
Two acetazolamide (AAZ) complexes with ruthenium(II) η6-p-cymene chloride were synthesised, characterised and tested for their inhibitory effects on several carbonic anhydrase (CA, EC 4.2.1.1) isoforms with pharmacological applications. Against human (h) isoform hCA I, the two complexes showed inhibition constants in the range of 8.5–23.4 nM (AAZ has a KI of 250 nM), against hCA II of 0.48–4.2 nM, whereas against hCA IX of 0.63–3.8 nM and against hCA XII of 0.04–0.52 nM, respectively. These highly effective ruthenium acetazolamide derivatives against the tumour-associated CA isoforms IX and XII warrant further in vivo studies, in hypoxic tumours overexpressing these enzymes.
1. Introduction
The carbonic anhydrases (CAs, EC 4.2.1.1) are a superfamily of metalloenzymes acting as efficient catalysts for the hydration of CO2 to bicarbonate and protons, as well as for the opposite reaction, the “dehydration” of bicarbonate in the presence of hydronium ions, leading to CO2Citation1–3. Since this equilibrium is involved in pH/CO2 homeostasis, respiration, secretion of electrolytes and several metabolic pathways, among which lipogenesis, such as urea biosynthesis, gluconeogenesis, etc., the activity of these enzymes is correlated with many physiological and pathological conditions, making them attractive drug targets for a variety of disordersCitation3–9. In fact, CAs are present not only in humans (with 15 different isoforms described to date) but they are also widespread in organisms all over the phylogenetic tree, with seven distinct genetic families reported, the α-, β-, γ-, δ-, η-, ζ- and θ-CAsCitation1,Citation2. CA inhibitors (CAIs) belonging to the sulfonamide (and sulfamate) types are clinically used for several decades as diureticsCitation4, antiglaucoma agentsCitation5, antiobesity drugsCitation6, and more recently, a number of studies showed that CA inhibition has profound antitumour effects by inhibition of hypoxia-inducible isoforms CA IX and XII, overexpressed in many hypoxic tumoursCitation7. Furthermore, several proof-of-concept studies demonstrated the involvement of some CA isoforms in neuropathic painCitation8 and arthritisCitation9, with the CAIs of sulfonamide/coumarinCitation10 types demonstrating significant effects in vivo, in animal models of these diseases. Thus, the field of drug design, synthesis and in vivo investigations of various types of CAIs is a highly dynamic one, with a large number of interesting new chemotypes acting on these widespread enzymes constantly emergingCitation9,Citation10. Among the clinically used sulfonamide CAIs are acetazolamide (AAZ), methazolamide (MZA), ethoxzolamide (EZA), saccharin (SAC), brinzolamide (BRZ) and dorzolamide (DRZ) – Citation1–3.
Figure 1. Clinically used sulfonamides with CA inhibitory activityCitation1–3.
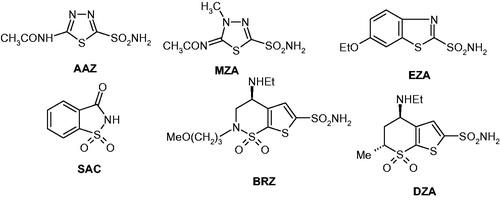
The sulfonamides are highly effective CAIs, with the heterocyclic and some aromatic compounds acting in the low nanomolar rangeCitation1–3, but their main drawback is that they are generally non-selective for the many CA isoforms which must be targeted for various pharmacological applicationsCitation4–9. Thus, alternative chemotypes for a diverse approach to CA inhibition were developedCitation1–7. One of them was that of obtaining coordination compounds in which the sulfonamides act as ligands to various transition or main group metal ions, leading to sulfonamide metal complexesCitation11. Originally investigated for obtaining transition metal ion complexes of acetazolamide AAZ, methazolamide MZA and ethoxzolamide EZA (the main sulfonamide, clinically used drugs belonging to this class of pharmacological agents)Citation11, this approach was subsequently extended to a large set of primary and secondary aromatic/heterocyclic sulfonamides, also including the clinical drugs saccharin (SAC), brinzolamide (BRZ) and dorzolamide (DRZ)Citation12–17. Other sulfonamides possessing a diverse scaffold but effective CA inhibitory properties were also included in such studies together with metal ions which may add a supplementary pharmacological activity, such as Pt(II), Pd(II) and Ru(II) for the antitumour effectsCitation11,Citation17,Citation18, zinc(II) for the antiglaucoma actionCitation14, Al(III) for antacid propertiesCitation13, Co(II), Ag(I) and Cu(II) for antifungal activityCitation13. Imaging tumours overexpressing some CA isoforms (e.g., CA IX and XII) with sulfonamide complexes incorporating isotopes of metal ions which emit positrons (for PET imaging), such as Ga(III), In(III) or Cu(II) were also investigatedCitation17, allowing interesting developments in the field.
Considering the interest of one of our groups for the investigation of Ru(II) complexes with various ligands resulting in versatile types of biological activities (antitumour, antibacterial, enzyme inhibitory, etc.)Citation19,Citation20, we report in this communication the preparation and investigation as CA inhibitors of organoruthenium(II) complexes of acetazolamide, which were found to possess subnanomolar affinity for many pharmacologically relevant isoforms, such as the cytosolic CA I and II and the transmembrane, tumour-associated CA IX and XII.
2. Materials and methods
2.1. Chemistry
The two acetazolamide organoruthenium acetazolamide complexes 1 and 2 were prepared as reported in Biancalana et al.Citation18 with small modifications described below. H NMR spectra were recorded on a Bruker Avance III 500 spectrometer (at room temperature and 500.10 MHz) using TMS as an internal standard in (CD3)2CO. Infrared spectra were recorded with a PerkinElmer Spectrum 100 FTIR spectrometer, equipped with a Specac Golden Gate Diamond ATR as a solid sample support. All NMR data processing was carried out using MestReNova, version 8.1.2.
Complex 1: [(η6-p-cymene)RuCl(μ-Cl)]2 (240 mg, 0.780 mmol) and acetazolamide AAZ (180 mg, 0.810 mmol, 1.03 eq.) were dissolved in 30 mL of a acetone solution for 12 h at room temperature. The solvent was removed and 5 mL of CHCl3 was added. The product 1 was filtered off and left to dry at 55 °C for 4 h. Yield: 47% (277 mg), red solid. 1H NMR (Acetone-d6, 500 MHz): δ = 11.97 (s, 1H, AAZ C(NH)C), 7.81 (s, 2H, –SO2NH2), 5.74 (d, 2H, J = 5.8 Hz, Ar–H cymene), 5.48 (d, 2H, J = 6.0 Hz, Ar–H cymene), 3.08 (hept, 1H, J = 7.0 Hz, cymene Ar–CH(CH3)2), 2.25 (s, 3H, AAZ Ar–CH3), 2.18 (s, 3H, cymene CH3), 1.27 (d, 3H, J = 7.0 Hz, cymene Ar–CH(CH3)2) ppm. IR selected bands (cm−1, ATR): 3146 (νN–H), 3060 (νC = C), 1709 (νC = O), 1521 (νC = N), 1368, 1353, 1311 (νasSO2), 1173 (νsSO2), 683, 620.
Complex 2: Complex 1 (200 mg, 0.38 mmol) was added to chromatography column with silica gel as stationary phase and acetone as mobile phase. The solvent was removed and 2 mL of CH2Cl2 and 30 mL of heptane was added to precipitate the product. The product was filtered off and left to dry at 55 °C for 4 h. Rf factor of complex 2 was found to be between 0.45 and 0.55. Yield: 60% (120 mg), yellow solid. 1H NMR (Acetone-d6, 500 MHz): δ = 12.02 (br, 1H, AAZ C(NH)C), 5.66 (m, 2H, Ar–H cymene), 5.43 (m, 2H, Ar–H cymene), 3.00 (s, 1H, –SO2NH), 2.96, (hept, 1H, J = 6.7 Hz, cymene Ar–CH(CH3)2), 2.38 (s, 3H, AAZ CH3), 2.16 (s, 3H, cymene Ar–CH3), 1.31 (d, J = 7.0 Hz, 6H, cymene Ar–CH(CH3)2). IR selected bands (cm−1, ATR): 3170 (νN–H), 2965 (νC–C), 1701 (νC = O), 1523 (νC = N), 1372, 1310 (νasSO2), 1289, 1155 (νsSO2), 920, 633.
2.2. CA enzyme inhibition assay
An Sx.18Mv-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the catalytic activity of various CA isozymes for CO2 hydration reactionCitation21. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH: 7.5, for α-CAs) or TRIS (pH: 8.3, for β- and γ-CAs) as buffers, 0.1 M Na2SO4 (for maintaining constant ionic strength), following the CA-catalysed CO2 hydration reaction for a period of 10 s at 25 °C. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5%–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitors (10 mM) were prepared in distilled-deionised water and dilutions up to 1 nM were done thereafter with the assay buffer. Enzyme and inhibitor solutions were pre-incubated together for 15 min (standard assay at room temperature) prior to assay, to allow for the formation of the enzyme–inhibitor complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation22. All CAs were recombinant proteins produced as reported earlier by our groupCitation23,Citation24.
3. Discussion and conclusion
Two of the organoruthenium AAZ complexes were described recently by Biancalana et al.Citation18 and were also isolated and characterised in this work. Herein, we would just like to mention some differences between our procedure and the one reported in the literatureCitation18. Complex 1 was prepared very similarly in both laboratories, from the sulfonamide ligand and [(η6-p-cymene)RuCl(μ-Cl)]2. We have performed the reaction for 12 h at room temperature (yield 47%), whereas Biancalana et al.Citation18 performed the reaction at room temperature for 3 days obtaining 70% yield. By refluxing 7 h, the yield was much higher (90%). The preparation of complex 2 was, however, much different between these two laboratories. Biancalana et al.Citation18 isolated product 2 via addition of base (NaOH) to the starting reaction mixture, or by previously preparing the acetazolamide sodium salt which was subsequently reacted with the organoruthenium derivative. In our case, a solution of complex 1 in acetone was added to a chromatography column filled with silica gel as stationary phase. The solvent was removed and addition of a mixture of dichloromethane and heptane led to the precipitation of product 2. We hypothesise that in our procedure, the acid–base reaction leading to the deprotonation of acetazolamide occurred in the column, being probably assisted by the chromatographic stationary phase. As far as we know, this is the first report in which a deprotonated acetazolamide complex is obtained without the use of a strong base such as sodium/potassium hydroxide or tertiary amines.
The structures of the two complexes 1 and 2 are shown in , and they were obtained by X-ray crystallography in both laboratories but reported first by Biancalana et al.Citation18
Figure 2. Structure of acetazolamide organoruthenium complexes 1 (left) and 2 (right). The X-ray crystallography for these compounds was reported in Biancalana et al.Citation18
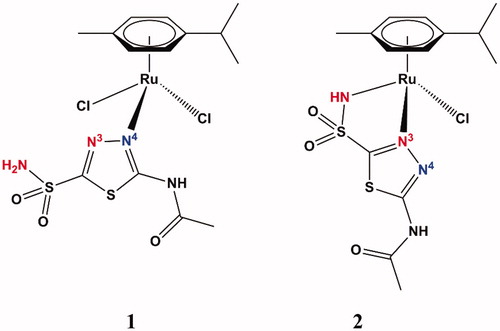
In complex 1, the sulfamoyl moiety of AAZ is not involved in the interaction with the metal ion, which apart the p-cymene ligand has two chloride anions and the endocyclic N4 ( – blue) from the 1,3,4-thiadiazole ring as ligands, in the pseudo-tetrahedral geometry characteristic of Ru(II) – . The second complex (2) is also pseudo-tetrahedral, with the Ru(II) ion being complexed to the cymene ring, one chloride anion and acetazolamide acting as bidentate ligand, with the deprotonated sulfonamide nitrogen atom and the endocyclic N3 ( – red) interacting with the metal ion (). Thus, as already documented earlierCitation11–16,Citation18, this heterocyclic sulfonamide may act as a very versatile ligand when interacting with metal ions for the formation of complexes.
Data of show that the two acetazolamide complexes are much more effective as inhibitors of four physiologically relevant isoforms, the cytosolic CA I and II and the transmembrane, tumour-associated CA IX and XII, involved in several pathologies such as glaucoma (CA II and XII)Citation3,Citation5, or tumoursCitation7 and arthritisCitation9 (CA IX and XII) as AAZ itself. Both complexes were several fold better CAIs compared to the parent ligand, but the complex 2, in which the sulfonamide moiety was deprotonated, acted as much better inhibitor compared to 1. It should be mentioned that AAZ itself is a highly effective, clinically used inhibitor ().
Table 1. Inhibition of human (h) CA isoforms hCA I, II, IX and XII with acetazolamide (AAZ) and its two Ru(II) derivatives 1 and 2, by a stopped flow, CO2 hydrase assay.
Against hCA I, the two complexes showed inhibition constants in the range of 8.5–23.4 nM (AAZ has a KI of 250 nM), against hCA II of 0.48–4.2 nM, whereas against hCA IX of 0.63–3.8 nM and against hCA XII of 0.04–0.52 nM.
Although these two complexes have such a high affinity for the enzyme, we were unable to determine the X-ray crystal structure of a CA isoform (such as CA II or I) in adduct with one of them, due to the fact that no good quality crystals were possible to obtain in various crystallisation conditions that we used. Thus, the inhibition mechanism with these two acetazolamide derivatives remains for the moment unknown. Biancalana et al.Citation18 have also discovered that 1 is involved in various equilibria in solution and one of the products is also 2. However, as seen from , stronger (bidentate) binding of ligand, obviously, results in higher activity.
Although Biancalana et al.Citation18 reported that compounds 1 and 2 were not cytotoxic to some cancer cell lines as well as to non-tumorigenic cells, we think that by considering the results of Can et al.Citation17a and Dilworth et al.Citation17b it is worth testing the antitumour effects of such derivatives in hypoxic tumour cell lines in which CA IX and XII are present. The negative results reported by Biancalana et al.Citation18 are due to the fact that the experiments (in human ovarian and kidney tumour cell lines) were performed in normoxia and not hypoxia. In fact, only in hypoxic conditions, there is a relevant overexpression of CA IX/XII with the consequent potent antitumour effect shown by the CAIs, such as, for example, the sulfonamide compound in Phase II clinical trials SLC-0111Citation25.
Disclosure statement
The authors do not report conflict of interest.
Additional information
Funding
Related Research Data
References
- (a) Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO2 capture. J Enzyme Inhib Med Chem 2013;28:229–30. (b) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (c) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. (d) Abbate F, Winum JY, Potter BV, et al. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with EMATE, a dual inhibitor of carbonic anhydrases and steroid sulfatase. Bioorg Med Chem Lett 2004;14:231–4. (e) Capasso C, Supuran CT. An overview of the alpha-, beta-and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32.
- (a) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12. (b) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21. (c) Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008-2018). Expert Opin Ther Pat 2018;28:729–40. (d) Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin Ther Pat 2018; 28:745–54.
- (a) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. (b) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. (c) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. (d) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. (e) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68.
- (a) Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005-2013). Expert Opin Ther Pat 2013; 23:681–91. (b) Temperini C, Cecchi A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Sulfonamide diuretics revisited—old leads for new applications. Org Biomol Chem 2008;6:2499–506.
- (a) Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16. (b) Borras J, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406.
- (a) Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35. (b) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25.
- (a) Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008-2013). Expert Opin Ther Pat 2013;23:737–49. (b) Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:E48. (c) Ward C, Langdon SP, Mullen P, et al. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat Rev 2013;39:171–9. (d) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8. (e) Casey JR, Morgan PE, Vullo D, et al. Carbonic anhydrase inhibitors. Design of selective, membrane-impermeant inhibitors targeting the human tumor-associated isozyme IX. J Med Chem 2004;47:2337–47.
- (a) Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother 2016;16:961–8. (b) Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. J Enzyme Inhib Med Chem 2016;31:894–9.
- (a) Margheri F, Ceruso M, Carta F, et al. Overexpression of the transmembrane carbonic anhydrase isoforms IX and XII in the inflamed synovium. J Enzyme Inhib Med Chem 2016;31:60–3. (b) Bua S, Di Cesare Mannelli L, Vullo D, et al. Design and synthesis of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the treatment of rheumatoid arthritis. J Med Chem 2017;60:1159–70.
- (a) Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors?. J Enzyme Inhib Med Chem 2018;33:485–95. (b) Di Fiore A, Maresca A, Supuran CT, De Simone G. Hydroxamate represents a versatile zinc binding group for the development of new carbonic anhydrase inhibitors. Chem Commun (Camb) 2012;48:8838–40. (c) Marques SM, Nuti E, Rossello A, et al. Dual inhibitors of matrix metalloproteinases and carbonic anhydrases: iminodiacetyl-based hydroxamate-benzenesulfonamide conjugates. J Med Chem 2008;51:7968–79. (d) Bozdag M, Carta F, Angeli A, et al. Synthesis of N'-phenyl-N-hydroxyureas and investigation of their inhibitory activities on human carbonic anhydrases. Bioorg Chem 2018;78:1–6.
- (a) Andruh M, Cristurean E, Stefan R, Supuran CT. Carbonic anhydrase inhibitors. Part 6. Novel coordination compounds of Pd(II), Pt(II) and Ni(II) with 6-ethoxybenzothiazole-2-sulfonamide. Rev Roum Chim 1991;36:727–32. (b) Luca C, Barboiu M, Supuran CT. Carbonic anhydrase inhibitors. Part 7. Stability constants of complex inhibitors and their mechanism of action. Rev Roum Chim 1991;36:1169–73. (c) Supuran CT, Manole G, Andruh M. Carbonic anhydrase inhibitors. Part 11. Coordination compounds of heterocyclic sulfonamides with lanthanides are potent inhibitors of isozymes I and II. J Inorg Biochem 1993;49:97–103. (d) Borras J, Alzuet G, Ferrer S, Supuran CT, Metal complexes of heterocyclic sulfonamides as carbonic anhydrases inhibitors. In: Supuran CT, Scozzafava A, Conway J, eds. Carbonic anhydrase – its inhibitors and activators. Boca Raton, FL: CRC Press; 2004:183–207.
- (a) Sumalan SL, Casanova J, Alzuet G, et al. Synthesis and characterization of metal(II)-8-quinolinsulfonamidato (sa) complexes (M = Co, Ni, Cu, and Zn). Crystal structure of [Zn(sa)2(NH3)]NH3 complex. Carbonic anhydrase inhibitory properties. J Inorg Biochem 1996;62:31–9. (b) Borràs J, Casanova J, Cristea T, et al. Complexes with biologically active ligands. Part 6 Ni(II) coordination compounds of hydrazine and heterocyclic sulfonamides as inhibitors of the zinc enzyme carbonic anhydrase. Met Based Drugs 1996;3:143–8.
- (a) Diaz JR, Fernández Baldo M, Echeverría G, et al. A substituted sulfonamide and its Co (II), Cu (II), and Zn (II) complexes as potential antifungal agents. J Enzyme Inhib Med Chem 2016;31:51–62. (b) Ilies MA, Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors. Part 91. Metal complexes of heterocyclic sulfonamides as potential pharmacological agents in the treatment of gastric Acid secretion imbalances. Met Based Drugs 2000;7:57–62. (c) Mastrolorenzo A, Supuran CT. Antifungal activity of Ag(I) and Zn(II) complexes of sulfacetamide derivatives. Met Based Drugs 2000;7:49–54.
- (a) Supuran CT, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors Part 72 synthesis and antiglaucoma properties of metal complexes of p-fluorobenzolamide. Met Based Drugs 1999;6:67–73. (b) Supuran CT, Scozzafava A, Briganti F, et al. Carbonic anhydrase inhibitors. Part 55 metal complexes of 1,3,4-thiadiazole-2-sulfonamide derivatives: in vitro inhibition studies with carbonic anhydrase isozymes I, II and IV. Met Based Drugs 1998;5:103–14. (c) Supuran CT, Scozzafava A, Jitianu A. Carbonic anhydrase inhibitors. Part 54: metal complexes of heterocyclic sulfonamides: a new class of antiglaucoma agents. Met Based Drugs 1997;4:307–15.
- (a) Mincione G, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 46 inhibition of carbonic anhydrase isozymes I, II and IV with trifluoromethylsulfonamide derivatives and their zinc(II) and copper(II) complexes. Met Based Drugs 1997;4:27–34. (b) Scozzafava A, Supuran CT. Complexes with biologically active ligands. Part 10 inhibition of carbonic anhydrase isozymes I and II with metal complexes of imidazo[2,1-b ]-1,3,4-thiadiazole-2-sulfonamide. Met Based Drugs 1997;4:19–26. (c) Jitianu A, llies MA, Briganti F, et al. Complexes with biologically active ligands. Part 9 metal complexes of 5-benzoylamino- and 5-(3-nitrobenzoyl-amino)-1,3,4-thiadiazole-2-sulfonamide as carbonic anhydrase inhibitors. Met Based Drugs 1997;4:1–7.
- (a) Supuran CT. Complexes with biologically active ligands. Part 4. Coordination compounds of chlorothiazide with transition metal ions behave as strong carbonic anhydrase inhibitors. Met Based Drugs 1996;3:79–83. (b) Supuran CT. Metal complexes of 1,3,4-thiadiazole-2,5-disulfonamide are strong dual carbonic anhydrase inhibitors, although the ligand possesses very weak such properties. Met Based Drugs 1995;2:331–6. (c) Supuran CT. Thienothiopyransulfonamides as complexing agents for the preparation of dual carbonic anhydrase inhibitors. Met Based Drugs 1995;2:327–30.
- (a) Can D, Spingler B, Schmutz P, et al. [(Cp-R)M(CO)3] (M = Re or 99mTc) arylsulfonamide, arylsulfamide, and arylsulfamate conjugates for selective targeting of human carbonic anhydrase IX. Angew Chem Int Ed Engl 2012;51:3354–7. (b) Dilworth JR, Pascu SI, Waghorn PA, et al. Synthesis of sulfonamide conjugates of Cu(II), Ga(III), In(III), Re(V) and Zn(II) complexes: carbonic anhydrase inhibition studies and cellular imaging investigations. Dalton Trans 2015;44:4859–73.
- Biancalana L, Batchelor LK, Ciancaleoni G, et al. Versatile coordination of acetazolamide to ruthenium(II) p-cymene complexes and preliminary cytotoxicity studies. Dalton Trans 2018;47:9367–84.
- (a) Kljun J, Bratsos I, Alessio E, et al. New uses for old drugs: attempts to convert quinolone antibacterials into potential anticancer agents containing ruthenium. Inorg Chem 2013;52:9039–52. (b) Kljun J, Scott AJ, Lanisnik RT, et al. Synthesis and biological evaluation of organoruthenium complexes with azole antifungal agents. First crystal structure of a tioconazole metal complex. Organometallics 2014;33:1594–601. (c) Turel I, Pečanac M, Golobič A, et al. Solution, solid state and biological characterization of ruthenium(III)-DMSO complexes with purine base derivatives. J Inorg Biochem 2004;98:393–401. (d) Seršen S, Kljun J, Kryeziu K, et al. Structure-related mode-of-action differences of anticancer organoruthenium complexes with β-diketonates. J Med Chem 2015;58:3984–96. (e) Gobec M, Kljun J, Sosic I, et al. Structural characterization and biological evaluation of a clioquinol-ruthenium complex with copper-independent antileukaemic activity. Dalton Trans 2014;43:9045–51. (f) Kljun J, Anko M, Traven K, et al. Pyrithione-based ruthenium complexes as inhibitors of aldo-keto reductase 1C enzymes and anticancer agents. Dalton Trans 2016;45:11791–800. (g) Mitrović A, Kljun J, Sosič I, et al. Clioquinol-ruthenium complex impairs tumour cell invasion by inhibiting cathepsin B activity. Dalton Trans 2016;45:16913–21.
- (a) Turel I, Kljun J, Perdih F, et al. First ruthenium organometallic complex of antibacterial agent ofloxacin. Crystal structure and interactions with DNA. Inorg Chem 2010;49:10750–2. (b) Hudej R, Kljun J, Kandioller W, et al. Synthesis and biological evaluation of the thionated antibacterial agent nalidixic acid and its organoruthenium(II) complex. Organometallics 2012;31:5867−74. (c) Traven K, Sinreih M, Stojan J, et al. Ruthenium complexes as inhibitors of the aldo-keto reductases AKR1C1-1C3. Chem Biol Interact 2015;234:349–59. (d) Traven K, Eleftheriadis N, Seršen S, et al. Ruthenium complexes as inhibitors of 15-lipoxygenase-1. Polyhedron 2015;101:306–13. (e) Ristovski S, Uzelac M, Kljun J, et al. Organoruthenium prodrugs as a new class of cholinesterase and glutathione-S-transferase inhibitors. ChemMedChem 2018;13:2166–2176.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Menchise V, De Simone G, Alterio V, et al. Carbonic anhydrase inhibitors: stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J Med Chem 2005;48:5721–7. (b) Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors—part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents in rabbits. Eur J Med Chem 1998;33:247–54. (c) Şentürk M, Gülçin İ, Beydemir Ş, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9. (d) Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47.
- (a) Krall N, Pretto F, Decurtins W, et al. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors . Angew Chem Int Ed Engl 2014;53:4231–5. (b) Rehman SU, Chohan ZH, Gulnaz F, Supuran CT. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzyme Inhib Med Chem 2005;20:333–40. (c) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: Structure-activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73. (d) Dubois L, Peeters S, Lieuwes NG, et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 2011;99:424–31. (e) Chohan ZH, Munawar A, Supuran CT. Transition metal ion complexes of Schiff-bases. Synthesis, characterization and antibacterial properties. Met Based Drugs 2001;8:137–43. (f) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal β-class (Cab) and γ-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–8.
- (a) Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8. (b) Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3. (c) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. (d) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11.
- (a) Bozdag M, Carta F, Ceruso M, et al. Discovery of 4-hydroxy-3-(3-(phenylureido)benzenesulfonamides as SLC-0111 analogues for the treatment of hypoxic tumors overexpressing carbonic anhydrase IX. J Med Chem 2018;61:6328–38. (b) Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res 2011;71:3364–76. (c) McDonald PC, Swayampakula M, Dedhar S. Coordinated regulation of metabolic transporters and migration/invasion by carbonic anhydrase IX. Metabolites 2018;8:20. (d) Boyd NH, Walker K, Fried J, et al. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017;2:92928.