
Abstract
The use of natural systems, such as outer membrane protein A (OmpA), phosphoporin E (PhoE), ice nucleation protein (INP), etc., has been proved very useful for the surface exposure of proteins on the outer membrane of Gram-negative bacteria. These strategies have the clear advantage of unifying in a one-step the production, the purification and the in vivo immobilisation of proteins/biocatalysts onto a specific biological support. Here, we introduce the novel Anchoring-and-Self-Labelling-protein-tag (ASLtag), which allows the in vivo immobilisation of enzymes on E. coli surface and the labelling of the neosynthesised proteins with the engineered alkylguanine-DNA-alkyl-transferase (H5) from Sulfolobus solfataricus. Our results demonstrated that this tag enhanced the overexpression of thermostable enzymes, such as the carbonic anhydrase (SspCA) from Sulfurihydrogenibium yellowstonense and the β-glycoside hydrolase (SsβGly) from S. solfataricus, without affecting their folding and catalytic activity, proposing a new tool for the improvement in the utilisation of biocatalysts of biotechnological interest.
1. Introduction
The term immobilised enzymes refers to ‘enzymes physically confined or localised in a certain defined region of space with retention of their catalytic activities, and which can be used repeatedly and continuously’Citation1. The immobilisation of enzymes on solid supports is historically very important for overcoming their general instability in harsh operational conditions and their low shelf-life, as well as the need for their recycling more timesCitation2. Furthermore, the physical separation of the biocatalyst from the reaction mixture avoids the protein contamination of the products. Although a reduction in reaction rates sometimes occurs, because the enzyme cannot mix freely with the substrate or a particular conformational change is needed for the biocatalyst efficiency, there are many examples of increased activity and stability of immobilised enzymesCitation3. Many chemical or physical methods for the enzyme immobilisation are currently available, from the physical adsorption to the covalent coupling on supports ()Citation3–6. Recently, the use of protein-tags based on an engineered version of the human O6-alkyl-guanine-DNA-alkyl-transferases (hAGT) is an effective alternative for the covalent immobilisation of proteins and enzymes ()Citation7–9. AGTs (or OGTs, MGMTs; E.C.: 2.1.1.63) are DNA repair enzymes, which irreversibly transfer the alkyl group from the damaged DNA containing O6-alkyl-guanines to their cysteine residue in the active siteCitation10–13. In 2003, Johnsson and his group demonstrated that most enzymes of this class display relatively low substrate specificity, making them reactive also with free O6-benzyl-guanines (O6-BG) nucleobasesCitation14. This led to the development of the so-called SNAP-tag™ technology, which uses derivatives of O6-BG potentially conjugated with an unlimited number of chemical groupsCitation15–18. This system allows the immobilisation on O6-BG-derivatised surface of the protein expressed in fusion with the SNAP-tagCitation18 (). However, all these approaches mainly depend on the high costs due to the isolation and purification of the biocatalysts. This limitation can be easily overcome by the heterologous expression of enzymes and their in vivo direct immobilisation on the surface of bacterial hosts, by the utilisation of transmembrane protein domains, as the ice nucleation protein (INP) of the Gram-negative bacterium Pseudomonas syringae ()Citation19,Citation20. This protein is composed of an N-terminal domain (N, 175 residues) structurally separated from a C-terminal domain (C, 49 residues) by a repetitive central domainCitation21. Both domains play a role in the anchoring of proteins to the outer membraneCitation21. The use of INP as anchoring carrier is considered of great interest in biotechnological applications, ranging from the development of bacterial cell surface-display systems for vaccine delivery to the fabrication of whole-cell biocatalysts and biosensorsCitation22–24. The N-terminal domain of INP (INPN) was recently and successfully used for the one-step procedure immobilisation ()Citation22–29. Moreover, Capasso et al. demonstrated that the amount of a thermostable carbonic anhydraseCitation30–33 fused to the INPN domain and expressed on the bacterial cell surface had a hydratase activity similar to that of the enzyme covalently immobilised onto magnetic nanoparticlesCitation30,Citation34. Here, we introduce a novel protein-tag system, (hereinafter Anchoring-and-Self-Labelling-protein-tag or ASLtag), which simultaneously allows the in vivo immobilisation of the enzyme of interest on the E. coli surface and its quantitative determination (). The ASLtag system is formed by the INPN domain fused to an engineered and thermostable variant of the alkylguanine-DNA-alkyl-transferase (H5) from the hyperthermophilic archaeon Sulfolobus solfataricusCitation35,Citation36. This enzyme was extensively characterised, suggesting its biotechnological role as thermostable alternative to the commercial SNAP-tag™ and its utilisation as protein-tag for heterologous expression of proteins of interest in E. coli and, for the first time, in thermophilic organisms as Thermus thermophilus and Sulfolobus islandicusCitation35–37. Thus, using the substrate of H5, a fluorescein derivative of the O6-BG (), we successfully estimated the expression of the ASLtag in E. coli cells, by in vitro gel-imaging techniques, as well as by in vivo fluorescent microscopy. Furthermore, we demonstrated that the activity and the stability of the enzymes of interest (SspCA, the α-carbonic anhydrase from Sulfurihydrogenibium yellowstonense; and SsβGly, the β-glycoside hydrolase from S. solfataricus) fused to the ASLtag and exposed on the surface of E. coli cells were not affected by the presence of this novel protein-tag.
Figure 1. Examples of enzyme immobilisation methods. Among the traditional methods for the irreversible immobilisation (a–c, e–f) of an enzyme (E, in blue), the recent introduction of the SNAP-tag™ technology (d, red semicircle) allowed an indirect immobilisation of protein of interest. The one step in vivo immobilisation of an enzyme (g) is possible when it is expressed as fusion protein with the N-terminal domain of the ice nucleation protein (INPN, in red). Because of the presence of a peptide leader upstream the coding sequence, the nascent polypeptide is translocated to the cell OM by the anchoring transmembrane INPN domain, leading to the immobilisation and exposition of the biocatalyst outside the cell.
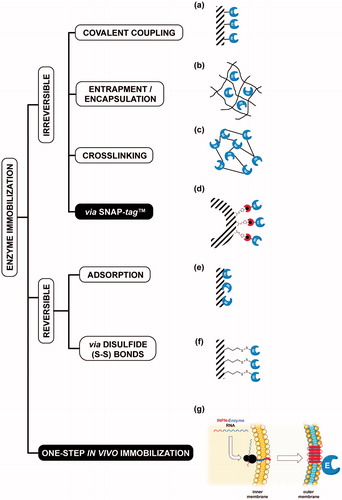
Figure 2. The ASLtag protein. The ASLtag gene is composed by the inpn ORF (in white) in frame fused to the ogtH5 gene (in green) in the pET22b expression vector. This tag can be further fused to a gene of interest (goi, in blue), for a one step procedure of the expression and immobilisation of an enzyme (E). The presence of the H5 moiety allows the quantitative estimation of the yield of E by the irreversible alkyl-transferase assay using a fluorescent O6-benzyl-guanine derivative (BG-FL). Between inpn and ogtH5, a spacer (in pink), a thrombin cleavage site (shown as an orange triangle) and a 6 × His-tag (in black) were inserted, for the easy separation and purification of any H5-E fusion protein.
2. Materials and methods
2.1. Reagents
All DNA restriction and modification enzymes and the fluorescent substrate for the OGT activity (SNAP-Vista Green™, hereinafter BG-FL) were purchased from New England Biolabs (Ipswich, MA); molecular biology kits for the plasmid preparations and DNA gel extractions (NucleoSpin® Gel and PCR Clean-up®) were from Macherey-Nagel GmbH (Germany); Lyophilised Thrombin Protease from GE Healthcare (Illinois, US). Eurofins Genomics (Germany) performed the oligonucleotides synthesis and the DNA sequencing service.
2.2. DNA constructs
To obtain the pET-ASLtag construct, we replaced the α-carbonic anhydrase (SspCA) gene with the ogtH5 gene in the previously described vector pET-22b/INPN-SspCACitation30. By the latter and the pQE-ogtH5 Citation35 plasmid as template, the DNA fragments relative to the INPN domain and H5 were respectively amplified with the INPN- and H5-Fwd/Rev oligonucleotides pairs (listed in ), under the following conditions: an initial denaturation at 95.0 °C for 5 min, 30 cycles of 30 s at 95.0 °C, 30 s at 50.0 °C and 30 s at 72.0 °C, followed by a final extension of 5 min at 72.0 °C. DNA products were fused to each other in a further PCR amplification, taking advantage of the total complementarity of the INPN-Rev to the H5-Fwd oligonucleotide, obtaining the final ASLtag DNA fragment. Subsequently, this fragment and the pET-22b/INPN-SspCA vector were digested with Hind III and Xba I restriction endonucleases, gel-purified, and ligated. The ligation mixture was used to transform the E. coli DH5α strain, and positive colonies were confirmed by colony PCR and DNA sequencing.
Table 1. Oligonucleotides used in this study.
The same cloning strategy was used to achieve the pET-ASLtag-SspCA construct: the ASLtag DNA fragment obtained this time using the H5-Rev2 oligonucleotide was used as template to further fuse to the SspCA gene (obtained by amplification with SspCA Fwd/Rev oligonucleotide pairs, ). Again, positive colonies after ligation and transformation were confirmed as above.
Finally, the pET-ASLtag-lacS plasmid preparation started with the achievement of the DNA fragment relative to the ogtH5 gene fused to the β-glycoside hydrolase from the thermophilic archaea Sulfolobus solfataricus (lacS), obtained by the Hind III/BamH I digestion from the pQE-ogtH5-lacS plasmidCitation36. The pET-ASLtag plasmid was similarly digested to obtain the pET22 recipient for the first ligation/transformation round. Positive blue colonies were selected by the hydrolase activity of the lacS gene product (SsβGly) on the Ampicillin selective Luria-Bertani (LB) Agar plates supplemented with 5-bromo-4-chloro-3-indolyl-β-D-glucopyranoside (X-Glc), and the insertion confirmed using PCR analysis. This intermediate vector was further digested with BamH I, treated with Alkaline Phosphatase Calf Intestinal (CIP) and ligated to the BamH I/BamH I INPN DNA fragment, derived from the digestion of the pET-ASLtag plasmid. In this case, positive blue colonies on LB-Amp-X-Glc agar plates were confirmed by Pst I restriction enzyme digestion analysis.
2.3. Determination of the protein expression by a fluorescent assay based on the H5 activity
All constructs were used to transform E. coli BL21(DE3) cells. Cultures were grown at 37.0 °C in LB selective medium supplemented with 100.0 mg/L ampicillin and 30.0 mg/L chloramphenicol; expression was induced with 1.0 mM isopropyl-thio-β-D-galactoside (IPTG) when an absorbance value of 0.5–0.6 A600 nm was reached, or in the ZY auto-induction medium (AI)Citation38, supplemented with the same selective antibiotics. After an overnight incubation, whole cells were collected and assayed by using the BG-FL fluorescent H5 substrate previously describedCitation35–37,Citation39 for a qualitative measurement of the protein expression, an aliquot of 1.0 mL of cells was centrifuged at 4000 × g and the pellet was resuspended in 50.0 μL of 5.0 μM BG-FL in phosphate-buffered saline (PBS 1×, 20.0 mM phosphate buffer, NaCl 150.0 mM, pH 7.3). After an incubation for 2.0 h at 37.0 °C, reactions were stopped by denaturing the samples in Leammli Buffer 1× and directly loaded on SDS-PAGE, followed by gel-imaging on a VersaDoc 4000™ system (Bio-Rad), by applying a blue LED/530 bandpass filter as excitation/emission parameters, respectively. Finally, the fluorescence intensity of each band was normalised to the intensity of the signal obtained from the Coomassie Blue staining analysis.
For a quantitative determination of the expression, whole cells were opportunely diluted to achieve an OD600nm of 1.0. By following the same above-mentioned assay, three different volumes of whole cells were loaded in the same gel with defined amounts (in the range of 0.8–50.0 pmols) of purified free H5 protein, after the reaction on BG-FL in the same conditions. The obtained values of the relative fluorescence as a function of the purified loaded H5 were fitted by a linear equation, whose parameters were then used for the estimation of the amount of expressed H5-derivated fusion proteins, assuming that the activity of the H5 moiety in the fusions is not affected by the presence of the other protein partner(s). Given that the concentration of E. coli cell cultures of 1.0 OD600nm is ca. 8.0 × 108 cells/mL, and the amount of wet cells is 1.7 g/L, it is possible to calculate the yield of expressed proteins in terms of pmol/mg of cellsCitation40.
2.4. Membranes fractionation
The E. coli outer (OM) and inner (IM) membranes were purified by following a procedure previously describedCitation27. Briefly, harvested bacterial cells were resuspended 1:20 (g/mL) in 25.0 mM Tris/HCl buffer, pH 8.0 and disrupted by sonication on ice (10 cycles of 10 s: 50 s on:off treatment). Cell extract was centrifuged at 120,000 × g for 1.0 h, and the supernatant containing the cytoplasmic fraction was discarded. Both IM and OM fractions were recovered in the pellet and resuspended in 20.0 mL of PBS 1×, containing 0.01 mM MgCl2 and 2.0% Triton X-100. After incubation at room temperature for 30.0 min, the solution was centrifuged as described above. Then, a defined amount of the OM fraction obtained was assayed for the H5 activity, leading to the quantitative determination of the total amount of fusion protein, as previously described.
2.5. Thrombin assay
The ASLtag on the OM was cleaved with the Thrombin Protease, in order to separate the H5 moiety from the INPN transmembrane domain. A suspension of E. coli whole cells was gently centrifuged at 3000 × g for 10.0 min at 4.0 °C and then resuspended in a half volume of PBS 1×. The sample was incubated with 30.0 U of Thrombin Protease at 25.0 °C overnight under gentle agitation in the presence of 5.0 μM of BG-FL, followed by the same centrifugation. Cells and supernatants were separately loaded on SDS-PAGE and analyzed by gel-imaging and Coomassie staining as described.
2.6. Microscopy analysis of E. coli cells
For in vivo imaging, E. coli BL21(DE3) cells transformed with pET-22b/INPN-SspCA or pET-ASLtag plasmids were IPTG-inducted, grown overnight at 37.0 °C and finally diluted until OD600nm of 1.0. An amount of cells corresponding to a volume of 1.0 mL of the culture was washed twice in PBS 1× and finally resuspended in 50.0 μL of the same buffer supplemented with 5.0 μM of the BG-FL substrate. After an incubation at 37.0 °C for 30.0 min, cells were washed twice, resuspended and again incubated for 30.0 min at 37.0 °C, to allow the external diffusion of the unreacted substrate. Labelling was first verified by fluorescence gel-imaging on SDS-PAGE and then spotted on poly-L-lysine coated slides for microscopy analysis.
Images were collected using a DM6 fluorescence microscope and Hamamatsu camera under the control of Leica LAS AF 6000 software; excitation and emission wavelengths used suitably for AlexaFluor488 dye were λex = 490 nm; λem = 525 nm, respectively.
2.7. β-glycoside hydrolase assay
The β-glycoside hydrolase assay was performed as previously describedCitation41 at different temperatures in 50 mM sodium phosphate buffer at pH 6.5, in the presence of 2 Np- and 4 Np-Glc substrates at 5.0 mM final concentration. OM fractions containing ASLtag and relative fusion proteins amounts ranging from 1.0 to 5.0 μg were used in each assay. For the correction of the spontaneous hydrolysis of the substrates, all the reactants except the enzyme (blank mixture), was taken into account. The enzymatic activity was calculated on the basis of the molar extinction coefficient (εM) values of 2- and 4-nitrophenol in 50 mM sodium phosphate buffer pH 6.5 at different temperatures, as previously reportedCitation41. We defined as one unit of enzyme activity the amount of enzyme hydrolyzing 1.0 μmol of substrate in 1.0 min, under the above-described conditions.
2.8. Carbonic anhydrase assay
CA activity assay was a modification of the procedure described by Capasso et al.Citation33. Briefly, the hydratase assay was performed at 0 °C using CO2 as substrate following the pH variation due to the catalyzed conversion of CO2 to bicarbonate. Bromothymol blue was used as pH indicator. The production of hydrogen ions during the CO2 hydration reaction lowers the pH of the solution leading to a colour transition of the dye. The time required for the colour change is inversely proportional to the amount of CA present in the sample. The Wilbur–Anderson units (WAU) were calculated according to the following definition: one WAU of CA activity is defined as the ratio (T0 − T)/T, where T0 (the time needed for the pH indicator color change for the uncatalyzed reaction) and T (the time needed for the pH indicator color change for the catalyzed reaction) are recorded as the time (in seconds) required for the pH to drop from 8.3 to the transition point of the dye (pH 6.8) in a control buffer and the presence of enzyme, respectively.
3. Results and discussion
3.1. Expression analysis and localisation of ASLtag in E. coli
The in vivo alkyl-transferase fluorescent assay of H5 on whole bacterial cells is a useful method to determine the heterologous expression of this protein and/or relative fusion proteins in the mesophilic E. coli and in the thermophilic species T. thermophilus and S. islandicus, without any purification procedureCitation35–37. The in vivo assay is possible since the bacterial cells are permeable to OGT fluorescent substrates (as the commercially available SNAP-Vista Green™ or SNAP-Cell TMR™); and the catalytic activity at mesophilic temperatures (25–37 °C) of H5 mutant is one order of magnitude higher than the SsOGT wild-type counterpart and comparable to the commercial SNAP-tag™Citation36. Despite GFPs utilisation, the covalent conjugation of H5 with the benzyl-fluorophore moiety of the substrate () allows the denaturation of the whole cells and the direct loading of the samples on SDS-PAGE for the gel-imaging analysis, as described in the Materials and MethodsCitation35–37,Citation39.
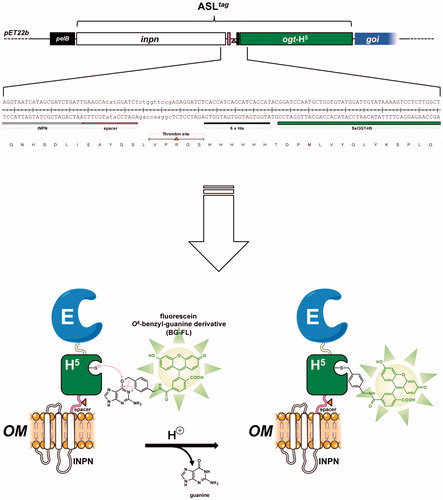
To evaluate the expression of the ASLtag, E. coli BL21(DE3)/pET-ASLtag cells were grown in LB medium and induced with IPTG or in the AI. In the latter case, although the expression level of the fusion protein was satisfactory, the presence of only the H5 signal (ca. 20% of the total fluorescence intensity) in the gel-imaging analysis is difficult to rationalise (). Probably, during the expression of the protein (e.g., in the advanced stage of the growth) could occur interruptions or failures in the translocation process of the ASLtag on the outer membrane of E. coli with following breaking/cleavage events, especially in the spacer region between INPN and H5 (). On the other hand, after the IPTG induction, a strong and clear signal at the expected molecular weight is visible, without any fragmentation (). This is an important result since, to date, the heterologous expression of SsOGT wild-type and relative variants in E. coli has been generally performed in the ABLE C strain, which keeps low the number of copies of the ogt-containing plasmidsCitation35,Citation36. As proof, transformed E. coli BL21(DE3) strain with the pQE-ogtH5 plasmidCitation36 showed a very low expression of free H5 (Supplementary Figure S1), whereas the fusion with the INPN domain and the consequent translocation on the outer cell membrane made the H5 expression possible in this strain. The comparative fluorescent analysis with a defined amount of free H5 enzyme, allowed us to quantitatively measure the heterologous expression of ASLtag as pmol/mg of the whole wet cells ( and ). The assay on H5 confirmed the anchoring function of the INPN trans-membrane protein: only the whole cells and the fraction containing the outer membrane displayed a fluorescent band corresponding to the ASLtag fusion protein, whereas the signal was missed in the lanes relative to the cytoplasmic and the inner membrane fractions (). Besides, the evidence of the INPN on the bacterial outer membrane was confirmed by treating the whole bacterial cells with the thrombin, too (). A cleavage site for this protease was localised between the INPN domain and the H5 moiety (). The fluorescent signal corresponding to the MW of the H5 protein was present only in the supernatant fraction when the protease was added at the same time with the BG-FL substrate (). Finally, we checked the OGT activity of ASLtag in living cells by microscope analysis, upon the labelling with the fluorescent substrate. The obtained images of living E. coli BL21(DE3) cells showed a strong and specific fluorescent signal only in those transformed with the ASLtag-containing plasmid (), indicating that the fusion protein is stable and proficient to labelling. This data suggest that ASLtag is suitable for localisation and analysis of membrane proteins, and provide an opportunity for further in vivo analyses of ASLtag-tagged proteins of interest under physiological conditions.
Figure 3. The ASLtag expression in E. coli. (a) E. coli BL21(DE3) strain transformed with the pET-ASLtag plasmid was grown in IPTG-inducted or in auto-induction medium (AI). After the in vivo OGT assay, a defined amount in micrograms of whole cells at OD600nm of 1.040 was directly loaded on SDS-PAGE, followed by gel-imaging fluorescence (F) and Coomassie staining (C) analyses. Open and closed green arrows indicate fluorescent signals of free H5 or H5-based fusion proteins, and the free BG-FL substrate, respectively. M: molecular weight marker. (b) Quantitative estimation of the ASLtag expression: defined amount of cells and purified H5 protein (in pmols) were loaded and analyzed on a SDS-PAGE (on the left). Fluorescent values obtained from H5 were fitted in a linear plot (on the right), as described in Materials and Methods. Obtained parameters allowed the quantitative determination of the amount of ASLtag in E. coli cells and shown in .
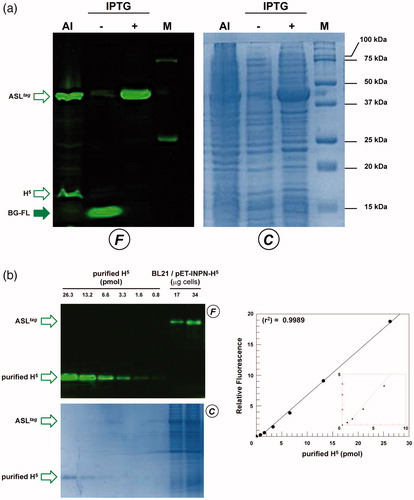
Figure 4. Localisation of ASLtag in E. coli. (a) gel-imaging and Coomassie staining analyses after SDS-PAGE of different loaded amount of the whole cells, the relative cytoplasmic fraction (FCE), the inner (IM) and the outer membrane (OM) fractions. (b) Cleavage of ASLtag by the Thrombin protease (T) on whole cells (W). After the H5 reaction during the protease treatment, cells were centrifuged, separating the supernatant (S) from the intact cells (C). (c) E. coli BL21(DE3) cells transformed with pET-22b/INPN-SspCA (Top) or with pET-ASLtag (bottom) were incubated with BG-FL and then analyzed at fluorescence microscopy. Images show bright field (BHF), AlexaFluor488 (green) and merged images. All used symbols are described in .
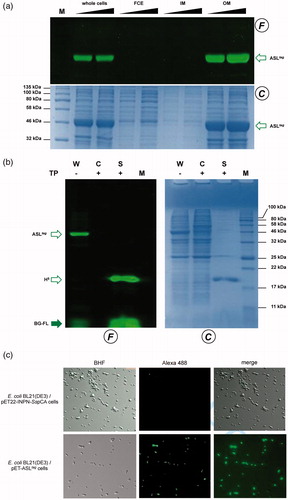
Table 2. Quantitative estimation of the heterologous expression of ASLtag and relative fusion proteins in E. coli BL21(DE3) whole wet cells.
3.2. In vivo immobilisation of thermostable enzymes by ASLtag
As described in the literature, it has been demonstrated that the monomeric α-carbonic anhydrase (SspCA) from the thermophilic bacterium S. yellowstonense can be actively expressed onto the outer membrane of E. coliCitation30. Following this strategy, we realised a plasmid expressing the ASLtag-SspCA construct by inserting the ogtH5 gene between the INPN and SspCA. The expression profile analyzed by following the H5 activity on BG-FL confirmed that in the AI multiple fluorescent signals are present (), mainly represented by the full-length ASLtag-SspCA (70.2 kDa; ca. 65% of the total fluorescence intensity) and lower band (ca. 35%) closer to 37 kDa than 50 kDa (). This signal is compatible to the ASLtag (42.0 kDa) as well as the H5-SspCA moiety (46.0 kDa), suggesting that both translation interruption and translocation failure events in this growth conditions can be not excluded. Again, we detected an optimal achievement of the full-length fusion protein under IPTG-based expression in LB medium (ca. 95%; ). In this condition and considering the INPN:H5:SspCA ratio as 1:1:1, the amount of the whole fusion protein expressed was estimated as ca. 60.0 pmol/g cells, which corresponds to ca. 1.5 μg of the sole immobilised carbonic anhydrase per mg of cells (). Preliminary assays on ASLtag-SspCA indicated that the presence of H5 does not hamper the hydratase activity of the SspCA, if compared with that of the previously expressed INPN-anchored enzymeCitation30 (data not shown).
Figure 5. Heterologous expression of thermostable enzymes fused to ASLtag. SDS-PAGE analysis of the ASLtag-SspCA (a) and ASLtag-SsβGly (b) expression. (a) Fluorescent signals on the F panel correspond to bands whose molecular weights are attributable to the protein fusions shown in the schemes. (b) 1.0 mM IPTG-inducted cells gave a very high fluorescent band (marked by an asterisk), presumably corresponding to the tetrameric form of SsβGly linked to 4 × ASLtag units, as shown. All used symbols are described in .
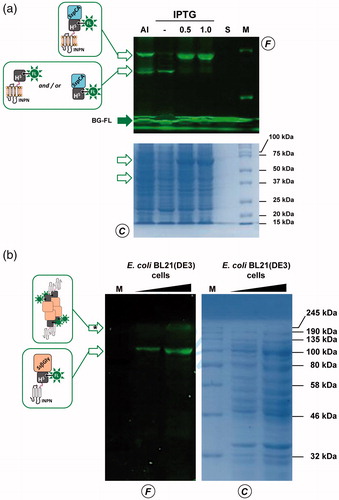
ASLtag system was also tested with another thermostable enzyme, the β-glycoside hydrolase (SsβGly) from the thermophilic archaea S. solfataricusCitation42. We previously demonstrated that the cytoplasmic H5-SsβGly fusion protein is stable and active for both the OGT and the β-glycosidase assays, suggesting that the presence of one enzyme does not interfere with the folding and activity of the otherCitation36. Interesting to note that anchoring this protein fusion to the bacterial outer membrane by the INPN domain is particularly challenging because SsβGly is active only in its tetrameric formCitation43,Citation44. The presence of blue colonies on LB agar plate in the presence of a glucoside chromogenic derivative (X-Glc), which is a preferred substrate of SsβGlyCitation41 but not of the E. coli LacZ (a β-galactosidase enzyme) was a first and convincing indication of the oligomerisation of this thermostable enzyme. Although in this case the amount of the expressed fusion protein was lower than the above examples ( and ), IPTG-inducted E. coli BL21(DE3)/pET-ASLtag-SsβGly cells displayed a fluorescent signal of expected molecular weight (98.8 kDa), corresponding to one monomer of SsβGly fused to one ASLtag unit. However, a higher band is clearly visible in the fluorescence analysis (marked with an asterisk in ), out of the molecular weight marker range used: as for the cytoplasmic H5-SsβGly fusion, it could suggest that it corresponds to a partially denatured part of the tetrameric form of ASLtag-SsβGly (ca. 400.0 kDa), which is particularly resistant to thermal denaturationCitation36,Citation42,Citation43,Citation45. Finally, an amount of 0.24 μg/mg of immobilised SsβGly on the OM fraction (on the basis of the calculated H5 pmol) was assayed on 2 Np- and 4 Np-Glc at three different temperatures. The results show an activity of 12.6 ± 0.7 and 8.8 ± 0.4 (50 °C), 30.3 ± 0.4 and 20.3 ± 0.9 (60 °C), 51.5 ± 0.9 and 30.8 ± 1.5 U/mg (70 °C), respectively, whereas OM fraction containing the sole ASLtag did not result in any β-glucosidase activity, as expected (data not shown). These values are correctly related to the activity of SsβGlyCitation45, clearly indicating that the formation of the quaternary structure on the E. coli OM occurs. However, since the 3D structure of this thermostable glycoside hydrolase showed that it is not laying on a surfaceCitation43, we hypothesised an invagination of the E. coli outer membrane to allow the assembly of four units of the ASLtag-SsβGly (Supplementary Figure S2).
4. Conclusions and perspectives
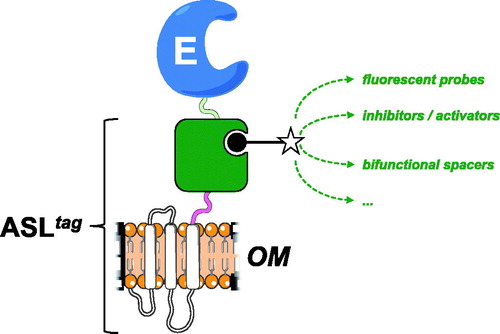
In the present work, we introduced and demonstrated the utility of a novel protein-tag, composed by the N-terminal domain of the INP protein fused to a DNA repair enzyme. From our results, it is readily apparent that ASLtag offers: (i) an easy expression and in vivo one-step procedure of enzyme immobilisation on biological supports (e.g., E. coli outer membrane); (ii) significantly reduces the costs of the enzyme purification and those of the immobilisation support, allowing a direct exposition of the enzyme to the solventCitation20,Citation30; (iii) an indirect labeling, by the reaction of a thermostable variant of the SNAP-tag™ (H5)Citation46,Citation47, which covalently links desired chemical groups conjugated to its benzyl-guanine substrateCitation14,Citation35. ASLtag favoured the expression of a monomeric protein (e.g., the thermostable SspCA) and an enzyme having a complex quaternary structure (e.g., the thermophilic SsβGly), without compromising their overall folding and enzymatic activity. Moreover, we showed that the utilisation of a fluorescein-derivative of the BG led to the localisation and the quantitative determination of the yield of the expressed ASLtag and the relative fusion proteins (). On the other hand, despite the GFPs utilisation limited only to all fluorescence-based applications, the possibility to conjugate different groups to the BG- for the H5 reactionCitation36 dramatically expands the biotechnological potential of this novel protein-tag. For example, it will be possible to modulate the activity of biocatalysts (by introducing inhibitors/activators), or connecting them with other proteins for the improvement of cascade reactions in the presence of bi-functional chemical groups ().
Figure 6. The biotechnological potential of the ASLtag. Any chemical group of interest (open star) conjugated to the BG-substrate (black closed circle) could be covalently bound to the H5 moiety (in green) of the ASLtag. This enhances the potential use of an immobilised enzyme (E) on the E. coli surface (OM), making available to it a series of molecules, e.g., fluorescent probes and enzymatic activity modulators, or bi-functional groups for cascade reactions with other biocatalysts.
Supplemental Material
Download PDF (245.7 KB)Acknowledgements
We are grateful to Maria Ciaramella for the constant and fruitful discussions during the execution of the experiments and the preparation of the manuscript. We are also grateful to Giovanni Del Monaco for technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Tosa T, Mori T, Fuse N, Chibata I. Studies on continuous enzyme reactions. I. Screening of carriers for preparation of water-insoluble aminoacylase. Enzymologia 1966;31:214–24.
- Zhou Z, Hartmann M. Progress in enzyme immobilization in ordered mesoporous materials and related applications. Chem Soc Rev 2013;42:3894–912.
- Mohamad NR, Che Marzuki NH, Buang NA, et al. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotech Biotech Equip 2015;29:205–20.
- Nguyen HH, Kima M. An overview of techniques in enzyme immobilization. App Sci Conv Tech 2017;26:157–63.
- Jakub ZJ, Meyer AS, Jesionowski T, Pinelo M. A general overview of support materials for enzyme immobilization: characteristics, properties, practical utility. Catalysts 2018;8:92.
- Sirisha VL, Jain A, Chapter JA. Nine – enzyme immobilization: an overview on methods, support material, and applications of immobilized enzymes. Adv Food Nut Res 2016;79:179–211.
- Engin S, Trouillet V, Franz CM. Benzylguanine thiol self-assembled monolayers for the immobilization of SNAP-tag proteins on microcontact-printed surface structures. Langmuir 2010;26:6097–101.
- Engin S, Fichtner D, Wedlich D, Fruk L. SNAP-tag as a tool for surface immobilization. Curr Pharm Des 2013;19:5443–8.
- Meldal M, Schoffelen S, Recent advances in covalent, site-specific protein immobilization. F1000Res. 2016;5. pii: F1000 Faculty Rev-2303. eCollection 2016.
- Pegg AE. Repair of O6-alkylguanine by alkyltransferases. Mutat Res 2000;462:83–100.
- Daniels DS, Mol CD, Arvai AS, et al. Active and alkylated human AGT structures: a novel zinc site, inhibitor and extrahelical base binding. Embo J 2000;19:1719–30.
- Daniels DS, Woo TT, Luu KX, et al. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat Struct Mol Biol 2004;11:714–20.
- Tubbs JL, Pegg AE, Tainer JA. DNA binding, nucleotide flip- ping, and the helix-turn-helix motif in base repair by O6-alkylguanine-DNA alkyltransferase and its implications for cancer chemotherapy. DNA Repair 2007;6:1100–15.
- Keppler A, Gendreizig S, Gronemeyer T, et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotech 2003;21:86–9.
- Gautier A, Juillerat A, Heinis C, et al. An engineered protein-tag for multi-protein labeling in living cells. Chem Biol 2008;15:128–36.
- Gronemeyer T, Chidley C, Juillerat A, et al. Directed evolution of O6-alkylguanine-DNA alkyltransferase for applications in protein labeling. Prot Eng Des Sel 2006;19:309–16.
- Yu Q, Griss R, Schena A, Johnsson K. Highly modular bioluminescent sensors for small molecules and proteins. Met Enz 2017;589:365–82.
- Huber W, Perspicace S, Kohler J, et al. SPR-based interaction studies with small molecular weight ligands using hAGT fusion proteins. Anal Biochem 2004;333:280–8.
- Cochet N, Widehem P. Ice crystallization by Pseudomonas syringae. Appl Microbiol Biotechnol 2000;54:153–61.
- Gurian-Sherman D, Lindow SE. Bacterial ice nucleation: significance and molecular basis. FASEB J 1993;7:1338–43.
- Graether SP, Jia Z. Modeling Pseudomonas syringae ice-nucleation proteina s a beta-helical protein. Biophys J 2001;80:1169–73.
- Samuelson P, Gunneriusson E, Nygren PA, Ståhl S. Display of proteins on bacteria. J Biotechnol 2002;96:129–54.
- Lee SY, Choi JH, Xu Z. Microbial cell-surface display. Trends Biotechnol 2003;21:45–52.
- Daugherty PS. Protein engineering with bacterial display. Curr Opin Struct Biol 2007;17:474–80.
- Jung H-C, Park J-H, Park S-H, et al. Expression of carboxymethylcellulase on the surface of Escherichia coli using Pseudomonas syringae ice nucleation protein. Enzyme Microb Technol 1998;22:348–54.
- Li L, Kang DG, Cha HJ. Functional display of foreign protein on surface of Escherichia coli using N-terminal domain of ice nucleation protein. Biotechnol Bioeng 2004;85:214–21.
- Li Q, Yu Z, Shao X, et al. Improved phosphate biosorption by bacterial surface display of phosphate-binding protein utilizing ice nucleation protein. FEMS Microbiol Lett 2009;299:44–52.
- Li Q, Yan Q, Chen J, et al. Molecular characterization of an ice nucleation protein variant (inaQ) from Pseudomonas syringae and the analysis of its transmembrane transport activity in Escherichia coli. Int J Biol Sci 2012;8:1097–108.
- Xu Y, Liu Q, Zhou L, et al. Surface display of GFP by Pseudomonas syringae truncated ice nucleation protein in attenuated Vibro anguillarum strain. Mar Biotechnol 2008;10:701–8.
- Del Prete S, Perfetto R, Rossi M, et al. A one-step procedure for immobilising the thermostable carbonic anhydrase (SspCA) on the surface membrane of Escherichia coli. J Enz Inhib Med Chem 2017;32:1120–8.
- Nakagawa S, Shtaih Z, Banta A, et al. Sulfurihydrogenibium yellowstonense sp. nov., an extremely thermophilic, facultatively heterotrophic, sulfur-oxidizing bacterium from Yellowstone National Park, and emended descriptions of the genus Sulfurihydrogenibium, Sulfurihydrogenibium subterraneum and Sulfurihydrogenibium azorense. Int J Syst Evol Microbiol 2005;55:2263–8.
- Akdemir A, Vullo D, De Luca V, et al. The extremo-alpha-carbonic anhydrase (CA) from Sulfurihydrogenibium azorense, the fastest CA known, is highly activated by amino acids and amines. Bioorg Med Chem Lett 2013;23:1087–90.
- Capasso C, De Luca V, Carginale V, et al. Biochemical properties of a novel and highly thermostable bacterial alpha-carbonic anhydrase from Sulfurihydrogenibium yellowstonense YO3AOP1. J Enzyme Inhib Med Chem 2012;27:892–789.
- Perfetto R, Del Prete S, Vullo D, et al. Production and covalent immobilisation of the recombinant bacterial carbonic anhydrase (SspCA) onto magnetic nanoparticles. J Enzyme Inhib Med Chem 2017;32:759–66.
- Perugino G, Vettone A, Illiano G, et al. Activity and regulation of archaeal DNA alkyltransferase: conserved protein involved in repair of DNA alkylation damage. J Biol Chem 2012;287:4222–31.
- Vettone A, Serpe M, Hidalgo A, et al. A novel thermostable protein-tag: optimization of the Sulfolobus solfataricus DNA-alkyl-transferase by protein engineering. Extremophiles 2016;20:13.
- Visone V, Han W, Perugino G, et al. In vivo and in vitro protein imaging in thermophilic archaea by exploiting a novel protein tag. PloS One 2017;12:e0185791.
- Studier FW. Protein production by auto-induction in high density shaking cultures. Prot Expr Purif 2005;41:207–34.
- Miggiano R, Valenti A, Rossi F, et al. Every OGT is illuminated… by fluorescent and synchrotron lights. Int J Mol Sci 2017;18:2613–31.
- Glazyrina J, Materne EM, Dreher T, et al. High cell density cultivation and recombinant protein production with Escherichia coli in a rocking-motion-type bioreactor. Microb Cell Fact 2010;9:42–52.
- Perugino G, Trincone A, Giordano A, et al. Activity of hyperthermophilic glycosynthases is significantly enhanced at acidic pH. Biochemistry 2003;42:8484–93.
- Moracci M, Capalbo L, Ciaramella M, Rossi M. Identification of two glutamic acid residues essential for catalysis in the beta-glycosidase from the thermoacidophilic archaeon Sulfolobus solfataricus. Protein Eng 1996;9:1191–5.
- Aguilar C, Sanderson I, Moracci M, et al. Crystal structure of the beta-glycosidase from the hyperthermophilic archeon Sulfolobus solfataricus: resilience as a key factor in thermostability. J Mol Biol 1997;271:789–802.
- Pouwels J, Moracci M, Cobucci-Ponzano B, et al. Activity and stability of hyperthermophilic enzymes: a comparative study on two archaeal-glycosidases. Extremophiles 2000; 4:157–64.
- Moracci M, Trincone A, Perugino G, et al. Restoration of the activity of active-site mutants of the hyperthermophilic-glycosidase from Sulfolobus solfataricus: dependence of the mechanism on the action of external nucleophiles. Biochemistry 1998; 37:17262–70.
- Perugino G, Miggiano R, Serpe M, et al. Structure-function relationships governing activity and stability of a DNA alkylation damage repair thermostable protein. Nuc Ac Res 2015;43:8801–16.
- Rossi F, Morrone C, Massarotti A, et al. Crystal structure of a thermophilic O6-alkylguanine-DNA alkyltransferase-derived self-labeling protein-tag in covalent complex with a fluorescent probe. Bioch Biophys Res Comm 2018;500:698–703.