
Abstract
Inhibition of the β-carbonic anhydrase (CA, EC 4.2.1.1) from pathogenic Candida glabrata (CgNce103) by 1H-indole-2,3-dione 3-[N-(4-sulfamoylphenyl)thiosemicarbazones] 4a–m was investigated. All the compounds were found to be potent inhibitors of CgNce103, with inhibition constants in the range of 6.4-63.9 nM. The 5,7-dichloro substituted derivative 4l showed the most effective inhibition (KI of 6.4 nM) as well as the highest selectivity for inhibiting CgNce103 over the cytosolic human (h) isoforms hCA I and II. A possible binding interaction of compound 4l within the active site of CgNce103 has been proposed based on docking studies.
1. Introduction
Candida species are yeasts that normally live on human skin, mucous membranes, and the gastrointestinal tract without causing infections. However, in immunocompromised patients these microorganisms can cause fungal infections of the mouth or throat, mucous membranes or vagina (candidiadis) or it may enter the blood stream to cause more serious candidemiaCitation1,Citation2. While many Candida species such as are responsible for these infections, Candida glabrata infections are becoming more frequentCitation3. The development of drug resistance against the clinically used antifungals is a very important medical problem. Compared to other Candida strains, C. glabrata infections are more difficult to treat because of the rapid development of drug resistance against many classical antifungal agentsCitation4,Citation5. The C. glabrata Carbonic Anhydrase CgNce103 enzyme may constitute a novel target for new classes of antifungals.
Carbonic anhydrases (CAs, EC 4.2.1.1) are a structurally diverse family of enzymes that catalyze the interconversion of carbondioxide (CO2) to bicarbonate (HCO3−). This reaction influences physiological pH values and the supply of HCO3− ions and as such many physiological, metabolic, and biosynthetic pathwaysCitation6–14. Inhibitors of these enzymes may constitute novel therapeutics against cancer or may have potential as antifungal drugsCitation6–16. Recently, the inhibition of bacterial CAs by sulfonamide derivatives have been shown to inhibit the growth of pathogenic microorganismsCitation17–25. CO2/HCO3− equilibration by fungal β-CAs plays a critical role in CO2 sensing and as such is an important mediator of fungal metabolism and pathogenesis. One such enzyme is β-CA of the opportunistic pathogen C. glabrata (CgNce103). This enzyme has an important role in the CO2-sensing of the fungal pathogensCitation26–32. CgNce103 is involved in the virulence and pathogenesis of C. glabrata due to its effects on the CO2/HCO3− concentrations and as such it constitutes a novel target for antifungal agents with a novel mechanism of actionCitation31–33.
Supuran et al. have evaluated several sulfonamides, sulfamates, sulfamides N-mono, and N,N-disubstituted dithiocarbamates against CgNce103 in their search for structurally novel inhibitorsCitation34–38. The results indicated that several of their compounds are potent CgNce103 inhibitorsCitation34,Citation35. As such, the development of selective and potent CgNce103 inhibitors may result in new antifungal drugs and could overcome the problem of developing resistance against currently used antifungal drugs.
In a recent study, we described the synthesis of novel 1H-indole-2,3-dione 3-thiosemicarbazone derivatives (compounds 4a–m) carrying a sulfamoyl group at 4-position of the phenyl ring, molecular modeling studies for target enzymes and the effects of the synthesised compounds on tumor-associated hCA IX and XII enzymesCitation39. All of the tested compounds showed selective inhibition in the low nanomolar range for hCA IX and XII enzymes over cytosolic isoforms hCA I and II enzymes. In this new study, these compounds were investigated for their inhibitions against Candida species CgNce103. Molecular modeling studies were conducted to rationalise the obtained inhibition values.
2. Experimental
2.1. CgNe103 enzyme inhibition assays
An Applied Photophysics (Leatherhead, UK) stopped-flow instrument has been used for assaying the CA-catalysed CO2 hydration activityCitation40. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM TRIS (pH 8.3) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionised water and dilutions up to 0.01 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3 and the Cheng-Prusoff equation, as reported earlier, and represent the mean from at least three different determinationsCitation36,Citation41–47.
2.2. Molecular docking studies
Three-dimensional structures for compounds 4a–m were generated in their lowest energy conformation (C=N double bond in Z isomer) using the MOE software package (v2018.0101, Chemical Computing Group Inc., Montreal, QC). The sulfonamide moiety was given a negative charge (R-SO2NH−), because this moiety binds to the active site Zn2+-ion. Subsequently, a steepest-descent energy minimisation protocol was applied using the MMFF94x force field. All ligands were docked into the active site of the CgNce103 homology model described in a previous study using the ChemScore scoring function (50 dockings per ligand; active site defined as all amino acids within 12 Å of centroid with coordinates x: 24.937, y: −20.463, z: −9.641) in the GOLD suite software package (v5.6.2, CCDC, Cambridge, UK)Citation36.
3. Results and discussion
3.1. CgNce103 enzyme inhibition assays
Compounds 4a–m and acetazolamide were tested in CgNce103 enzyme inhibition assays. The compounds inhibited CgNce103 with KI values in the range of 6.4–63.9 nM (). Compounds 4g (R = 5-I), 4h, 4j, 4k and 4l (R = 5,7-diCl) showed the best inhibitory effects against CgNce103 with KI values 19.8, 19.8, 15.0, 12.8, and 6.4 nM, respectively. Among them, compound 4l has the highest activity and the best selectivity for CgNce103 over hCA I and II. The KI values of compound 4l for hCA I and II are respectively 87-fold and 122-fold higher compared to CgNce103.
Table 1. CgNce103 enzyme inhibition data (KI, nM) for compounds 4a–m.
The unsubstituted compound 4a showed the highest measured KI value (KI: 63.9 nM), while the lowest KI value was measured for the 5,7-dichloro substituted compound 4l (KI: 6.4 nM). As such, there is only approximately 10-fold difference in the highest and lowest measured KI value amongst compounds 4a–m. This narrow activity window makes it rather difficult to suggest structure–activity relationships for these compounds. Together with the fact that the compounds only differ in their substituents on the 5 and 7 positions, we expect that the binding interactions of the compounds with the CgNce103 active site is very similar.
3.2. Molecular docking studies
Docking studies were performed to unravel putative ligand–enzyme binding interactions for this series of compounds. To this end, three-dimensional structures for compounds 4a–m were generated in their lowest energy conformation (C=N double bond in Z isomer) and docked into the active site of the CgNce103 homology model as previously describedCitation36. In short, the crystal structure of Saccharomyces cerevisiae CA Nce103 (pdb code: 3eyx; 2.04 Å), which shows 52.3% sequence identity to CgNce103, was used as a template to construct the homology model for CgNce103 using the MOE software packageCitation36. The CgNce103 sequence was obtained from the National Center for Biotechnology Information (NCBI; GenBank: CAG59355.1; 219 amino acids). The template backbone was fixed during the homology model construction, The homology model with the highest contact score was selected and a steepest-descent energy minimisation protocol was applied using the AMBER12:EHT force field. To this end, all heavy atoms of the active site residues, the zinc ion, the zinc-binding residues, and the protein backbone were fixed and the other parts were minimised using a controlled release of position restraints. The minimised structure was used in the docking studies.
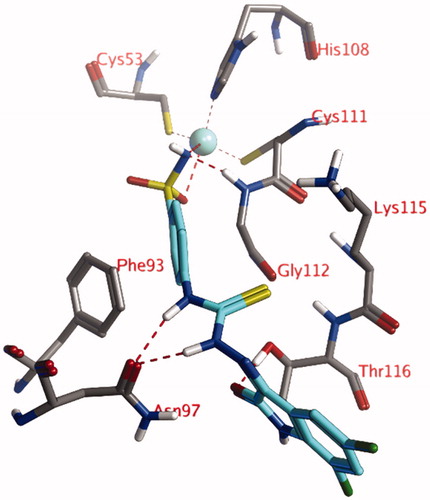
The docked pose of compound 4l, the compound with the lowest measured KI value, shows an interaction of the anionic sulfonamide moiety (R-SO2NH-) with the active site zinc ion (). The phenyl group adjacent to this sulfonamide moiety forms hydrophobic interactions (edge-to-face π–π stacking) with the aromatic side chain of Phe93. The thiosemicarbazone group forms hydrogen bonds with the side chain of Asn97, while the ligand’s carbonyl group forms a hydrogen bond to the side chain of Thr116. All other molecules of the tested series adopt very similar poses as described for compound 4l, as the substituents points toward the solvent and do not form an interaction with the protein.
Figure 1. The docked pose of compound 4l (turquoise) within the active site of CgNce103. Hydrogen bonds and interactions with the Zn(II) ion are indicated in red dashed lines.
4. Conclusions
In this study, novel 1H-indole-2,3-dione 3-thiosemicarbazone derivatives 4a–m carrying a sulfamoyl group at the 4-position of the phenyl ring were synthesised and tested against CgNce103 of the opportunistic pathogen C. glabrata. The compounds showed KI values in the range 6.4–63.9 nM against CgNce103 and between 2-fold and 120-fold higher KI values for the wide-spread human carbonic anhydrase isoforms I and II. Compound 4l has the highest activity and the best selectivity for CgNce103 over hCA I and II. The selectivity rates of 4l for CgNce103 over hCA I and II were found to be 4-fold and 111-fold higher than acetazolamide, respectively. Docking studies have suggested a possible binding pose for these compounds in the active site of CgNce103. These compounds may have the potential to serve as leads for developing new antifungal drugs with a novel mechanism of action which could overcome the problem of developing resistance against the classical antifungal agents.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Kim J, Sudbery P. Candida albicans, a major human fungal pathogen. J Microbiol 2011;49:171–7.
- Fournier P, Schwebel C, Maubon D. Antifungal use influences Candida species distribution and susceptibility in the intensive care unit. J Antimicrob Chemother 2011;66:2880–6.
- Diekema D, Arbefeville S, Boyken L, et al. The changing epidemiology of healthcare-associated candidemia over three decades. Diagn Microbiol Infect Dis 2012;73:45–8.
- Panackal AA, Gribskov JL, Staab JF, et al. Clinical significance of azole antifungal drug cross-resistance in Candida glabrata. J Clin Microbiol 2006;44:1740–3.
- Redding SW, Kirkpatrick WR, Saville S, et al. Multiple patterns of resistance to fluconazole in Candida glabrata isolates from a patient with oropharyngeal candidiasis receiving head and neck radiation. J Clin Microbiol 2003;41:619–22.
- Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32.
- Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin Ther Pat 2018;28:745–54.
- Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol 2011;2:34.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008–2018). Expert Opin Ther Pat 2018;28:729–40.
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007;15:4336–50.
- Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21.
- Vullo D, Lehneck R, Poggeler S, Supuran CT. Sulfonamide inhibition studies of two beta-carbonic anhydrases from the ascomycete fungus Sordaria macrospora, CAS1 and CAS2. J Enzyme Inhib Med Chem 2018;33:390–6.
- da Silva Cardoso V, Vermelho AB, Ricci Junior E, et al. Antileishmanial activity of sulphonamide nanoemulsions targeting the beta-carbonic anhydrase from Leishmania species. J Enzyme Inhib Med Chem 2018;33:850–7.
- Borras J, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406.
- Angeli A, Abbas G, Del Prete S, et al. Selenides bearing benzenesulfonamide show potent inhibition activity against carbonic anhydrases from pathogenic bacteria Vibrio cholerae and Burkholderia pseudomallei. Bioorg Chem 2018;79:319–22.
- Carta F, Scozzafava A, Supuran CT. Sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2012;22:747–58.
- Vullo D, Del Prete S, Di Fonzo P, et al. Comparison of the Sulfonamide Inhibition Profiles of the beta- and gamma-Carbonic Anhydrases from the Pathogenic Bacterium Burkholderia pseudomallei. Molecules 2017;22:421.
- Chohan ZH, Scozzafava A, Supuran CT. Unsymmetrical 1,1'-disubstituted ferrocenes: synthesis of Co(ii), Cu(ii), Ni(ii) and Zn(ii) chelates of ferrocenyl -1-thiadiazolo-1'-tetrazole, -1-thiadiazolo-1'-triazole and -1-tetrazolo-1'-triazole with antimicrobial properties. J Enzyme Inhib Med Chem 2002;17:261–6.
- Del Prete S, Vullo D, Osman SM, et al. Sulfonamide inhibition profiles of the beta-carbonic anhydrase from the pathogenic bacterium Francisella tularensis responsible of the febrile illness tularemia. Bioorg Med Chem 2017;25:3555–61.
- Supuran CT, Scozzafava A, Mastrolorenzo A. Bacterial proteases: current therapeutic use and future prospects for the development of new antibiotics. Expert Opin Ther Pat 2001;11:221–59.
- Supuran CT, Capasso C. An Overview of the Bacterial Carbonic Anhydrases. Metabolites 2017;7:56.
- Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25.
- Chohan ZH, Supuran CT, Scozzafava A. Metal binding and antibacterial activity of ciprofloxacin complexes. J Enzyme Inhib Med Chem 2005;20:303–7.
- Ohndorf UM, Schlicker C, Steegborn C, Crystallographic studies on carbonic anhydrases from fungal pathogens for structure-assisted drug development. In: Supuran CT, Winum JY, Wang B, eds. Drug design of zinc–enzyme inhibitors: functional, structural, and disease applications. Hoboken (NJ): John Wiley & Sons; 2009: 323–34.
- Hall RA, Mühlschlegel FA, Fungal and nematode carbonic anhydrases: their inhibition in drug design. In Supuran CT, Winum JY, Wang B, eds. Drug design of zinc-enzyme inhibitors. Functional, structural, and disease applications. Hoboken (NJ): John Wiley & Sons; 2009: 301–22.
- Lehneck R, Poggeler S, Fungal carbonic anhydrases and their inhibition. In Supuran CT, Capasso C, eds. Zinc enzyme inhibitors - enzymes from microorganisms. Vol. 22. Topics in Medicinal Chemistry. Switzerland: Springer International Publishing; 2017:95–110.
- Schlicker C, Hall RA, Vullo D, et al. Structure and inhibition of the CO2-sensing carbonic anhydrase Can2 from the pathogenic fungus Cryptococcus neoformans. J Mol Biol 2009;385:1207–20.
- Vullo D, Leewattanapasuk W, Muhlschlegel FA, et al. Carbonic anhydrase inhibitors: inhibition of the beta-class enzyme from the pathogenic yeast Candida glabrata with sulfonamides, sulfamates and sulfamides. Bioorg Med Chem Lett 2013;23:2647–452.
- Annunziato G, Giovati L, Angeli A, et al. Discovering a new class of antifungal agents that selectively inhibits microbial carbonic anhydrases. J Enzyme Inhib Med Chem 2018;33:1537–44.
- Akdemir A, Güzel-Akdemir Ö, Karalı N, Supuran CT. Isatin analogs as novel inhibitors of Candida spp. beta-carbonic anhydrase enzymes. Bioorg Med Chem 2016;24:1648–52.
- Monti SM, Maresca A, Viparelli F, et al. Dithiocarbamates are strong inhibitors of the beta-class fungal carbonic anhydrases from Cryptococcus neoformans, Candida albicans and Candida glabrata. Bioorg Med Chem Lett 2012;22:859–62.
- Güzel-Akdemir Ö, Angeli A, Demir K, et al. Novel thiazolidinone-containing compounds, without the well-known sulphonamide zinc-binding group acting as human carbonic anhydrase IX inhibitors. J Enzyme Inhib Med Chem 2018;33:1299–308.
- Karali N, Akdemir A, Goktas F, et al. Novel sulfonamide-containing 2-indolinones that selectively inhibit tumor-associated alpha carbonic anhydrases. Bioorg Med Chem 2017;25:3714–8.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Berrino E, Bozdag M, Del Prete S, et al. Inhibition of α-, β-, γ-, and δ-carbonic anhydrases from bacteria and diatoms with N'-aryl-N-hydroxy-ureas. J Enzyme Inhib Med Chem 2018;33:1194–8.
- Bua S, Berrino E, Del Prete S, et al. Synthesis of novel benzenesulfamide derivatives with inhibitory activity against human cytosolic carbonic anhydrase I and II and Vibrio cholerae α- and β-class enzymes. J Enzyme Inhib Med Chem 2018;33:1125–36.
- Akocak S, Lolak N, Bua S, et al. Synthesis and biological evaluation of novel N,N'-diaryl cyanoguanidines acting as potent and selective carbonic anhydrase II inhibitors. Bioorg Chem 2018;77:245–51.
- Abo-Ashour MF, Eldehna WM, Nocentini A, et al. Novel hydrazido benzenesulfonamides-isatin conjugates: Synthesis, carbonic anhydrase inhibitory activity and molecular modeling studies. Eur J Med Chem 2018;157:28–36.
- Melis C, Meleddu R, Angeli A, et al. Isatin: a privileged scaffold for the design of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:68–73.
- Eldehna WM, Abo-Ashour MF, Nocentini A, et al. Novel 4/3-((4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylidene)amino) benzenesulfonamides: Ssnthesis, carbonic anhydrase inhibitory activity, anticancer activity and molecular modelling studies. Eur J Med Chem 2017;139:250–62.
- Supuran CT. Bortezomib inhibits bacterial and fungal beta-carbonic anhydrases. Bioorg Med Chem 2016;24:4406–9.