
Abstract
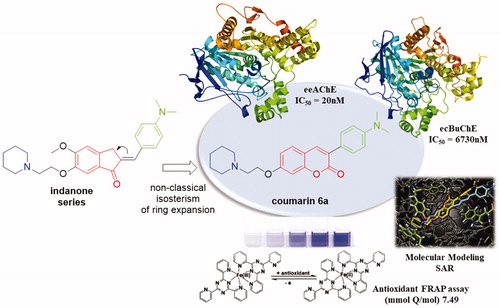
A series of 3-substituted-7-aminoalcoxy-coumarin was designed and evaluated as cholinesterase inhibitors and antioxidants. All compounds were effective in inhibiting AChE with potencies in the nanomolar range. The 3-(4-(dimethylamino)phenyl)-7-aminoethoxy-coumarin (6a) was considered a hit, showing good AChE inhibition potency (IC50 = 20 nM) and selectivity (IC50 BuChE/AChE = 354), quite similar to the reference drug donepezil (IC50 = 6 nM; IC50 BuChE/AChE = 365), also presenting antioxidant properties, low citotoxicity and good-predicted ADMET properties. The mode of action (mixed-type) and SAR analysis for this series of compounds were described by means of kinetic and molecular modeling evaluations.
Graphical Abstract
Introduction
Alzheimer's disease (AD) is the most frequent case of age-related neurodegenerative dementia, characterized by progressive loss of memory and other cognitive functionsCitation1,Citation2. AD is a heterogeneous disease, driven by the interaction between multiple deleterious factors. However, the exact mode of how these factors contribute to impair neuronal functions and neuronal survival still remains undetermined. One of the main markers of AD is the accumulation of β-amyloid plaques (Aβ) in nerve cells. In healthy brain, these aggregates of proteins are degraded and eliminatedCitation3. However, in AD the aggregates accumulate to form insoluble plaquesCitation3. Another characteristic is the presence of insoluble neurofibrillary filaments that is associated with tau protein (PTau)Citation4. In AD, however, PTau becomes hyperphosphorylated, denaturing and resulting in its dissociation of microtubules, followed by formation of neurofibrillary filaments that aggregate, acting as physical barriers to microtubulesCitation4. In addition, the occurrence of glial cell neuroinflammation, synaptic loss, and specific neuronal death is common in ADCitation5 and can be aggravated by oxidative stressCitation6.
The knowledge of neurotransmitter disorders in AD has led to the approval of drugs with symptomatic effectsCitation7. The cholinergic hypothesis of AD states that the degeneration of cholinergic neurons in basal forebrain nuclei causes disorders in the presynaptic cholinergic terminals in the hippocampus and neocortex, which are regions of extreme importance for memory disorders and other cognitive symptomsCitation8. Because of neurodegeneration, the activity of cholinergic neurons and levels of neurotransmitter ACh are reduced. One approach to improve cholinergic neurotransmission is to increase the availability of ACh by inhibition of acetylcholinesteraseCitation9.
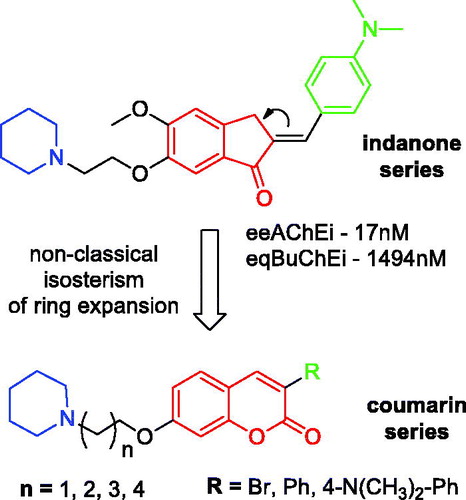
Acetyl (AChE) and butyrylcholinesterase (BuChE) inhibitors are the main drugs for the clinical treatment of AD in the initial to moderate stageCitation10. Galantamine and donepezil are selective inhibitors of AChE, whereas rivastigmine inhibits AChE and BuChE with similar affinities. Selective AChE inhibitors have demonstrated better therapeutic effects when compared to nonselective inhibitorsCitation11 since BuChE is also associated with drug metabolism and detoxification, lipoprotein metabolism and diseasesCitation12. Thus, our objectives herein were the design, synthesis and pharmacological evaluation of novel 3-substituted-7-aminoalcoxy-coumarins as selective inhibitors of AChE and antioxidant, based on a previously described indanone seriesCitation13.
Materials and methods
General procedure for the synthesis of 2a–d
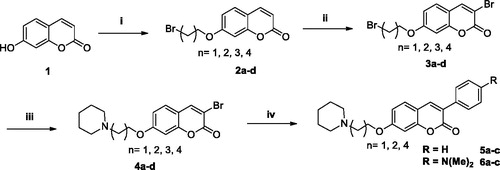
In a reactional borosilicate tube, 10–15 mmol of dibromoalkanes (4–6 eq.) and 5 mmol (2 eq.) of K2CO3 were solubilized in 2 ml of acetone (Scheme 2). To this stirred suspension a solution of 2.5 mmol of 7-hydroxycoumarin (1) in 8 ml of acetone was added dropwise. Thereafter, the reactional tube was sealed and the reaction was kept at 60 °C and stirred for 6–12 h. After reaction completion, acetone was evaporated and the crude reaction partitioned with distilled water and ethyl acetate. The final slurry was precipitated in hexanes under ultrasound irradiation and filtered off.
General procedure for the synthesis of 3a–d
To a stirred solution of 1.7 mmol of the respective O-alkyl coumarin derivative (2a–d), 5 mmol (3 eq.) of sodium acetate in 8 ml of glacial acetic acid and 2.1 mmol (1.3 eq.) of Br2 were slowly added (Scheme 2). The reaction was stirred at room temperature for 2 h. After reagent consumption, the reaction mixture was poured to a beaker containing crushed ice. The formed precipitate was filtered off under vacuum and purified by silica gel column chromatography (hexanes: dichloromethane mixture, 50–90% gradient elution).
General procedure for the synthesis of 4a–d
In a reactional vessel, 1.3 mmol of the respective 3-bromo-7-(bromoalkoxy)coumarin derivatives (3a–d) and 3.9 mmol (3 eq.) of piperidine were dissolved in 8 ml CH3CN (Scheme 2). The reaction was kept under stirring at 60 °C for 3–8 h. Acetonitrile was evaporated in a rotary evaporator and the respective products purified by silica gel column chromatography (dichloromethane: methanol, 0–25% gradient elution).
General procedure for the synthesis of 5a–c and 6a–c
In a reaction borosilicate tube, 0.14 mmol of the corresponding derivative (4a, 4b and 4d), 0.20 mmol (1.4 eq.) of appropriate phenylboronic acid and 0.42 mmol (3 eq.) of K2CO3 were solubilized in 4 ml of a solvent mixture (water: ethanol: toluene (2:1:1)) (Scheme 2). The reaction was degassed with N2 then 0.01 mmol (7 mol%) of Pd(PPh3)4 catalyst added. The reaction tubes were sealed and the mixtures were subjected to magnetic stirring and heating at 65 °C for 3–5 h. At the end of the reaction, the solvent mixture was evaporated in a rotary evaporator and the respective products purified by silica gel column chromatography (dichloromethane: methanol, 0–25% mixture gradient elution).
Cholinesterase inhibition and kinetics assays
Activity of enzymes and inhibition kinetics were determined using a Bio-Rad iMark microplate reader based on a modification of the Ellman method.Citation14,Citation15 Compounds were dissolved in DMSO. The assay solution which contained 60 µL 5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB) at 1.1 mM, 30 µL AChE/BuChE at 0.20 U/mL (initial concentration) and 150 µL tested compound solution with different concentrations. Absorbance was then recorded at λ = 415 nm. After 10-min incubation at 30 °C, 24 µL acetylthiocholine iodide/S-butyrylthiocholine iodide (at 2.75 mM for activity inhibition assay and 2.75–0.44 mM for kinetic study assay) were added and the absorbance recorded after a 10-min incubation (for activity inhibition assay) or after 0–20 min incubation (for kinetic study assay) at 30 °C.
Molecular modeling
For EeAChE (Electrophorus electricus), the PDB structure 1C2O was used; for EqBuChE (Equus caballus), a 3 D homology model was necessarily built from a sequence available in the UniProtKB/Swiss-Prot (entry Q9N1N9) with the automated mode of the protein structure homology-modeling server, Swiss-ModelCitation16, using as template the human BuChE (PDB 4TPK)Citation17. Spartan’14 program [Wavefunction, Inc.] was utilized to construct and optimize the inhibitors with the PM6 methodCitation18. The program GOLD 5.6 (CCDC Software Ltd., Cambridge, UK) was used to for the docking study with the GoldScore scoring functionCitation19.
Evaluation of the antioxidant activity by the ferric reducing ability of plasma (FRAP) method
A 0.5 ml solution of coumarin compounds in methanol (50 µM final concentration) was mixed with 4.5 ml of the FRAP reagent. After 10 min of incubation at 37 °C, absorbance at 593 nm was measured using methanol as blank.Citation20,Citation21 The calibration curve was prepared with quercetin and the results expressed as: antioxidant index based on quercetine (Q) (mmol Q/mol). The analyses were performed in triplicate.
Murine neuroblastoma cell (N2a) culture and cell viability assay
N2a cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 0.1% gentamicin in a 5% CO2 atmosphere. N2a cells were transferred to a 96-well plate (∼10,000 cells/cm2) and incubated for 24 h, before treatment with the compounds at 10 or 50 µM. Cell viability was evaluated by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) assay.
Results and discussion
Compounds design and synthesis
The design of the novel alkylamino-coumarin derivatives (Scheme 1) was based on the structural requirements for mixed-type selective AChE inhibition present in alkylamino-indanone inhibitor recently describedCitation13, as well as on the widespread use of coumarins for this pharmacological activityCitation22,Citation23. The coumarin series was based on: 1- the maintenance of the cyclic alkylamino group, which is responsible for the interaction with the cationic catalytic site (CAS) of AChE, exploring different lengths of methylene linkers (2–6); 2-exchange of the indanone nucleus by the coumarin through non-classical isosterism of ring expansionCitation24; 3- use of hydrophobic groups at position 3 of coumarin, targeting interactions with the peripheral anionic site (PAS) of AChE.
Scheme 1. Design of alkylamino-coumarin cholinesterase inhibitors series.
The synthesis of the desired compounds started with the 7-OH-coumarin (1). In the first step, through an O-alkylation reaction of 1 with diverse dibromo-alkanes, the 7-bromoalkoxy-coumarin products (2a–d) were obtained in yields of 68–78%. The second step consisted of a bromination reaction of the bromoalkoxy-coumarins (2a–d) using Br2 in buffered medium of sodium acetate/acetic acid at room temperature, furnishing the brominated derivatives (3a–d) in yields ranging between 79–84%. These intermediates (3a–d) were then subjected to amination reactions with piperidine in acetonitrile, leading to the formation of the desired 7-amino-alkoxy-3-bromo-coumarin derivatives (4a–d) as yellow solids in 95–99% yields after purification by flash chromatography. From the 7-amino-alkoxy-3-bromo-coumarin derivatives with 2, 3 and 5 methylene spacers (4a, 4b, 4d), Suzuki cross coupling reactions were then carried out using Pd(PPh3)4 catalyst and phenyl and 4-dimethylamino-phenyl boronic acids to obtain the final arylated 3-substituted coumarins (5a–c and 6a–c) in yields ranging between 70–75% after purification by flash chromatography (Scheme 2).
Scheme 2. Reagents and conditions: (i) Br(CH2)nBr (n = 2–5), K2CO3, acetone, 60° C, 2-8h, 68–78%; (ii) Br2, AcOH, NaOAc, r.t., 2h, 79–84%; (iii) piperidine, acetonitrile, 60° C, 2-5h, 95–99%; (iv) Ph-B(OH)2 or 4-(Me)2N-Ph-B(OH)2, Na2CO3, Pd(PPh3)4, H2O, EtOH, PhMe, 80° C, 3h, 70–75%.
Cholinesterase inhibitory activity, biological profile, and SAR analyses
The inhibitory activities of the coumarin compounds (4a–d, 5a–c and 6a–c) on AChE and BuChE were determined by the Ellman’s methodCitation14,Citation15 using donepezil as the reference compound. As depicted in , compounds presented potent inhibitory activities against AChE with IC50 values varying from 0.02 to 0.92 µM for compounds 6a and 6c respectively. On the other hand, the tested coumarins were not so efficient in inhibiting BuChE with IC50 ranging from 0.90 to 15.87 µM, demonstrating a good selectivity for AChE. The inhibition behavior of the simplest bromo-coumarins (4a–d) was quite similar of that related in the literature for AChECitation13, the bigger the methylene chain the lower the activity. However, we were surprised by compound 4d with a five-methylene spacer link that was equipotent to 4a (IC50=0.14 µM for 4d and IC50=0.18 µM for 4a). Conversely, the inhibitions of BuChE was in general inverse to those of AChE, and compounds with longest linker chains were more potent in inhibiting BuChE (IC50=8.37 µM for 4a and IC50=5.00 µM for 4d). By this way, we decided to evaluate the 3-aryl substituted coumarins with 2, 3 and 5 methylene spacers in the 7-amino-alkoxy group. In general, the substitution of bromine for phenyl (5a–c) or 4-dimethylamine-phenyl (6a–c) led to compounds with better potencies on the inhibition of both AChE and BuChE, and a reduction in the selectivity index (IC50 BuChE/AChE). However, one compound behavior itself differently and presented an interesting profile, the 4-dimethylamine-phenyl substituted coumarin (6a) with the best inhibition of AChE (IC50=0.02 µM) and selectivity (IC50 BuChE/AChE = 354), quite similar to the reference drug donepezil (IC50 AChE = 0.007 µM and selectivity = 365) ().
Table 1. AChE and BuChE inhibitory activities of coumarin compounds.
The antioxidant evaluation of coumarin compounds showed that only 6a–c presented activity in Ferric Ion Reduction Method (FRAP) with values from 2.42 to 7.49 mmol Q/mol (). Series 4a–d and 5a–c did not demonstrate any considerable result, similar to other 7-alkoxy coumarins described in the literatureCitation25. Probably, the antioxidant effect is coming from dimethylamino-phenyl moiety and this feature could be explored in a forthcoming series.
Aiming at discovery the mode of action of coumarins described herein, the most potent compounds from the bromo and aryl 3-substituted coumarins, i.e. 4d and 6a, were selected for kinetic studies. The linear Lineweaver–Burk equation of the Michaelis–Menten was applied to evaluate the inhibition profile. Increasing concentrations of both compounds were able to increase Km and decrease Vmax, presenting a mixed-type inhibition in AChE as well as in BuChE, as exemplified in for compound 6a (complete analysis in Supplementary material). The competitive inhibitory constant (Ki) and the noncompetitive constant (Ki’) for 6a and 4d are described in Table 3 at Supplementary material. As example, the best Ki values against AChE were obtained for compound 6a: Ki = 0.001 µM (competitive) and Ki’=0.010 µM (noncompetitive).
Figure 1. Lineweaver-Burk plots of EeAChE inhibition kinetics of compound 6a. Inset: concentrations used for 6a are depicted with [I] graphic symbol.
![Figure 1. Lineweaver-Burk plots of EeAChE inhibition kinetics of compound 6a. Inset: concentrations used for 6a are depicted with [I] graphic symbol.](/cms/asset/84b9c8f9-15fc-4b06-9148-c79d5df96bfa/ienz_a_1571270_f0001_c.jpg)
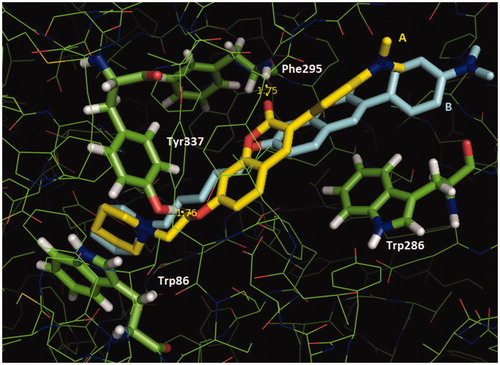
With the complete inhibitory profile of the target compounds, we proceeded with a molecular modeling evaluation to understand the importance of changing the methylene size spacer and nature of substituents in position 3 of coumarins. Thus, we selected compounds 4a and 6a, the strongest 2-methylene spacer bromo and aryl substituted coumarins inhibitors of AChE; and 6c, the weakest inhibitor. Docking results in EeAChE and EqBuChE are presented in Table 4 at Supplementary material. All inhibitors were generally predicted as better ligands of EeAChE, being 6a (Goldscore = 78.1) better than 6c (Goldscore = 71.4) and 4a (Goldscore = 64.9), whereas 6c (Goldscore = 67.9) was better than 6a (Goldscore = 63.0) and 4a (Goldscore = 57.6) as a ligand of EqBuChE, in qualitative accordance to our experimental results. The molecular docking results of compound 4a, 6a and 6c showed that all were able to occupy the peripheral (PAS) and the catalytic (CAS) sites simultaneously in the EeAChE () (and Figure 5 at Supplementary material), as previewed by kinetic evaluations. In the CAS, they interact similarly by means of their protonated piperidinyl group with Trp86 (a cation-π interaction). In the PAS, both 6a and 6c molecules were involved in π-stacking interactions with Trp286, which was more effective for 6c, involving its coumarin ring (). On the other hand, 4a was only capable of doing weak hydrophobic interactions with Trp286 (Figure 5 at Supplementary material). The presence of a narrower spacer in 6a makes its coumarin ring to be best located in the gorge, where it is involved in H-bonds with Tyr337 and the peptide group of Phe295. These H-bonds, that had no counterparts in the 6c/enzyme complex, were probably the reason for the most effective interaction between compounds with short spacers and EeAChE, which could be related to their greater inhibitory action over the enzyme.
Figure 2. Superposition of the interaction poses of compounds 6a (A, carbon atoms in yellow) and 6c (B, carbon atoms cyan) with EeAChE obtained by molecular docking (Goldscore function). H-bond distances (Å) are shown in yellow. Figure generated with PyMol 0.99 (DeLano Scientific LLC).
Cell cytotoxicity and in silico ADMET physico-chemical profile analysis
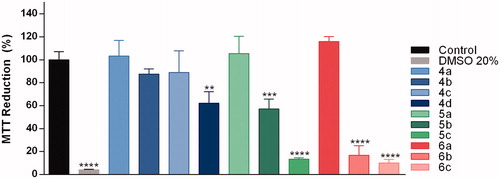
In order to accede the drugability of tested coumarins, we first proceeded with the cytotoxicity evaluation against N2a cells (neuroblastoma), after 48 h incubation at concentrations of 10 and 50 µM (Figure 3 and Supplementary material). The most potent compounds in inhibiting AChE, i.e. 4a, 5a, and 6a, were not cytotoxic at the maximum tested concentration (50 µM) (). As a rule, long methylene chains (three or five spacers) in phenyl-substituted coumarins (5b, 5c, 6b and 6c) could not be useful for further developments due to increase in toxicity.
Figure 3. Neuroblastoma cell viability after compound treatment. Samples containing compounds were added to the culture 48 h before MTT addition. The compounds were tested at the final concentration of 50 µM. MTT reduction was evaluated as described in Experimental Procedures. Data are expressed as the percentage of MTT reduction relative to the value for control cells (cells without treatment). Error bars represent standard deviations. **p < .01; ***p < .001; ****p < .0001.
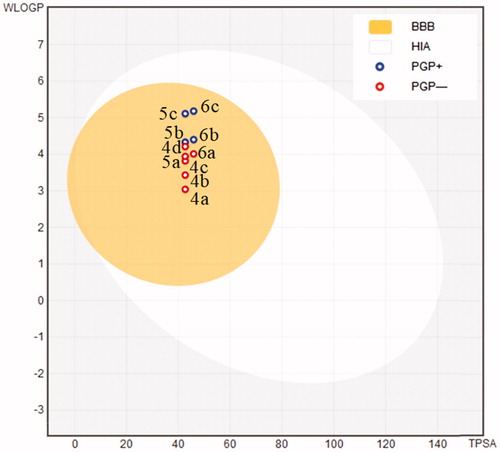
Finally, in silico evaluations showed a good ADMET profile for coumarin compounds. Parameters as topological polar surface area (TPSA), consensus Log P, Log S, human intestinal absorption (HIA), blood–brain barrier permeation (BBB), and P‐glycoprotein (P-gP) substrate and drug-likeness profile (Supplementary material)Citation26. TPSA values and consensus Log P ranged from 42.68 to 45.92 and 3.35 to 5.01, respectively. The moderate polarity (PSA < 79 Å2) and relative lipophilicic characteristics put our compounds in the yellow compartment of BOILED‐Egg model (), having a high probability to access the CNSCitation27, which is fundamental for the distribution of central‐acting molecules. Additionally, the most potent compounds 4a, 5a and 6a were not considered as P-gP substrate and having a good drug-likeness profile with no one violation on the LipinskiCitation28, GhoseCitation29, VeberCitation30, EganCitation31 and MueggeCitation32 rules.
Figure 4. BOILED-Egg ADMET modelCitation27 for coumarin compounds 4a–d, 5a–c, and 6a–c. (HIA) gastrointestinal absorption; (BBB) brain penetration; (PGP+) substrate for P‐glycoprotein; (PGP-) Not a substrate for P‐glycoprotein.
Conclusions
The designed and synthesized coumarin compounds were able to potently inhibit cholinesterases in the nanomolar range. In general, compounds with narrow methylene linkers were more potent and selective for AChE (with IC50 and selectivity of up to 20 nM and 354 times, respectively), and less toxic as well. The introduction of aromatic substituents in position 3 of coumarins led to compounds with better potencies on the inhibition of both AChE and BuChE. As highlighted, compound 6a could be elected as a hit for in vivo studies, showing good AChE inhibition potency and selectivity (IC50=20 nM and 354 times), antioxidant properties, low cytotoxicity and good predict ADMET profile.
Supplemental Material
Download PDF (2.3 MB)Acknowledgments
The authors would like to thank Marco E. F. de Lima for furnishing donepezil standard and Marina Amaral from Laboratório de Apoio ao Desenvolvimento Tecnológico (LADETEC, UFRRJ) for the mass spectrometry analysis.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Prince M, Ali GC, Guerchet M, et al. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimers Res Ther 2016;8:23.
- Alzheimer’s Disease International. World Alzheimer Report 2015: The global impact of dementia: an analysis of prevalence, incidence, costs and trends. London: Alzheimer’s Disease International (ADI); 2015. Available at: https://www.alz.co.uk/research/world-report-2015.
- Hardy J. Testing times for the “amyloid cascade hypothesis”. Neurobiol Aging 2002;23:1073–74.
- Goedert M. Tau protein and neurodegeneration. Semin Cell Dev Biol 2004;15:45–9.
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell 2012;148:1204–22.
- Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer's disease. Oxid Med Cell Longev 2013;2013:316523.
- Anand R, Gill KD, Mahdi AA, et al. Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology 2014;76:27–50.
- Terry AV, Buccafusco JJ. The cholinergic hypothesis of age and Alzheimer's disease-related cognitive deficits: recent challenges and their implications for novel drug development. J Pharmacol Exp Ther 2003;306:821–27.
- Ferri CP, Prince M, Brayne C, et al. Global prevalence of dementia: a Delphi consensus study. Lancet 2005;366:2112–7.
- Mushtaq G, Greig NH, Khan JA, et al. Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer's disease and type 2 diabetes mellitus. CNS Neurol Disord Drug Targets 2014;13:1432–9.
- Kumar A, Singh A, Ekavali E. A review on Alzheimer's disease pathophysiology and its management: an update. Pharmacol Rep 2015;67:195–203.
- Li Q, Yang H, Chen Y, et al. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer's disease. Eur J Med Chem 2017;132:294–309.
- Huang L, Miao H, Sun Y, et al. Discovery of indanone derivatives as multi-target-directed ligands against Alzheimer's disease. Eur J Med Chem 2014;87:429–39.
- Ellman GL, Courtney KD, Andres V, et al. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95.
- Torres JM, Lira AF, Silva DR, et al. Structural insights into cholinesterases inhibition by harmane β-carbolinium derivatives: a kinetics-molecular modeling approach. Phytochemistry 2012;81:24–30.
- Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 2014;42:W252–8.
- Brus B, Kosak U, Turk S, et al. Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J Med Chem 2014;57:8167–79.
- Stewart JJP. Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J Mol Model 2007;13:1173–213.
- Jones G, Willett P, Glen RC, et al. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–48.
- Sant’ana LDO, Sousa JLM, Salgueiro FB, et al. Characterization of monofloral honeys with multivariate analysis of their chemical profile and antioxidant activity. J Food Sci 2012;71:c135–40.
- Benzie IF, Strain J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power: the FRAP assay”. Anal Biochem 1996;239:70–6.
- Pereira TM, Franco D, Vitorio F, et al. Coumarin compounds in medicinal chemistry: some important examples from the last years. Curr Top Med Chem 2018;18:124–48.
- Sonmez F, Zengin Kurt B, Gazioglu I, et al. Design, synthesis and docking study of novel coumarin ligands as potential selective acetylcholinesterase inhibitors. J Enzyme Inhib Med Chem 2017;32:285–97.
- Lima LM, Barreiro EJ. Bioisosterism: a useful strategy for molecular modification and drug design. Curr Med Chem 2005;12:23–49.
- Malhotra S, Tavakkoli M, Edraki N, et al. Neuroprotective and antioxidant activities of 4-methylcoumarins: development of structure–activity relationships. Biol Pharm Bull 2016;39:1544–8.
- Antoine D, Olivier M, Vincent Z. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017;7:42717.
- Antoine D, Vincent Z. A boiled‐egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016;11:1117–21.
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001;46:3–26.
- Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem 1999;1:55–68.
- Veber DF, Stephen RJ, Hung-Yuan C, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002;45:2615–23.
- Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem 2000;43:3867–77.
- Muegge I, Heald SL, Brittelli D. Simple selection criteria for drug-like chemical matter. J Med Chem 2001;44:1841–6.