
Abstract
Famotidine, an antiulcer drug belonging to the H2 antagonists class of pharmacological agents, was recently shown to potently inhibit human (h) and bacterial carbonic anhydrases (CAs, EC 4.2.1.1). We investigated the inhibitory effects of famotidine against all classes of CAs from the pathogenic bacteria Vibrio cholerae, Burkholderia pseudomallei and Mycobacterium tuberculosis Rv3273 β-CA, as well as the CAs from the nonpathogenic bacteria/cyanobacteria Sulfurihydrogenibium yellowstonensis, S. azorense, Pseudoalteromonas haloplanktis, Colwellia psychrerythraea and Nostoc commune. The δ- and ζ-CAs from the diatom Thalassiosira weissflogii, the fungal enzymes from Cryptococcus neoformans, Candida glabrata and Malassezia globosa, as well as the protozoan enzymes from Trypanosoma cruzi and Plasmodium falciparum, were also investigated. Anopheles gambiae β-CA was also investigated. All these enzymes were effectively inhibited by famotidine, with affinities between the low nanomolar to the micromolar range. The best inhibition was observed against C. glabrata β-CA and TweCAζ, with KIs ranging between 13.6 and 22.1 nM.
1. Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are ubiquitous metalloenzymes all over the phylogenetic tree, with seven distinct genetic families described so far, the α-, β-, γ-, δ-, ζ-, η- and Θ-CAsCitation1–13, and the probability to discover other such families is quite high. This is mainly due to the fact that these enzymes catalyse a simple but essential chemical reaction, the interconversion of carbon dioxide and water, with formation of bicarbonate and protons: CO2+H2O ⇌ HCO3–+H+Citation1,Citation14–18. In tissues/organisms where CAs are present, they are involved in pH regulation (a tightly controlled process in all living organisms) and metabolic processes connected to carboxylation/decarboxylation reactionsCitation1–13. In plants and some bacteria (e.g. cyanobacteria), CAs also play a function in photosynthesis, for example assuring a carbon concentrating mechanism (CCM) which enhances the concentration of CO2 available for 1,5-bisphosphate carboxylase/oxygenase (RUBISCO)Citation19–22. Diatoms are also encoding for several classes of CAs. In fact, the δ- and ζ-CAs were discovered in these organismsCitation4,Citation6, in which they probably play essential functions, poorly understood at this momentCitation21.
In the genome of many pathogenic bacteria (e.g. Helicobacter pylori, the Gram-negative bacterium responsible of gastric ulcersCitation10; Vibrio cholerae, the Gram-negative bacterium provoking cholera)Citation9, Brucella suis, the non-motile Gram-negative coccobacillus responsible of brucellosisCitation1,Citation2, Mycobacterium tuberculosis, the obligate pathogenic bacterium responsible for tuberculosisCitation8, Burkholderia pseudomaleiCitation14, and many others) CAs belonging to at least three classes were found, and many of them were shown to be essential for growth of the pathogenCitation17–19. CA inhibitors (CAIs) targeting such pathogenic enzymesCitation8–14,Citation17–19 were reported in the last decadeCitation5,Citation8,Citation11,Citation13–15 and are considered as an alternative to clinically used antibacterials, for which a wide range of resistance was reported in the last decadesCitation5. Furthermore, pathogenic fungi such as Candida glabrataCitation5,Citation22, C. albicansCitation5, Cryptococcus neoformansCitation5,Citation22 or Malassezia globosaCitation23 also encode for β-CAs which were demonstrated to be druggable, with many interesting inhibitors belonging to various classes reported, some of which also showed some efficacy in vivoCitation5,Citation22,Citation23. Pathogenic protozoans, such as Plasmodium falciparum or Trypanosoma cruzi also encode for CAs belonging to various classes (η-CA for the first pathogenCitation7, and α-CA for the second oneCitation24) which were shown to be inhibited efficiently by sulphonamide CAIs, with antiprotozoans effects in vitro and ex vivoCitation24. A β-CA was isolated and characterised from the malaria-transmitting mosquito Anopheles gambiae, AgaCACitation25, which was proposed as a new drug target for controlling the spread of insect-transmitted diseases. Thus, although the anti-infective field of CA inhibitors (CAIs) was scarcely investigated up until recently, the data presented above and intense research efforts in the last decade demonstrate a great potential for this class of derivatives in the field of anti-infectives targeting bacterial, fungal and protozoan infection as well as the insect pest control.
Recently, our group demonstrated that a widely used antiulcer drug, famotidine (FAM), belonging to the histamine receptor H2 antagonists class, also shows significant CA inhibitory propertiesCitation26. Famotidine was in fact assayed as inhibitor of all the catalytically active human (h) CA isoforms, hCA I-XIV and of the two bacterial CAs from H. pylori. Considering the interesting results reported earlier against these enzymes, here we report an inhibition study with famotidine of 20 CAs belonging to the α-η classes from bacteria, cyanobacteria, diatoms, fungi, protozoans and insects.
2. Material and methods
2.1. Chemistry
Famotidine (FAM) and acetazolamide (AAZ) were commercially available, highest purity reagents from Sigma-Aldrich (Milan, Italy).
2.2. CA enzyme inhibition assay
An Sx.18Mv-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the catalytic activity of various CA isozymes for CO2 hydration reactionCitation27. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.5, for α-, δ-, ζ- and η-CAs) or TRIS (pH 8.3, for β- and γ-CAs) as buffers, 0.1 M Na2SO4 (for maintaining constant ionic strength), following the CA-catalysed CO2 hydration reaction for a period of 10 s at 25 °C. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used to determine the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitors (10 mM) were prepared in distilled–deionised water and dilutions up to 1 nM were done thereafter with the assay buffer. Enzyme and inhibitor solutions were pre-incubated together for 15 min (standard assay at room temperature) prior to assay, in order to allow the formation of the enzyme–inhibitor complex. The inhibition constants were obtained by non-linear least-squares methods, using GraphPad PRISM 3 (GraphPad Software, La Jolla, CA) and the Cheng–Prusoff equation, as reported earlierCitation26,Citation28,Citation29. All CAs were recombinant proteins produced as reported earlier by our groupsCitation6,Citation9–14,Citation17–23.
3. Results and discussion
The X-ray crystal structures of FAM bound to hCA I () and hCA II () show why this compound effectively inhibits these enzymesCitation26. Indeed, FAM has a KI of 922 nM against hCA I and of 58 nM against hCA IICitation26.
Figure 1. hCA I adduct of famotidine (FAM). (a) Overall structure. (b) Active site details, with the Zn(II) ion (gray sphere), its three His ligands and the inhibitor in green.
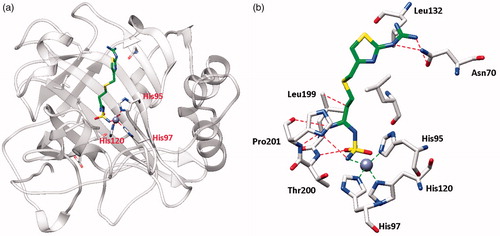
Figure 2. hCA II adduct of famotidine (FAM). (a) Overall structure. (b) Active site details, with the Zn(II) ion (gray sphere), its three His ligands and the inhibitor in green.
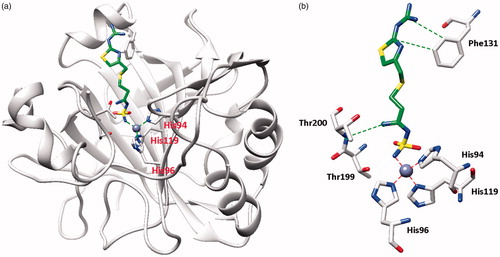
From the X-ray crystal structures of famotidine bound to hCA I () and hCA II () shown above, it can be seen that the inhibitor makes favorable contacts when bound to both enzymes. The deprotonated sulfamide moiety was coordinated to the zinc ion as the fourth ligand, with the metal in a tetrahedral geometry (the remaining three ligands being residues His95, 97 and 120 in hCA I, and His94, 96 and 119 in hCA II, respectively). The main difference between the binding of the drug to the two isoforms resides in the interaction with the amino acid residue in position 131. Phe131 in hCA II is essential for collocating the inhibitor within the lipophilic half of the active site cavityCitation26. On the other hand, the presence of Ser131 in hCA I only led to a loose van der Waals interaction with the famotidine scaffold, which did not force it towards the lipophilic side of the active site, leading thus to two opposite orientations of the scaffold (not shown in , see Ref.Citation26 for details) and few hydrophobic interactions with the active site. These characteristics reflect the loss of inhibitory potency of famotidine against hCA I compared to hCA II. However, as reported earlierCitation26, FAM is an effective inhibitor of the α- and β-CAs from H. pylori, with KIs of 20.7–49.8 NmCitation26.
Thus, we decided to test the inhibitory effects of FAM (with acetazolamide, AAZ, as standard) against a multitude of CAs of bacterial, diatom, fungal, protozoan and insect origin (). The following data may be noted from the inhibition constants reported in :
Table 1. Inhibition data against bacterial, diatom, fungal, protozoan, insect and human CAs with famotidine (FAM) and acetazolamide (AAZ) by a stopped flow CO2 hydrase assayCitation22.
The β-CAs from the bacterium V. cholerae (VchCAβ) and the fungus M. globosa (MgaCA) were quite insensitive to FAM as an inhibitor, with inhibition constants in the high micromolar range (of 42–83 µM). Acetazolamide (AAZ) was also a poor inhibitor of MgCA with a KI of 40 µM.
Low micromolar inhibitory effects of FAM were observed against the following enzymes: VchCAγ (V. cholerae), Rv3273 (M. tuberculosis), BpsCAβ/γ (B. pseudomalei), NcoCA (N. commune) and TcCA (T. cruzi). These enzymes were inhibited with KIs ranging between 1265 and 8293 nM, and they belong to the β- and γ-CA classes, except for TcCA which is an α-CA.
More effective inhibitory effects of FAM were observed for the following enzymes: SspCA and SazCA (from extremophilic bacteria and belonging to the α-CA class), the δ-CA TweCAδ from the diatom T. weissflogii, Can2 from C. neoformans (fungus), PfCA (from P. falciparum) and AgaCA (from the mosquito A. gambiae). FAM showed KIs in the range of 107–408 nM, and it should be mentioned that these CAs belong to many diverse genetic families (α-, β-, δ- and η-CAs).
The following enzymes were inhibited by FAM with KIs < 100 nM: VchCAα (V. cholerae); PhaCA (P. haloplanktis); CpsCA (C. psychrerythraea); TweCAζ (from the diatom T. weissflogii, with Zn(II) and Cd(II) at the active site, respectively); and CgNce103 (from C. glabrata). KIs in the range of 13.6–90.7 nM were measured for these enzymes belonging to the α-, β-, γ- and ζ-CA classes (). The enzymes which were the most effectively inhibited by FAM were CgNce103 and the zinc-containing ζ-CA TweCAζ (KIs of 13.6–22.1 nM). FAM is in fact a more effective CAI than AAZ against TweCAζ and almost as effective as AAZ against the C. glabrata β-CA.
The inhibition pòrofiles of FAM and AAZ are rather different against this panel of CAs ().
4. Conclusions
Drug repurposing (or repositioning) started to be considered as an interesting source of new therapeutic/pharmacological agents also in the field of CAIsCitation30. Famotidine, an antiulcer drug discovered as a histamine H2-receptors antagonist, was recently demonstrated to potently inhibit some human and H. pylori CAs belonging to the α- and β-CA genetic families. Here, we prove that famotidine exerts quite interesting CA inhibitory action against bacterial, cyanobacterial, diatom, fungal and protozoan enzymes belonging to the α-, β-, γ-, δ-, ζ- and η-CA classes. For some pathogenic enzymes such as those from V. cholerae, C. albicans and P. falciparum as well as the mosquitoes involved in malaria transmission (A. gambiae), the drug showed efficacy in the range of 13.6–397 nM, making it a possible lead or a possible agent for more detailed, in vivo investigations.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- (a) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. (b) Capasso C, Supuran CT. An overview of the alpha-, beta-and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32. (c) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. (d) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- (a) Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. (b) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. (c) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68. (d) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12. (e) Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21. (f) Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008-2018). Expert Opin Ther Pat 2018;28:729–40. (g) Supuran CT, Capasso C. Biomedical applications of prokaryotic carbonic anhydrases. Exp Opin Ther Pat 2018;28:745–54.
- (a) Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO(2) capture. J Enzyme Inhib Med Chem 2013;28:229–30. (b) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (c) Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005 - 2013). Expert Opin Ther Pat 2013;23:681–91. (d) Temperini C, Cecchi A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Sulfonamide diuretics revisited—old leads for new applications? Org Biomol Chem 2008;6:2499–506. (e) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: Structure-activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73.
- (a) Xu Y, Feng L, Jeffrey PD, et al. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008;452:56–61. (b) Ferry JG. The gamma class of carbonic anhydrases. Biochim Biophys Acta 2010;1804:374–81. (c) Tripp BC, Bell CB III, Cruz F, et al. A role for iron in an ancient carbonic anhydrase. J Biol Chem 2004;279:6683–7. (d) Innocenti A, Zimmerman SA, Scozzafava A, et al. Carbonic anhydrase activators: activation of the archaeal beta-class (Cab) and gamma-class (Cam) carbonic anhydrases with amino acids and amines. Bioorg Med Chem Lett 2008;18:6194–8.
- (a) Capasso C, Supuran CT. Bacterial carbonic anhydrases in zinc enzyme inhibitors. In C.T. Supuran, C. Capasso, eds. Volume 1: Zinc Enzyme Inhibitors: enzymes from microorganisms. Springer International Publishing, Top Med Chem 2017;22:135–52. (b) Capasso C, Supuran CT. Protozoan carbonic anhydrases in zinc enzyme inhibitors. In C.T. Supuran, C. Capasso, eds. Volume 1: Zinc Enzyme Inhibitors: enzymes from microorganisms. Springer International Publishing, Top Med Chem 2017;22:114–34. (c) Lehneck R, Poggeler S. Fungal carbonic anhydrases and their inhibition in zinc enzyme inhibitors. In C.T. Supuran, C. Capasso, eds. Volume 1: Zinc enzyme Inhibitors: enzymes from microorganisms. Springer International Publishing, Top Med Chem 2017;22:95–110.
- (a) Del Prete S, Vullo D, De Luca V, et al. Biochemical characterization of the δ-carbonic anhydrase from the marine diatom Thalassiosira weissflogii, TweCA. J Enzyme Inhib Med Chem 2014;29:906–11. (b) Del Prete S, Vullo D, Scozzafava A, et al. Cloning, characterization and anion inhibition study of the δ-class carbonic anhydrase (TweCA) from the marine diatom Thalassiosira weissflogii. Bioorg Med Chem 2014;22:531–7.
- (a) Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum–the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96. (b) De Simone G, Di Fiore A, Capasso C, Supuran CT. The zinc coordination pattern in the η-carbonic anhydrase from Plasmodium falciparum is different from all other carbonic anhydrase genetic families. Bioorg Med Chem Lett 2015;25:1385–9. (c) Del Prete S, De Luca V, Supuran CT, Capasso C. Protonography, a technique applicable for the analysis of η-carbonic anhydrase activity. J Enzyme Inhib Med Chem 2015;30:920–4. (d) Del Prete S, De Luca V, De Simone G, et al. Cloning, expression and purification of the complete domain of the η-carbonic anhydrase from Plasmodium falciparum. J Enzyme Inhib Med Chem 2016;31:54–9.
- (a) Nishimori I, Minakuchi T, Maresca A, et al. The β-carbonic anhydrases from Mycobacterium tuberculosis as drug targets. Curr Pharm Des 2010;16:3300–9. (b) Nishimori I, Minakuchi T, Vullo D, et al. Carbonic anhydrase inhibitors. Cloning, characterization, and inhibition studies of a new beta-carbonic anhydrase from Mycobacterium tuberculosis. J Med Chem 2009;52:3116–20.
- (a) Del Prete S, Isik S, Vullo D, et al. DNA cloning, characterization, and inhibition studies of an α-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. J Med Chem 2012;55:10742–8. (b) Del Prete S, Vullo D, De Luca V, et al. Anion inhibition profiles of alpha-, beta- and gamma-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorg Med Chem 2016;24:3413–7. (c) Del Prete S, Vullo D, De Luca V, et al. Comparison of the sulfonamide inhibition profiles of the alpha-, beta- and gamma-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorg Med Chem Lett 2016;26:1941–6. (d) Ferraroni M, Del Prete S, Vullo D, et al. Crystal structure and kinetic studies of a tetrameric type II beta-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Acta Cryst D Biol Cryst 2015;71:2449–56.
- (a) Nishimori I, Onishi S, Takeuchi H, Supuran CT. The alpha and beta classes carbonic anhydrases from Helicobacter pylori as novel drug targets. Curr Pharm Des 2008;14:622–30. (b) Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704. (c) Capasso C, Supuran CT. An overview of the selectivity and efficiency of the bacterial carbonic anhydrase inhibitors. Curr Med Chem 2015;22:2130–9.
- (a) Annunziato G, Giovati L, Angeli A, et al. Discovering a new class of antifungal agents that selectively inhibits microbial carbonic anhydrases. J Enzyme Inhib Med Chem 2018;33:1537–44. (b) Güzel-Akdemir Ö, Angeli A, Demir K, et al. Novel thiazolidinone-containing compounds, without the well-known sulphonamide zinc-binding group acting as human carbonic anhydrase IX inhibitors. J Enzyme Inhib Med Chem 2018;33:1299–308. (c) Murray AB, Aggarwal M, Pinard M, et al. Structural mapping of anion inhibitors to β-carbonic anhydrase psCA3 from Pseudomonas aeruginosa. ChemMedChem 2018;13:2024–9.
- (a) Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16. (b) Borras J, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406. (c) Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35. (d) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25.
- (a) Annunziato G, Angeli A, D’Alba F, et al. Discovery of new potential anti-infective compounds based on carbonic anhydrase inhibitors by rational target-focused repurposing approaches. ChemMedChem 2016;11:1904–14. (b) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. (c) Del Prete S, Vullo D, De Luca V, et al. Sulfonamide inhibition studies of the beta-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Bioorg Med Chem 2016;24:1115–20.
- (a) Berrino E, Bozdag M, Del Prete S, et al. Inhibition of α-, β-, γ-, and δ-carbonic anhydrases from bacteria and diatoms with N’-aryl-N-hydroxy-ureas. J Enzyme Inhib Med Chem 2018;33:1194–8. (b) Bua S, Berrino E, Del Prete S, et al. Synthesis of novel benzenesulfamide derivatives with inhibitory activity against human cytosolic carbonic anhydrase I and II and Vibrio cholerae α- and β-class enzymes. J Enzyme Inhib Med Chem 2018;33:1125–36. (c) Angeli A, Abbas G, Del Prete S, et al. Selenides bearing benzenesulfonamide show potent inhibition activity against carbonic anhydrases from pathogenic bacteria Vibrio cholerae and Burkholderia pseudomallei. Bioorg Chem 2018;79:319–22.
- (a) Wani TV, Bua S, Khude PS, et al. Evaluation of sulphonamide derivatives acting as inhibitors of human carbonic anhydrase isoforms I, II and Mycobacterium tuberculosis β-class enzyme Rv3273. J Enzyme Inhib Med Chem 2018;33:962–71. (b) Stefanucci A, Angeli A, Dimmito MP, et al. Activation of β- and γ-carbonic anhydrases from pathogenic bacteria with tripeptides. J Enzyme Inhib Med Chem 2018;33:945–50. (c) Modak JK, Liu YC, Machuca MA, et al. Structural basis for the inhibition of Helicobacter pylori alpha-carbonic anhydrase by sulfonamides. PloS One 2015;10:e0127149. (d) Buzás GM, Supuran CT. The history and rationale of using carbonic anhydrase inhibitors in the treatment of peptic ulcers. In memoriam Ioan Puşcaş (1932-2015). J Enzyme Inhib Med Chem 2016;31:527–33.
- (a) Nishimori I, Vullo D, Minakuchi T, et al. Carbonic anhydrase inhibitors: cloning and sulfonamide inhibition studies of a carboxyterminal truncated alpha-carbonic anhydrase from Helicobacter pylori. Bioorg Med Chem Lett 2006;16:2182–8. (b) Kohler S, Ouahrani-Bettache S, Winum JY. Brucella suis carbonic anhydrases and their inhibitors: towards alternative antibiotics? J Enzyme Inhib Med Chem 2017;32:683–7. (c) Akocak S, Lolak N, Vullo D, et al. Synthesis and biological evaluation of histamine Schiff bases as carbonic anhydrase I, II, IV, VII, and IX activators. J Enzyme Inhib Med Chem 2017;32:1305–12.
- (a) De Luca V, Del Prete S, Carginale V, et al. Cloning, characterization and anion inhibition studies of a γ-carbonic anhydrase from the Antarctic cyanobacterium Nostoc commune. Bioorg Med Chem Lett 2015;25:4970–5. (b) Vullo D, De Luca V, Del Prete S, et al. Sulfonamide inhibition studies of the γ-carbonic anhydrase from the Antarctic cyanobacterium Nostoc commune. Bioorg Med Chem 2015;23:1728–34. (c) De Luca V, Del Prete S, Vullo D, et al. Expression and characterization of a recombinant psychrophilic γ-carbonic anhydrase (NcoCA) identified in the genome of the Antarctic cyanobacteria belonging to the genus Nostoc. J Enzyme Inhib Med Chem 2016;31:810–7.
- (a) Vullo D, De Luca V, Scozzafava A, et al. The extremo-α-carbonic anhydrase from the thermophilic bacterium Sulfurihydrogenibium azorense is highly inhibited by sulfonamides. Bioorg Med Chem 2013;21:4521–5. (b) Di Fiore A, Capasso C, De Luca V, et al. X-ray structure of the first ‘extremo-α-carbonic anhydrase’, a dimeric enzyme from the thermophilic bacterium Sulfurihydrogenibium yellowstonense YO3AOP1. Acta Crystallogr D Biol Crystallogr 2013;69:1150–9. (c) Luca VD, Vullo D, Scozzafava A, et al. An α-carbonic anhydrase from the thermophilic bacterium Sulphurihydrogenibium azorense is the fastest enzyme known for the CO2 hydration reaction. Bioorg Med Chem 2013;21:1465–9.
- (a) De Luca V, Vullo D, Del Prete S, et al. Cloning, characterization and anion inhibition studies of a γ-carbonic anhydrase from the Antarctic bacterium Colwellia psychrerythraea. Bioorg Med Chem 2016;24:835–40. (b) Vullo D, De Luca V, Del Prete S, et al. Sulfonamide inhibition studies of the γ-carbonic anhydrase from the Antarctic bacterium Colwellia psychrerythraea. Bioorg Med Chem Lett 2016;26:1253–9.
- (a) De Luca V, Vullo D, Del Prete S, et al. Cloning, characterization and anion inhibition studies of a new γ-carbonic anhydrase from the Antarctic bacterium Pseudoalteromonas haloplanktis. Bioorg Med Chem 2015;23:4405–9. (b) Vullo D, De Luca V, Del Prete S, et al. Sulfonamide inhibition studies of the γ-carbonic anhydrase from the Antarctic bacterium Pseudoalteromonas haloplanktis. Bioorg Med Chem Lett 2015;25:3550–5.
- (a) Alterio V, Langella E, Viparelli F, et al. Structural and inhibition insights into carbonic anhydrase CDCA1 from the marine diatom Thalassiosira weissflogii. Biochimie 2012;94:1232–41. (b) Viparelli F, Monti SM, De Simone G, et al. Inhibition of the R1 fragment of the cadmium-containing zeta-class carbonic anhydrase from the diatom Thalassiosira weissflogii with anions. Bioorg Med Chem Lett 2010;20:4745–8.
- (a) Monti SM, Maresca A, Viparelli F, et al. Dithiocarbamates are strong inhibitors of the beta-class fungal carbonic anhydrases from Cryptococcus neoformans, Candida albicans and Candida glabrata. Bioorg Med Chem Lett 2012;22:859–62. (b) De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- (a) Del Prete S, De Luca V, Vullo D, et al. A new procedure for the cloning, expression and purification of the β-carbonic anhydrase from the pathogenic yeast Malassezia globosa, an anti-dandruff drug target. J Enzyme Inhib Med Chem 2016;31:1156–61. (b) Nocentini A, Vullo D, Del Prete S, et al. Inhibition of the β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa with monothiocarbamates. J Enzyme Inhib Med Chem 2017;32:1064–70. (c) Nocentini A, Bua S, Del Prete S, et al. Natural polyphenols selectively inhibit β-carbonic anhydrase from the dandruff-producing fungus Malassezia globosa: activity and modeling studies. ChemMedChem 2018;13:816–23.
- (a) Nocentini A, Cadoni R, Dumy P, et al. Carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani chagasi are inhibited by benzoxaboroles. J Enzyme Inhib Med Chem 2018;33:286–9. (b) Vermelho AB, da Silva Cardoso V, Ricci Junior E, et al. Nanoemulsions of sulfonamide carbonic anhydrase inhibitors strongly inhibit the growth of Trypanosoma cruzi. J Enzyme Inhib Med Chem 2018;33:139–46. (c) Supuran CT. Inhibition of carbonic anhydrase from Trypanosoma cruzi for the management of Chagas disease: an underexplored therapeutic opportunity. Future Med Chem 2016;8:311–24.
- (a) Vullo D, Syrjänen L, Kuuslahti M, et al. Anion inhibition studies of a beta carbonic anhydrase from the malaria mosquito Anopheles gambiae. J Enzyme Inhib Med Chem 2018;33:359–63. (b) Syrjänen L, Kuuslahti M, Tolvanen M, et al. The β-carbonic anhydrase from the malaria mosquito Anopheles gambiae is highly inhibited by sulfonamides. Bioorg Med Chem 2015;23:2303–9.
- Angeli A, Ferraroni M, Supuran CT. Famotidine, an antiulcer agent, strongly inhibits Helicobacter pylori and human carbonic anhydrases. ACS Med Chem Lett 2018;9:1035–8.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- (a) Menchise V, De Simone G, Alterio V, et al. Carbonic anhydrase inhibitors: stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J Med Chem 2005;48:5721–7. (b) Supuran CT, Mincione F, Scozzafava A, et al. Carbonic anhydrase inhibitors—part 52. Metal complexes of heterocyclic sulfonamides: a new class of strong topical intraocular pressure-lowering agents in rabbits. Eur J Med Chem 1998;33:247–54. (c) Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8. (d) Şentürk M, Gülçin İ, Beydemir Ş, et al. In vitro inhibition of human carbonic anhydrase I and II isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–9.
- (a) Krall N, Pretto F, Decurtins W, et al. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5. (b) Rehman SU, Chohan ZH, Gulnaz F, Supuran CT. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzyme Inhib Med Chem 2005;20:333–40. (c) Chohan ZH, Munawar A, Supuran CT. Transition metal ion complexes of Schiff-bases. Synthesis, characterization and antibacterial properties. Met Based Drugs 2001;8:137–43. (d) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal β-class (Cab) and γ-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–8.
- (a) Lee HS, Bae T, Lee JH, et al. Rational drug repositioning guided by an integrated pharmacological network of protein, disease and drug. BMC Syst Biol 2012;6:80. (b) Mori M, Cau Y, Vignaroli G, et al. Hit recycling: discovery of a potent carbonic anhydrase inhibitor by in silico target fishing. ACS Chem Biol 2015;10:1964–9.