
Abstract
Diabetes mellitus (DM) is a global disease with a high incidence of type 2 diabetes. Current studies have shown that insulin enhancers play an important role in the treatment of type 2 diabetes and have great importance in the improvement of type 2 diabetes. In this research, Rosiglitazone was taken as the lead compound, and the structure was modified by using the bioisostere principle, and a new class of 2,4-thiazolanedione compound was designed and synthesised. The novel series of compounds were studied for their biological activities in vitro and in vivo. In vitro tests, the biological activities showed that the target compounds have good selective activation of peroxisome-proliferator-activated receptor γ (PPARγ), such as the compounds 6a, 6e, 6f, 6g and 6i, especially the compound 6e to PPARγ was EC50 = 0.03 ± 0.01 μmol/L in vitro. Then, in vivo biological activities’ test results showed that the tendency of increasing in blood sugar had an obvious inhibiting effect, and had a significant insulin hypoglycaemic effect of enhancing and extending the exogenous. In addition, the results of cytotoxicity tests and acute toxicity tests (LD50) showed that these compounds belong to the low toxicity compounds.
1. Introduction
Diabetes mellitus (DM) is a kind of disease with carbohydrate, protein and fat metabolism disorder caused by multiple aetiologies, which has a high incidence, only after those of cardiovascular disease and tumourCitation1–3. Based on the 16th International Diabetes Federation (IDF) conference in 1997, diabetes could be divided into two broad categories: insulin-dependent diabetes mellitus (IDDM, I diabetes) and non-insulin-dependent diabetes mellitus (NIDDM, 2 diabetes)Citation4–8. The IDDM patients of pancreatic islet β-cells in the body were damagedCitation9–14, the level of insulin in the blood plasma was far lower than the normal value, and most of the patients were under 30 years oldCitation15–19. The disease (1) was mainly treated with insulinCitation20–22 () and its analoguesCitation23–25. The NIDDM patients had less insulin resistance and secretion disorder, normal or slightly lower insulin level in plasma, and the internal target tissue of the body was not sensitive to insulin responseCitation26–30. Due to the relative insufficiency of insulin, hyperglycaemia caused, and most of the patients were over 40 years old. Insulin resistance, especially in the liver and muscle tissue insulin resistance of type 3, plays a very important role in the pathogenesis of diabetesCitation31–33. The NIDDM patients account for about more than 90% of the patients, and the main drugs used for treatment were promoter to insulin secretion, insulin enhancers and α-glucosidase inhibitorsCitation34–36 (). In addition to the main drugs delivery was injection way of insulin, the other was used oral way for all kinds of type 2 diabetes drugs, so it was also called oral medications.
Figure 1. The protein structure of insulinCitation20–22.
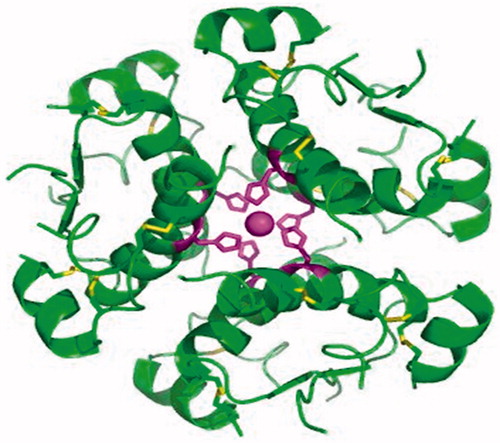
Figure 2. The structure of NIDDM drugs.
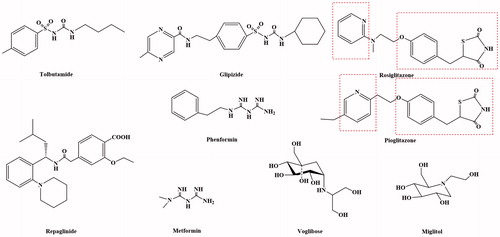
In recent years, the research has shown that the occurrence and development of insulin resistance in type 2 plays an extremely important role. The main reason for insulin resistance was that the binding of insulin antibodies and insulin inhibits the transport of insulin targets, the insulin receptors on the target cells were reduced in hyperinsulinaemia, and the acidosis made the body to become less sensitive to insulin. Therefore, the development and use of drugs that could improve patients' insulin sensitivity and improve the state of insulin resistance were of great significance for the treatment of diabetes. The insulin enhancers used clinically include biguanides (MetforminCitation37 and PhenforminCitation38–42) and thiazolidinediones (RosiglitazoneCitation43–45 and PioglitazoneCitation46–51) (). At present, thiazolidinedione hypoglycaemic drugs were the main type of insulin enhancers. These drugs have all characteristics of thiazolidinedione in terms of chemical structure and could also be regarded as derivatives of phenylpropionic acid. The mechanism of action of these drugs could increase the sensitivity of insulin to the target tissue of the receptor, reduce the production of liver sugars and enhance the glucose uptake in peripheral tissues. The target of its action was the peroxidase-proliferator activated receptor (PPAR) of nucleusCitation52–58. The PPAR had three types: PPARα, PPARβ and PPARγ (). The thiazolidinediones drug could be activated to PPARγ, increase the sensitivity of fat cells, liver cells and skeletal muscle cells to insulin, and promote the uptake, transport and oxidation of insulin target cells to blood sugar. At the same time, it can reduce the content of blood sugar and free fatty acids. In addition, Rosiglitazone also increases glucose uptake by glucose transporters 1 and 4. Rosiglitazone could also improve atherosclerosis and correct lipid disorders. The main adverse effects of Rosiglitazone were elevated liver transaminase levels, mild oedema and anaemiaCitation59.
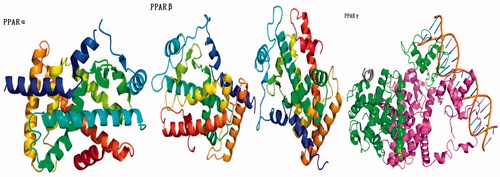
Figure 3. The protein structure of PPARCitation52–58.
By combining the advantages and disadvantages of thiazolidinediones drug in the clinical application, a new type of thiazolidinedione structure was designed with Rosiglitazone as the lead compound and modified by the bioisostere principle (). The newly designed target compounds retain the basic skeleton structure of Rosiglitazone from a chemical perspective, and mainly optimise the structure of benzene ring and its substituents to achieve the design of compounds with good biological activity, low toxic and side effects. These target compounds were synthesised by five steps of acylchlorination, amidation, condensation, knoevenagel and addition reaction (Scheme 1). The synthetic route was characterised by simple operation, high total yield and mild reaction conditions.
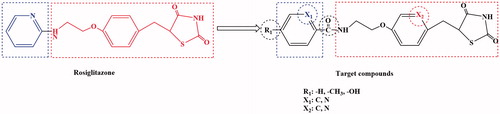
Figure 4. Design of 2,4-thiazolidinedione compounds.
Scheme 1. The synthetic route of 2,4-thiazolidinedione compounds.
2. Results and discussion
2.1. The design and synthesis of target compounds
Rosiglitazone was a thiazolanedione hypoglycaemic agent, which belongs to insulin enhancers that were used to treat type 2 diabetes. A recent studyCitation43–45 showed that Rosiglitazone has a mechanism of action of PPARγ and increases glucose uptake by glucose transporters 1 and 4. The Rosiglitazone could also improve atherosclerosis and correct lipid disorders. The main adverse effects of Rosiglitazone were elevated liver transaminase levels, mild oedema and anaemia. It was against these shortcomings, the Rosiglitazone as the lead compound, designed and synthesised of a new class of 2,4-thiazolanedione compounds for the treatment of type 2 diabetes which was very necessary. The novel series of compounds were designed, and retain the basic skeleton of Rosiglitazone in terms of its chemical structure, namely 2,4-thiazolidinedione and six-membered ring structure (). According to the structure–activity relationship (SAR), the target compounds should have the same treatment effect as Rosiglitazone or be superior to its effect. In the design of the target compounds, the six-membered ring and substituents on the ring were modified. The specific structure modification was selection C atom and N atom on the six-membered ring by using the modified bioisostere principle, and the substituents on the ring selection –H, –CH3 and –OH. Such structural modification was designed to change the logP, pKa and spatial structures of the target compounds in order to obtain the target compounds with good biological activity and little toxic and side effects. In terms of synthetic route, we selected a synthetic route with simple operation, mild reaction conditions and high total yield. The synthesis of the target compounds was of five steps: acylchlorination, amidation, condensation, knoevenagel and addition reaction (Scheme 1). A total of 12 compounds were synthesised and they were analysed by 1H NMR, 13C NMR, MS and elemental analysis. The yield of the target compounds 6a–6f was 69.8–84.2%.
2.2. The biological activities
2.2.1. The biological activity screening in vitro
Drug in vitro experiment was one of the basic contents to evaluate drugs activity, and it was a necessary approach to research drugs. Drugs’ efficacy was often affected by some physical and chemical properties of compounds, such as lipid–water partition coefficient (log p), and dissociation degree (pKa). In this part of the research, the physical and chemical properties of 2,4-thiazolidinedione compounds were studied that were very necessary to study the pharmacodynamics (). In , we could find that target compounds were fat-soluble. The fat-soluble compounds were favourable for drugs absorption, and drugs were easy to enter the cells. This compound the log p was from 1.45 to 3.26. The pKa will affect the absorption and delivery of the drugs, different environmental (acid and alkalinity) medicine may be different, sometimes exists in the form of ions, sometimes exists in the form of molecules. Usually in the form of ion drugs has no effect. Therefore, it was necessary to study the dissociation degree (pKa) of drugs to study the pharmacodynamics. In , we could find that target compounds the pKa were from 8.58 to 10.43, and these compounds could be absorbed in the intestinal tract. After the physical and chemical properties were studied, the biological activity was studied. The in vitro biological activity evaluation was based on the concentration of 50% of maximal effect (EC50), and PPARα, PPARβ and PPARγ were selected (). In , we can find that these compounds had a good biological activity to PPARγ, but inactive to PPARα and PPARβ. These data show that these kinds of target compounds have good selectivity. Especially, as compounds 6a, 6e, 6f, 6g and 6i had better biological activity in vitro, which of compound 6e to PPARγ was EC50 = 0.03 ± 0.01 μmol/l. The in vitro biological activity evaluation was based on the concentration of 50% of maximal effect (EC50), and PPARα, PPARβ and PPARγ were selected (). In , we can found that these compounds had a good biological activity to PPARγ, but inactive to PPARα and PPARβ. These data showed that these kinds of target compounds have good selectivity. Especially, as compounds 6a, 6e, 6f, 6g and 6i had better biological activity in vitro, which of compound 6e to PPARγ was EC50 = 0.03 ± 0.01 μmol/l. After that, the target compounds 6a, 6e, 6f, 6g and 6i had good biological activity and were preliminarily screened out for further study on PPARγ receptor agonist activity (). The experimental results showed that the five target compounds had good biological activity in vitro. According to the structure–activity relationship (SAR), different R1 (–H, –CH3 and –OH) substituents have little effect on EC50, which was mainly affected by the two six-membered ring structures in the target compounds. As could be seen from the data of in vitro experiments, when both six-membered rings were benzene rings, the target compounds had a good biological activity to PPARγ. In addition, when the first six-membered ring was pyridine ring and the second six-membered ring was benzene ring, it also shows a good biological activity to PPARγ.
Figure 5. In vitro activities of PPARγ agonists activity.
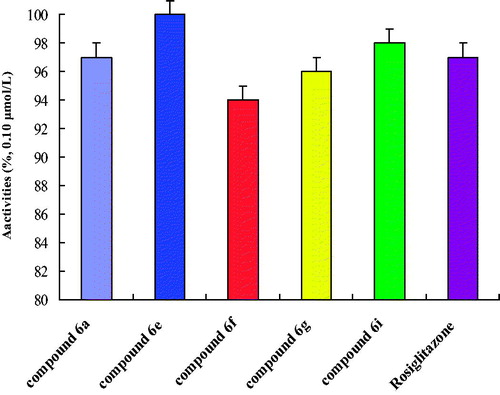
Table 1. In vitro the activation of test compounds on PPARα, PPARβ and PPARγ.
2.2.2. The evaluation of biological activity in vivo
The compounds 6a, 6e, 6f, 6g and 6i were selected from the in vivo activity test, and it was used for in vivo activity evaluation. In vivo experiments, glucose tolerance test, insulin tolerance test, cell survival experiment and acute toxicity were used as the test indicators (, Citation3 and Citation4). In , we chose dose 10 mg kg−1 d−1 and used oral administration. The Rosiglitazone was used as the positive reference substance and DMSO was used as the blank control. The mice were given the drug for 10 d, blood was collected from the tail vein of the mice before and after the sugar load for 1 h and 2 h, and the blood glucose level of the mice before and after the sugar load was measured by the glucometer. The experimental results showed that the compounds 6a, 6e, 6f, 6g and 6i had good resistance activation to glucose. In , experimental results showed that the compounds 6a, 6e, 6f, 6g and 6i had good resistance activation to insulin. In , the results of cell survival experiments show that in the compounds 6a, 6e, 6f, 6g and 6i in the 10−3–10−2 mol L−1, no obvious cytotoxic effect was observed. In , the acute toxicity test results show the oral administration of the compounds, and it belongs to low toxicity compounds.
Table 2. The oral glucose tolerance test.
Table 3. The effect of compounds on blood glucose under insulin load.
Table 4. The cytotoxicity and acute toxicity tests.
3. Conclusion
We report here the design, synthesis and evaluation of a series of novel PPARγ selective activators containing 2,4-thiazolanedione compounds. Rosiglitazone is used as the lead compound, a total of 12 compounds (6a–6l) were designed and synthesised, and used the bioisostere principle to modify the lead compound. We selected five steps to synthesis the target compounds, and the route with high total yield, mild conditions and simple operation. Structure–activity relationship (SAR) studies led to the identification of PPARγ selective activators (the compounds 6a, 6e, 6f, 6g and 6i) with stronger potency and efficacy to activate PPARγ. The compound 6e to PPARγ was EC50 = 0.03 ± 0.01 μmol/L in vitro. The in vivo biological activity test results showed that the tendency of increasing the blood sugar had obvious inhibition, and could significantly enhance and extend the exogenous insulin hypoglycaemic effect. In addition, the results of cytotoxicity test and acute toxicity test showed that these compounds belong to low toxicity compounds.
4. Experimental
4.1. Chemistry section
4.1.1. A general method for synthesis of compounds 2a to 2f
The benzoic acid (1a, 12.03 g, 0.10 mol) was put into 500 ml round bottom flask, then 200 ml of methylene chloride (CH2Cl2) was used as the solvent and 10 drops of N,N’-dimethyl formamide (DMF) used as a catalyst for the reaction were added. The flask was placed in the ice water bath (≤10 °C), and a magnetic mixer was used to stir until the reaction liquid became clarified. The thionyl chloride (SOCl2, 11.25 ml, 0.15 mol) was constantly dropped into the flask under stirring, and the drop speed rate and the reaction temperature were controlled (≤10 °C and ≥20 min). After the reactants were added, the reaction lasted for 6–10 h under reflux. The methylene chloride and excessive thionyl chloride were removed under vacuum. The mixture was dried to get the crude product of benzoyl chloride (2a). The crude product was obtained by atmospheric distillation, the product was collected at 198 °C, and the pure product of benzoyl chloride (2a) as a colourless transparent liquid was obtained. The general method was used to synthesise compounds 2c and 2e as all were colourless transparent liquid. And the general method was used to synthesise crude compounds 2b, 2d and 2f, the crude products were recrystallised with hexane, filtered and dried in vacuum to give pure products of compounds 2b, 2d and 2f as all were of white crystal.
4.1.2. A general method for synthesis of compounds 3a to 3f
The benzoyl chloride (2a, 14.05 g, 0.10 mol) was put into 500 ml round bottom flask, then 200 ml of methylene chloride (CH2Cl2) was added as a solvent for the reaction. The flask was placed in the ice water bath (0–10 °C), and a magnetic mixer was used to stir until the reaction liquid became clarified. The ethanolamine (6.11 g, 0.10 mol) was constantly dropped into the flask under stirring, and the drop speed rate and the reaction temperature were controlled (0–10 °C and ≥20 min). After the reactants were added, the reaction lasted 6 h under standing. The methylene chloride (CH2Cl2) was removed under vacuum. The mixture was dried to get the crude product of N-(2-hydroxyethyl) benzamide (3a). The crude product was recrystallised with toluene, filtered and dried in vacuum to give pure product of 4-chloro-N-(hydroxymethyl) benzamide (3a) as a white crystal. The general method was used to synthesis compound 3b to 3f as all were of white crystal.
4.1.3. A general method for synthesis of compounds 4a to 4l
The compound 3a (16.51 g, 0.10 mol) and the solid potassium hydroxide (5.61 g, 0.01 mol) were put into 500 ml round bottom flask, and 100 ml tetrahydrofuran (THF) was added as the reaction solvent. Under room temperature with magnetic stirring, the 4-chlorobenzaldehyde (14.05 g, 0.10 mol) was added at a constant pressure by a drop funnel, and the rate of droplet acceleration was controlled. When the dripping was completed, under the condition of nitrogen protection, the reaction was refluxed for 10 h. When the reaction was complete, the reaction solution was poured into the ice water, then the ethyl acetate extract was added three times, and added with the superstratum extract. The extract was washed twice with saturated sodium chloride solution and dried with anhydrous calcium chloride. Decompression distillation and recovery of ethyl acetate were done to obtain the crude product of compound 4a. The crude product was recrystallized with toluene filtered, and dried in vacuum to give pure product of compound 4a with a white crystal. The general method was used to synthesise compounds 4b to 4l as all were of white crystal.
4.1.4. A general method for synthesis of compounds 5a to 5l
The compound 4a (26.93 g, 0.10 mol) and 2,4-thiazolanedione (11.71 g, 0.10 mol) were put into 500 ml round bottom flask. The 100 ml of glacial acetic acid was added as the reaction solvent. Ten drops of hexahydropyridine were added as the reaction catalyst, and then 30 ml of toluene was added as the water solvent. The water divider was connected that divided water for 6 h. When the water was separated, the reaction liquid was cooled and then refrigerated for 24 h. The precipitate was precipitated, filtered and the precipitate was washed with ether for three times, and the mixture was vacuum dried to get the crude product of compound 5a. The crude product was recrystallised with toluene to give pure product of compound 5a which is of a white crystal. The general method was used to synthesise compounds 5b to 5l as all were of white crystal.
4.1.5. A general method for synthesis of compounds 6a to 6l
The compound 5a (37.04 g, 0.10 mol) was put into 500 ml round bottom flask, then 200 ml of methanol was added as the solvent. The magnesium powder (24.00 g, 0.10 mol) was added to a round bottom flask, and the refluxing reaction was performed for 12 h with the protection of nitrogen. After the reflux reaction was completed, it was immediately filtrated, the filtrate was collected, and the filtrate was poured into the ice water. The extract was extracted three times with ethyl acetate, combined with the superstratum extract, and washed twice with saturated sodium bicarbonate and saturated sodium chloride solution, and dried with anhydrous calcium chloride after washing. Decompression distillation and recovery of solvent methanol were done to obtain the crude product of compound 5a. The crude product was recrystallized with methanol to give pure product of compound 6a which is of a white powder. The general method was used to synthesise compounds 6b to 6l as all were of white powder.
N-(2–(4-((2,4-dioxothiazolidin-5-yl) methyl) phenoxy)ethyl) benzamide (6a): white powder, yield 84.2%, m.p. 164–166 °C; 1H NMR (300 MHz, DMSO) δ: 3.32 (2H, d, J = 6.1 Hz, –CH2–), 3.48 (2H, t, J = 7.2 Hz, –CH2–), 4.14 (2H, t, J = 7.2 Hz, –CH2–), 4.46 (1H, t, J = 6.1 Hz, –CH–), 6.92 (2H, ddd, J = 8.8, 1.8, 0.5 Hz, Ph-H), 7.11 (2H, ddd, J = 8.8, 1.0, 0.5 Hz, Ph-H), 7.48 (2H, dddd, J = 8.5, 7.5, 1.4, 0.4 Hz, Ph-H), 7.63 (1H, tt, J = 7.5, 1.5 Hz, Ph-H), 7.85 (2H, dddd, J = 8.5, 1.6, 1.5, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 39.4, 39.7, 49.8, 61.2, 115.2, 127.6, 127.9, 128.5, 128.9, 133.7, 1, 158.4, 168.1, 169.1, 175.8; HR-ESI-MS m/z: calcd for C19H18N2O4S{(M + H)+} 370.0986, found 370.4231; Anal. calcd for C19H18N2O4S: C, 61.61; H, 4.90; N, 7.56; O, 17.28; S, 8.65; found: C, 61.62; H, 4.91; N, 7.54; O, 17.27; S, 8.66%.
N-(2-((6-((2,4-dioxothiazolidin-5-yl) methyl) pyridin-3-yl) oxy) ethyl) benzamide (6b): white powder, yield 82.7%, m.p. 171–173 °C; 1H NMR (300 MHz, DMSO) δ: 3.14 (2H, d, J = 6.3 Hz, –CH2–), 3.48 (2H, t, J = 6.5 Hz, –CH2–), 4.15 (2H, t, J = 6.5 Hz, –CH2–), 4.46 (1H, t, J = 6.3 Hz, –CH–), 7.03 (1H, dd, J = 8.0, 0.5 Hz, Py-H), 7.32 (1H, dd, J = 8.0, 1.6 Hz, Py-H), 7.46 (2H, dddd, J = 8.5, 7.5, 1.4, 0.4 Hz, Ph-H), 7.65 (1H, tt, J = 7.5, 1.5 Hz, Ph-H), 7.78 (2H, dddd, J = 8.5, 1.6, 1.5, 0.4 Hz, Ph-H), 8.23 (1H, dd, J = 1.6, 0.5 Hz, Py-H); 13C NMR (75 MHz, DMSO) δ: 30.4, 39.4, 49.8, 61.2, 122.1, 127.8, 128.6, 129.7, 133.7, 142.6, 151.4, 160.4, 168.1, 169.2, 175.7; HR-ESI-MS m/z: calcd for C18H17N3O4S{(M + H)+} 371.0943, found 371.4112; Anal. calcd for C18H17N3O4S: C, 58.21; H, 4.61; N, 11.31; O, 17.23; S, 8.63; found: C, 58.22; H, 4.60; N, 11.32; O, 17.21; S, 8.64%.
N-(2-(4-((2,4-dioxothiazolidin-5-yl) methyl) phenoxy) ethyl) picolinamide (6c): white powder, yield 80.5%, m.p. 174–176 °C; 1H NMR (300 MHz, DMSO) δ: 2.88 (2H, d, J = 6.1 Hz, –CH2–), 3.50 (2H, t, J = 7.1 Hz, –CH2–), 4.17 (2H, t, J = 7.1 Hz, –CH2–), 4.49 (1H, t, J = 6.1 Hz, –CH–), 7.01 (2H, ddd, J = 8.8, 1.8, 0.5 Hz, Ph-H), 7.12 (2H, ddd, J = 8.8, 1.0, 0.5 Hz, Ph-H), 7.56 (1H, ddd, J = 7.6, 5.2, 1.7 Hz, Py-H), 7.94–8.02 (2H, 8.01 (ddd, J = 8.2, 1.7, 0.5 Hz, Py-H), 7.97 (ddd, J = 8.2, 7.6, 1.9 Hz, Py-H)), 8.71 (1H, ddd, J = 5.2, 1.9, 0.5 Hz, Py-H); 13C NMR (75 MHz, DMSO) δ: 39.2, 39.7, 49.8, 60.8, 115.2, 122.6, 123.4, 127.7, 134.0, 137.2, 148.3, 151.7, 158.4, 164.5, 169.0, 175.6; HR-ESI-MS m/z: calcd for C18H17N3O4S{(M + H)+} 371.0942, found 371.4111; Anal. calcd for C18H17N3O4S: C, 58.21; H, 4.61; N, 11.31; O, 17.23; S, 8.63; found: C, 58.22; H, 4.60; N, 11.33; O, 17.22; S, 8.62%.
N-(2-((6-((2,4-dioxothiazolidin-5-yl) methyl) pyridin-3-yl) oxy) ethyl) picolinamide (6d): white powder, yield 73.9%, m.p. 189–191 °C; 1H NMR (300 MHz, DMSO) δ: 3.12 (2H, d, J = 6.3 Hz, –CH2–), 3.49 (2H, t, J = 6.9 Hz, –CH2–), 4.18 (2H, t, J = 6.9 Hz, –CH2–), 4.42 (1H, t, J = 6.3 Hz, –CH–), 7.02 (1H, dd, J = 8.0, 0.5 Hz, Py-H), 7.33 (1H, dd, J = 8.0, 1.6 Hz, Py-H), 7.57 (1H, ddd, J = 7.6, 5.2, 1.7 Hz, Py-H), 7.92–8.02 (2H, 8.01 (ddd, J = 8.2, 1.7, 0.5 Hz, Py-H), 7.97 (ddd, J = 8.2, 7.6, 1.9 Hz, Py-H)), 8.27 (1H, dd, J = 1.6, 0.5 Hz, Py-H), 8.70 (1H, ddd, J = 5.2, 1.9, 0.5 Hz, Py-H); 13C NMR (75 MHz, DMSO) δ: 30.4, 39.2, 49.9, 60.8, 122.0, 122.6, 123.5, 129.7, 137.4, 142.6, 148.4, 151.4, 151.8, 160.4, 164.5, 169.0, 175.7; HR-ESI-MS m/z: calcd for C17H16N4O4S{(M + H)+} 372.0891, found 372.3992; Anal. calcd for C17H16N4O4S: C, 54.83; H, 4.33; N, 15.05; O, 17.18; S, 8.61; found: C, 54.81; H, 4.34; N, 15.06; O, 17.17; S, 8.62%.
N-(2-(4-((2,4-dioxothiazolidin-5-yl) methyl) phenoxy) ethyl)-4-methylbenzamide (6e): white powder, yield 75.1%, m.p. 170–172 °C; 1H NMR (300 MHz, DMSO) δ: 2.32 (3H, s, –CH3), 2.88 (2H, d, J = 6.1 Hz, –CH2–), 3.48 (2H, t, J = 7.2 Hz, –CH2–), 4.15(2H, t, J = 7.2 Hz, –CH2–), 4.47 (1H, t, J = 6.1 Hz, –CH–), 7.01 (2H, ddd, J = 8.8, 1.8, 0.5 Hz, Ph-H), 7.06–7.20 (4H, 7.16 (ddd, J = 8.5, 1.2, 0.5 Hz, Ph-H), 7.11 (ddd, J = 8.8, 1.0, 0.5 Hz, Ph-H)), 7.86 (2H, ddd, J = 8.5, 1.7, 0.5 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 21.4, 39.2, 39.7, 49.8, 60.8, 115.2, 127.5, 127.9, 128.6, 131.0, 139.7, 158.5, 168.3, 169.0, 175.6; HR-ESI-MS m/z: calcd for C20H20N2O4S{(M + H)+} 384.1142, found 384.4503; Anal. calcd for C20H20N2O4S: C, 62.48; H, 5.24; N, 7.29; O, 16.65; S, 8.34; found: C, 62.46; H, 5.25; N, 7.28; O, 16.66; S, 8.35%.
N-(2-((6-((2,4-dioxothiazolidin-5-yl) methyl) pyridin-3-yl) oxy) ethyl)-4-methylbenzamide (6f): white powder, yield 74.2%, m.p. 177–179 °C; 1H NMR (300 MHz, DMSO) δ: 2.32 (3H, s, –CH3), 3.12 (2H, d, J = 6.3 Hz, –CH2–), 3.48 (2H, t, J = 6.5 Hz, –CH2–), 4.18 (2H, t, J = 6.5 Hz, –CH2–), 4.42 (1H, t, J = 6.3 Hz, –CH–), 7.01 (1H, dd, J = 8.0, 0.5 Hz, Py-H), 7.16 (2H, ddd, J = 8.5, 1.2, 0.5 Hz, Ph-H), 7.35 (1H, dd, J = 8.0, 1.6 Hz, Py-H), 7.86 (2H, ddd, J = 8.5, 1.7, 0.5 Hz, Ph-H), 8.26 (1H, dd, J = 1.6, 0.5 Hz, Py-H); 13C NMR (75 MHz, DMSO) δ: 21.2, 30.6, 39.4, 49.8, 60.8, 122.0, 127.5, 128.6, 129.4, 131.0, 139.7, 142.6, 151.5, 160.2, 168.1, 169.0, 175.6; HR-ESI-MS m/z: calcd for C19H19N3O4S{(M + H)+} 385.1094, found 385.4381; Anal. calcd for C19H19N3O4S: C, 59.21; H, 4.97; N, 10.90; O, 16.60; S, 8.32; found: C, 59.23; H, 4.96; N, 10.91; O, 16.60; S, 8.30%.
N-(2-(4-((2,4-dioxothiazolidin-5-yl) methyl) phenoxy) ethyl)-5-methylpicolinamide (6g): white powder, yield 73.1%, m.p. 180–182 °C; 1H NMR (300 MHz, DMSO) δ: 2.32 (3H, s, –CH3), 3.12 (2H, d, J = 6.3 Hz, –CH2–), 3.48 (2H, t, J = 6.5 Hz, –CH2–), 4.18(2H, t, J = 6.5 Hz, –CH2–), 4.42 (1H, t, J = 6.3 Hz, –CH–), 7.01 (1H, dd, J = 8.0, 0.5 Hz, Py-H), 7.16 (2H, ddd, J = 8.5, 1.2, 0.5 Hz, Ph-H), 7.35 (1H, dd, J = 8.0, 1.6 Hz, Py-H), 7.86 (2H, ddd, J = 8.5, 1.7, 0.5 Hz, Ph-H), 8.26 (1H, dd, J = 1.6, 0.5 Hz, Py-H); 13C NMR (75 MHz, DMSO) δ: 17.6, 39.2, 39.7, 49.8, 60.8, 115.0, 122.0, 127.8, 132.5, 134.0, 137.3, 152.0,152.8, 158.6, 164.3, 169.0, 175.6; HR-ESI-MS m/z: calcd for C19H19N3O4S{(M + H)+} 385.1095, found 385.4382; Anal. calcd for C19H19N3O4S: C, 59.21; H, 4.97; N, 10.90; O, 16.60; S, 8.32; found: C, 59.23; H, 4.96; N, 10.90; O, 16.60; S, 8.31%.
N-(2-((6-((2,4-dioxothiazolidin-5-yl) methyl) pyridin-3-yl)oxy) ethyl)-5-methylpicolinamide (6h): white powder, yield 73.1%, m.p. 189–191 °C; 1H NMR (300 MHz, DMSO) δ: 2.26 (3H, s, –CH3), 3.12 (2H, d, J = 6.3 Hz, –CH2–), 3.49 (2H, t, J = 6.9 Hz, –CH2–), 4.18 (2H, t, J = 6.9 Hz, –CH2–), 4.42 (1H, t, J = 6.3 Hz, –CH–), 7.01 (1H, dd, J = 8.0, 0.5 Hz, Py-H), 7.35 (1H, dd, J = 8.0, 1.6 Hz, Py-H), 7.86–7.94 (2H, 7.92 (dd, J = 8.2, 0.6 Hz, Py-H), 7.91 (dd, J = 8.2, 2.0 Hz, Py-H)), 8.26 (1H, dd, J = 1.6, 0.5 Hz, Py-H), 8.46 (1H, dd, J = 2.0, 0.6 Hz, Py-H); 13C NMR (75 MHz, DMSO) δ: 17.6, 30.4, 39.2, 49.8, 60.8, 122.0, 122.2, 129.7, 132.5, 137.2, 142.6, 151.4, 152.0, 152.6 160.4, 164.5, 169.0, 175.6; HR-ESI-MS m/z: calcd for C18H18N4O4S{(M + H)+}386.1048, found 386.4261; Anal. calcd for C18H18N4O4S: C, 55.95; H, 4.70; N, 14.50; O, 16.56; S, 8.30; found: C, 55.93; H, 4.71; N, 14.51; O, 16.55; S, 8.31%.
N-(2-(4-((2,4-dioxothiazolidin-5-yl) methyl) phenoxy) ethyl)-4-hydroxybenzamide (6i): white powder, yield 80.3%, m.p. 192–194 °C; 1H NMR (300 MHz, DMSO) δ: 2.87(2H, d, J = 6.1 Hz, –CH2–), 3.47 (2H, t, J = 7.2 Hz, –CH2–), 4.12 (2H, t, J = 7.2 Hz, –CH2–), 4.45 (1H, t, J = 6.1 Hz, –CH–), 6.97–7.03 (4H, 6.99 (ddd, J = 8.8, 1.8, 0.5 Hz, Ph-H), 7.01 (ddd, J = 8.6, 1.1, 0.4 Hz, Ph-H)), 7.12 (2H, ddd, J = 8.8, 1.0, 0.5 Hz, Ph-H), 7.98 (2H, ddd, J = 8.6, 1.7, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 39.2, 39.7, 49.8, 60.8, 115.0, 115.7 127.5, 127.9, 133.0,134.1, 157.7, 158.6, 168.3,169.0, 175.7; HR-ESI-MS m/z: calcd for C19H18N2O5S{(M + H)+} 386.0933, found 386.4221; Anal. calcd for C19H18N2O5S: C, 59.06; H, 4.70; N, 7.25; O, 20.70; S, 8.30; found: C, 59.04; H, 4.71; N, 7.26; O, 20.71; S, 8.29%.
N-(2-((6-((2,4-dioxothiazolidin-5-yl) methyl) pyridin-3-yl)oxy) ethyl)-4-hydroxybenzamide (6j): white powder, yield 79.1%, m.p. 197–199 °C; 1H NMR (300 MHz, DMSO) δ: 3.12(2H, d, J = 6.3 Hz, –CH2–), 3.47 (2H, t, J = 6.5 Hz, –CH2–), 4.18 (2H, t, J = 6.5 Hz, –CH2–), 4.42 (1H, t, J = 6.3 Hz, –CH–), 6.99 (1H, dd, J = 8.0, 0.5 Hz, Py-H), 7.01 (2H, ddd, J = 8.6, 1.1, 0.4 Hz, Ph-H), 7.35 (1H, dd, J = 8.0, 1.6 Hz, Py-H), 7.98 (2H, ddd, J = 8.6, 1.7, 0.4 Hz, Ph-H), 8.27 (1H, dd, J = 1.6, 0.5 Hz, Py-H); 13C NMR (75 MHz, DMSO) δ: 30.5, 39.2, 49.8, 60.8, 115.6, 122.0, 127.6, 129.7, 133.0,142.6, 151.6, 157.9, 160.4, 168.3, 169.0, 175,7; HR-ESI-MS m/z: calcd for C18H17N3O5S{(M + H)+}387.0887, found 387.4101; Anal. calcd for C18H17N3O5S: C, 55.81; H, 4.42; N, 10.85; O, 20.65; S, 8.28; found: C, 55.83; H, 4.41; N, 10.84; O, 20.665; S, 8.27%.
N-(2-(4-((2,4-dioxothiazolidin-5-yl) methyl) phenoxy) ethyl)-5-hydroxypicolinamide (6k): white powder, yield 77.2%, m.p. 201–203 °C; 1H NMR (300 MHz, DMSO) δ: 2.88(2H, d, J = 6.1 Hz, –CH2–), 3.48 (2H, t, J = 7.1 Hz, –CH2–), 4.17 (2H, t, J = 7.1 Hz, –CH2–), 4.46 (1H, t, J = 6.1 Hz, –CH–), 7.01 (2H, ddd, J = 8.8, 1.8, 0.5 Hz, Ph-H), 7.10 (2H, ddd, J = 8.8, 1.0, 0.5 Hz, Ph-H), 7.71 (1H, dd, J = 8.1, 1.8 Hz, Py-H), 7.87 (1H, dd, J = 8.1, 0.5 Hz, Py-H), 8.46 (1H, dd, J = 1.8, 0.5 Hz, Py-H); 13C NMR (75 MHz, DMSO) δ: 39.2, 39.7, 49.8, 60.8, 114.7, 115.1, 122.0, 127.9, 134.0, 135.3, 151.3, 151.9, 158.6, 164.3, 169.0, 175.6; HR-ESI-MS m/z: calcd for C18H17N3O5S{(M + H)+}387.0887, found 387.4101; Anal. calcd for C18H17N3O5S: C, 55.81; H, 4.42; N, 10.85; O, 20.65; S, 8.28; found: C, 55.83; H, 4.41; N, 10.84; O, 20.665; S, 8.27%.
N-(2-((6-((2,4-dioxothiazolidin-5-yl) methyl) pyridin-3-yl) oxy) ethyl)-5-hydroxypicolinamide (6l): white powder, yield 70.6%, m.p. 211–213 °C; 1H NMR (300 MHz, DMSO) δ: 3.11 (2H, d, J = 6.3 Hz, –CH2–), 3.47 (2H, t, J = 6.9 Hz, –CH2–), 4.18 (2H, t, J = 6.9 Hz, –CH2–), 4.42 (1H, t, J = 6.3 Hz, –CH–), 7.00 (1H, dd, J = 8.0, 0.5 Hz, Py-H), 7.35 (1H, dd, J = 8.0, 1.6 Hz, Py-H), 7.70 (1H, dd, J = 8.1, 1.8 Hz, Py-H), 7.88 (1H, dd, J = 8.1, 0.5 Hz, Py-H), 8.27 (1H, dd, J = 1.6, 0.5 Hz, Py-H), 8.46 (1H, dd, J = 1.8, 0.5 Hz, Py-H); 13C NMR (75 MHz, DMSO) δ: 30.4, 39.2, 49.8, 60.8, 114.8, 119.8,122.1, 129.0, 135.2, 142.6, 151.3, 151.5, 151.9, 160.2, 164.3, 169.0,175.6; HR-ESI-MS m/z: calcd for C17H16N4O5S{(M + H)+}387.0887, found 387.4101; Anal. calcd for C17H16N4O5S: C, 52.57; H, 4.15; N, 14.43; O, 20.60; S, 8.25; found: C, 52.59; H, 4.15; N, 14.45; O, 20.61; S, 8.24%.
4.2. Biological section
4.2.1. Screening of biological activity of compounds in vitro
According to the literatureCitation60, the cells containing PPAR (α, β and γ) were obtained and transfected into 96-well plates the day before for cells culture. Transfection was performed according to the instructions of human PPAR enzyme-linked immunoassay. Various compounds (6a–6l) were dissolved in DMSO and were added after the cells adhered to the wall. The Rosiglitazone was used as the positive control group and DMSO was used as the blank control group during the experiment. After 24 h, the drugs were added, and 20 μl of MTT was added to each well for another 6 h, and the cell liquid was collected by removing the culture medium, and 150 μl of DMSO was added to each hole. And the absorbance value (A) of each hole was measured under the wavelength of 460 nm. The concentration for 50% of maximal effect (EC50, the concentration that can cause the maximum effect of 50%) as an indicator of in vitro screening. EC50 was one of the drugs’ safety indicators, and, in general, the EC50 values were higher and the drug was safer.
4.2.2. In vivo animal study
(1) Glucose tolerance test: The healthy mice (20 ± 2 g) were selected. After 1 week of acclimation, mice with no body surface damage, good general condition and flexible reaction were selected for the experiment. The test compounds were dissolved with DMSO, and were given a dose volume of 0.2 ml 10 g−1, and a dose of 10 mg kg−1, and given by gavage (oral) once a day. The selected mice were randomly divided into 10, each male and female, with Rosiglitazone as the positive control group and DMSO as the blank group. The mice were given the drug for 10 d, and after the last dose, the mice fasted for 12 h. The next day, blood was collected from the tail vein of the mice before and after the sugar load for 1 h and 2 h, and the blood glucose level of the mice before and after the sugar load was measured by the glucometer. (2) Insulin tolerance test: The healthy mice were randomly divided into 10, each male and female, with Rosiglitazone as the positive control group and DMSO as the blank group, given the drug continuously for 15 d. The 12 h of fasting after the last administration, blood was taken from the orbital venous plexus (used to measure the blood glucose level after fasting). Each group was immediately given a subcutaneous injection of 0.2 μg kg−1 of insulin. After 1 h and 2 h of injection, blood was taken again. (3) Cell survival experiment: The HUVEC cells were inoculated on the 96-well plate at the initial concentration of 3 × 103 cells/holes, and 100 μl RPMI 1640 medium were added to each hole. At 5% CO2 and 37 °C cell cultivation in 24 h, every pore solution in different concentrations. The six replicas were performed at each concentration, with Rosiglitazone as the positive control group. And determine the hole under 460 nm wavelength absorbance value (A).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Kitabchi A, Umpierrez G, Miles J, et al. Hyperglycemic crises in adult patients with diabetes. Diabetes Care 2009;32:1335–43.
- Vos T, Flaxman A, Naghavi M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2163–96.
- Kenny C. When hypoglycemia is not obvious: diagnosing and treating under-recognized and undisclosed hypoglycemia. Primary Care Diabetes 2014;8:3–11.
- Cukierman T, Gerstein HC, Williamson JD. Cognitive decline and dementia in diabetes–systematic overview of prospective observational studies. Diabetologia 2005;48:2460–9.
- Yang Y, Hu X, Zhang Q, Zou R. Diabetes mellitus and risk of falls in older adults: a systematic review and meta-analysis. Age Ageing 2016;45:761–7.
- Chiang JL, Kirkman MS, Laffel LMB, Peters AL. Type 1 diabetes through the life span: a position statement of the American Diabetes Association. Diabetes Care 2014;37:2034–54.
- Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010;362:800–11.
- Haw J, Galaviz K, Straus A, et al. Long-term sustainability of diabetes prevention approaches: a systematic review and meta-analysis of randomized clinical trials. JAMA Intern Med 2017;177:1808–17.
- Verrotti A, Scaparrotta A, Olivieri C, et al. Seizures and type 1 diabetes mellitus: current state of knowledge. Eur J Endocrinol 2012;167:749–58.
- Rosberger D. Diabetic retinopathy: current concepts and emerging therapy. Endocrinol Metab Clin North Am 2013;42:721–45.
- Polisena J, Tran K, Cimon K, et al. Home telehealth for diabetes management: a systematic review and meta-analysis. Diabetes Obes Metab 2009;11:913–30.
- MacIsaac R, Jerums G, Ekinci E. Glycemic control as primary prevention for diabetic kidney disease. Adv Chronic Kidney Dis 2018;25:141–8.
- Arguedas J, Leiva V, Wright J. Blood pressure targets for hypertension in people with diabetes mellitus. Cochrane Database Syst Rev 2013;10:CD008277.
- Wild S, Roglic G, Green A, et al. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 2004;27:1047–53.
- Laios K, Karamanou M, Saridaki Z, et al. Aretaeus of Cappadocia and the first description of diabetes. Hormones 2012;11:109–13.
- Stewart W, Ricci J, Chee E, et al. Lost productive time and costs due to diabetes and diabetic neuropathic pain in the US workforce. J Occup Environ Med 2007;49:672–9.
- Cheng J, Zhang W, Zhang X, et al. Effect of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers on all-cause mortality, cardiovascular deaths, and cardiovascular events in patients with diabetes mellitus: a meta-analysis. JAMA Intern Med 2014;174:773–85.
- Pignone M, Alberts M, Colwell J, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes: a position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Diabetes Care 2010;33:1395–402.
- Frachetti K, Goldfine A. Bariatric surgery for diabetes management. Curr Opin Endocrinol Diabetes Obes 2009;16:119–24.
- Kung Y, Du Y, Huang W, et al. Total synthesis of crystalline bovine insulin. Sci Sin 1965;14:1710–6.
- Marglin A, Merrifield R. The synthesis of bovine insulin by the solid phase method. J Am Chem Soc 1966;88:5051–2.
- Blundell TL, Cutfield JF, Cutfield SM, et al. Atomic positions in rhombohedral 2-zinc insulin crystals. Nature 1971;231:506–11.
- Serena G, Camhi S, Sturgeon C, et al. The role of gluten in celiac disease and Type 1 diabetes. Nutrients 2015;7:7143–62.
- Nathan D, Cleary P, Backlund J, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005;353:2643–53.
- Mottalib A, Kasetty M, Mar J, et al. Weight management in patients with Type 1 diabetes and obesity. Curr Diabetes Rep 2017;17:92.
- Ripsin C, Kang H, Urban R. Management of blood glucose in type 2 diabetes mellitus. Am Fam Physician 2009;79:29–36.
- Malik V, Popkin B, Bray G, et al. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: a meta-analysis. Diabetes Care 2010;33:2477–83.
- Emadian A, Andrews R, England C, et al. The effect of macronutrients on glycaemic control: a systematic review of dietary randomised controlled trials in overweight and obese adults with type 2 diabetes in which there was no difference in weight loss between treatment groups. Br J Nutr 2015;114:1656–66.
- Grams J, Garvey W. Weight loss and the prevention and treatment of Type 2 diabetes using lifestyle therapy, pharmacotherapy, and Bariatric Surgery: mechanisms of action. Curr Obes Rep 2015;4:287–302.
- Pozzilli P, Strollo R, Bonora E. One size does not fit all glycemic targets for type 2 diabetes. J Diabetes Investig 2014;5:134–41.
- Krentz A, Bailey C. Oral antidiabetic agents: current role in type 2 diabetes mellitus. Drugs 2005;65:385–411.
- Schulman A, Genio F, Sinha N, et al. "Metabolic" surgery for treatment of type 2 diabetes mellitus. Endocr Pract 2009;15:624–31.
- Colucci R. Bariatric surgery in patients with type 2 diabetes: a viable option. Postgrad Med 2011;123:24–33.
- Dixon J, Roux C, Rubino F, et al. Bariatric surgery for type 2 diabetes. Lancet 2012;379:2300–11.
- Meisinger C, Thorand B, Schneider A, et al. Sex differences in risk factors for incident type 2 Diabetes Mellitus: the MONICA Augsburg Cohort Study. JAMA Intern Med 2002;162:82–9.
- Maria R, Pala L, Mannucci E. Role of insulin in the type 2 diabetes therapy: past, present and future. Int J Endocrinol Metab 2013;11:137–44.
- Shapiro S, Parrino V, Freedman L. Hypoglycemic agents. I. Chemical properties of β-phenethylbiguanide. A new hypoglycemic agent. J Am Chem Soc 1959;81:2220–5.
- McKendry J, Kuwayti K, Rado P. Clinical experience with DBI (Phenformin) in the management of diabetes. Can Med Assoc J 1959;80:773–8.
- Fimognari F, Corsonello A, Pastorelli R, et al. Older age and phenformin therapy: a dangerous association. Intern Emerg Med 2008;3:401–3.
- Ching C, Lai C, Poon W, et al. Hazards posed by a banned drug-phenformin is still hanging around. Hong Kong Med J 2008;14:50–4.
- Weinberg B, Samuel E, Navdeep S. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 2015;11:9–15.
- Pryor R, Cabreiro F. Repurposing metformin: an old drug with new tricks in its binding pockets. Biochem J 2015;471:307–22.
- Gold M, Alderton C, Zvartau H, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer's disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 2010;30:131–46.
- Lewis J. Will 2008 mark the start of a new clinical trial era in gastroenterology. Gastroenterology 2008;134:1289.
- Cantello BCC, Cawthorne MA, Haigh D, et al. The synthesis of BRL 49653 – a novel and potent antihyperglycaemic agent. Bioorganic 1994;4:1181–4.
- Scheen A. Outcomes and lessons from the PROactive study. Diabetes Res Clin Pract 2012;98:175–86.
- Belfort R, Harrison S, Brown K, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 2006;355:2297–307.
- Filipova E, Uzunova K, Kalinov K, et al. Pioglitazone and the risk of bladder cancer: a meta-analysis. Diabetes Ther 2017;8:705–26.
- Colca J, McDonald W, Waldon D, et al. Identification of a novel mitochondrial protein ("mitoNEET") cross-linked specifically by a thiazolidinedione photoprobe. Am J Physiol Endocrinol Metab 2004;286:E252–60.
- Colle R, Larminat D, Rotenberg S, et al. Pioglitazone could induce remission in major depression: a meta-analysis. Neuropsychiatr Dis Treat 2017;13:9–16.
- Doyle C, McDougle C. Pharmacotherapy to control behavioral symptoms in children with autism. Expert Opin Pharmacother 2012;13:1615–29.
- Berger J, Moller D. The mechanisms of action of PPARs. Annu Rev Med 2002;53:409–35.
- Dreyer C, Krey G, Keller H, et al. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992;68:879–87.
- Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990;347:645–50.
- Meirhaeghe A, Amouyel P. Impact of genetic variation of PPARgamma in humans. Mol Genet Metab 2004;83:93–102.
- Zoete V, Grosdidier A, Michielin O. Peroxisome proliferator-activated receptor structures: ligand specificity, molecular switch and interactions with regulators. Biochim Biophys Acta 2007;1771:915–25.
- Chittiboyina A, Venkatraman M, Mizuno C, et al. Design and synthesis of the first generation of dithiolane thiazolidinedione- and phenylacetic acid-based PPARgamma agonists. J Med Chem 2006;49:4072–84.
- Madhavan G, Chakrabarti R, Vikramadithyan R, et al. Synthesis and biological activity of novel pyrimidinone containing thiazolidinedione derivatives. Biol Med Chem 2002;10:2671–80.
- Atanasov A, Wang J, Kramer M, et al. Honokiol: a non-adipogenic PPARγ agonist from nature. Biochim Biophys Acta 2013;1830:4813–9.
- Li Z, Liao C, Ko B, et al. Design, synthesis, and evaluation of a new class of noncyclic 1, 3-dicarbonyl compounds as PPARα selective activators. Biol Med Chem Lett 2004;14:3507–11.